
Abstract
Cytokines serve as a major mechanism of communication between immune cells and are the functional molecules at the end of immune pathways. Abnormalities in cytokines are involved in a wide variety of diseases, including chronic inflammation, autoimmune diseases, and cancer. Cytokines are not only direct targets of therapeutics but also important biomarkers for assessing drug efficacy and safety. Traditionally, enzyme-linked immunosorbent assays (ELISA) were most popular for identifying and quantifying cytokines. However, ELISA is expensive, labor intensive, and low throughput. Here, we report the development of a miniaturized Luminex (Austin, TX) assay platform to establish a panel of high-throughput, multiplexed assays for measuring cytokines in human whole blood. The miniaturized 384-well Luminex assay uses <25% of the assay reagents compared with the 96-well assay. The development and validation of the 384-well Luminex cytokine assays enabled high-throughput screening of compounds in primary cells using cytokines as physiologically relevant readouts. Furthermore, this miniaturized multiplexed technology platform allows for high-throughput biomarker profiling of biofluids from animal studies and patient samples for translational research.
Introduction
Cytokines are important in cell-to-cell communication and cellular signaling. They play key roles in regulating immune responses such as inflammation and defense against microbial pathogens.1,2 The accumulation of cytokine knowledge led to the realization of their value in drug development, either as direct drug targets or as biomarkers.3,4 Cytokines form a complex network with both paracrine and autocrine functions. Many cytokines have closely related or overlapping biological functions, which makes it difficult to interpret the biological significance of the quantity of single cytokines. 5 Therefore, efficient measurement of the levels of multiple cytokines in a variety of samples is becoming more and more important. Information about cytokine profiles allows researchers to determine signaling networks in response to a particular stimulus in cell lines, primary cells, or biological fluids in a disease state. 6
Enzyme-linked immunosorbent assay (ELISA) is a classic method to measure cytokines. 7 It has greatly helped advance our understanding of the biology of cytokines. However, a conventional ELISA usually requires over 100 µL of sample per analyte to be tested. It is also cost ineffective and labor intensive to do multiple ELISA experiments to measure different cytokines from the same sample.
For these reasons, several multiplex technologies have been developed to measure multiple cytokines at the same time in the same assay well. 8 These technologies enable the determination of cytokine profiles in biological samples derived from cell culture, animals, or patients. Several formats exist for multiplexed cytokine assays. Bead-based assays such as the Luminex xMAP Multiplexing Technology (Luminex, Austin, TX), FlowCytomix (eBioscience, San Diego, CA), and BD Cytometric Bead Array 9 rely on different populations of prestained microbeads with unique fluorescence signatures, which can be distinguished by a flow cytometer. Each microbead population is precoated with different capture antibodies that can bind to their target analyte, which then forms a sandwich with labeled secondary antibodies for detection. A second type of technology involves microspotting different capture antibodies to specific positions in each well of a microplate. These assays work like an ELISA in principle, except that light is detected by an imaging system and the intensity of light emitted from individual spots is determined. Examples of this format include Q-Plex (Quansys Biosciences, Logan, UT), SearchLight (Aushon Biosystems, Bellerica, MA), and FAST Quant (Whatman, Pittsburgh, PA). In addition, a plate-based electrochemiluminescent assay was developed by Mesoscale Discovery (MSD, Rockville, MA). Like ELISA, MSD uses a pair of antibodies to capture the analyte. Unlike ELISA, MSD detects signal by electrochemiluminescence, which uses labels that emit light when electrochemically stimulated. In combination with patterned arrays, MSD can be used to measure several cytokines in the same well. In addition, new multiplex technologies are still being developed. For example, SomaLogic (Boulder, CO) developed the SOMAmer protein-binding reagents for the detection of multiple proteins at the same time. CyPlex (Wallingford, CT) is a cartridge-based technology that provides multiplexed test results without multiplexing assays and, therefore, minimizes cross-reactivity. These technologies are being evaluated for different applications in industry and academia.
Since their introduction, much effort has been put into the validation and comparison of these multiplexed cytokine assays.10–13 Many studies have been done to compare the multiplexed assays with the “gold-standard” ELISA assays. Generally, the multiplexed assays show good quantitative agreement.14,15 Over the years, these multiplexed assays are becoming more and more popular, with Luminex being one of the most widely used platforms. A few studies have been done to compare Luminex and other multiplex technologies. The Luminex assays generally have good sensitivity, precision, and accuracy.10,12,13,16 However, with the traditional 96-well Luminex assay, the cost per well is still too high to use the technology for high-throughput structure-activity relationship (SAR) screening in the lead optimization space and for biomarker profiling for translational research.
Therefore, Luminex multiplexed cytokine assays were miniaturized to a 384-well format to increase throughput and reduce sample volume and cost. Using a 4-plex kit from Millipore (Billerica, MA) as an example, a side-by-side comparison was done in 96-well and 384-well formats. Focus was placed on assay sensitivity (lower limit of quantification [LLOQ]), precision (percent coefficient of variation [%CV]), and accuracy (recovery). The 384-well format assay uses <25% of the reagents recommended per well for a 96-well plate. Our data suggest miniaturization to 384-well format can largely keep the sensitivity and accuracy of the Luminex assays with acceptable precision. The development and validation of the 384-well Luminex cytokine assays enabled us to screen compounds using cytokines as physiologically relevant readouts in primary cells. Furthermore, the 384-well format was also suitable in larger multiplexing studies (i.e., 41-plex) for high-throughput biomarker profiling in a variety of biofluids from animal efficacy studies and patient samples.
Materials and Methods
Reagents
The Human Cytokine/Chemokine Magnetic Bead Panel kit was from Millipore (HCYTOMAG-60K). Four cytokines representing four major T helper lineages—IFN-γ (Th1), IL-5 (Th2), IL-10 (Treg), and IL-17A (Th17)—were chosen for condition optimization and whole-blood screening assays. The entire 41-plex panel was used to evaluate performance of the 384-well method with human plasma. All reagents were provided with the kit and were prepared according to the manufacturer’s recommendations. Briefly, the antibody-bead vials were sonicated for 30 s and vortexed for 1 min. Then, 60 µL of bead solutions from each antibody bead vial was added to the mixing bottle, and 2.76 mL of bead diluents was added to bring the final volume to 3.0 mL to make the working bead mix. Standards, quality controls, and serum matrix were reconstituted according to manufacturer’s guidance and were frozen as aliquots at −70 °C. Before each experiment, reconstituted standards were thawed out on ice and serially diluted 1 to 5 in assay buffer as recommended by the manufacturer. Assay buffer was used for background wells. Two quality controls (recombinant protein supplied by the manufacturer and reconstituted in artificial serum) were also included in the study. Each standard point had three replicates, while background and controls had six replicates in each experiment.
Luminex Cytokine Assays in 96-Well Format
Assays in the 96-well format were conducted on filter plates based on the manufacturer’s recommendations. In total, 200 µL of wash buffer was added into each well of a 96-well filter plate (cat. no. MSHVN4B10; Millipore). The plate was sealed and mixed on a plate shaker for 10 min at room temperature (20–25 °C). Wash buffer was removed through a multiwell plate vacuum manifold (cat. no. 5017; Pall Corporation, Port Washington, NY). The bottom of the plate was dried by blotting with a paper towel. Then, 25 µL of each standard, control, and sample was added into each well, 25 µL of serum matrix was added to each standard and controls well, and 25 µL of assay buffer was added to each sample well. The working bead mix was vortexed immediately before use. Next, 25 µL of the mixed beads was added to each well. The plate was then sealed, wrapped with aluminum foil, and incubated with agitation on a plate shaker for 2 h at room temperature. After incubation, liquid was removed from the plate by vacuum. The plate was washed twice with 200 µL of wash buffer each time. At the end of each wash, wash buffer was removed by vacuum. After the second wash, the bottom of the plate was dried by tapping on a paper towel, and 25 µL of detection antibodies was added into each well. The plate was then sealed, wrapped in aluminum foil, and incubated with agitation on a plate shaker for 1 h at room temperature. Next, 25 µL of streptavidin-phycoerythrin was added to each well containing the 25 µL of detection antibodies. The plate was shaken for an additional 30 min at room temperature. Liquid was removed from the plate by vacuum. The plate was washed twice with 200 µL of wash buffer each time. At the end of each wash, wash buffer was removed by vacuum. The bottom of the plate was dried by tapping on a paper towel. Then, 150 µL of sheath fluid (cat. no. 40-50000; Luminex) was added to each well. The beads were resuspended on a plate shaker for 5 min and read on a Bio-Plex 3D instrument (Bio-Rad, Hercules, CA). The instrument was set to collect at least 50 beads per analyte. The raw data were measured as mean fluorescence intensity (MFI).
Automated Luminex Cytokine Assays in 384-Well Format
In total, 50 µL of wash buffer was added into each well of a black-sided clear-bottom 384-well plate (781096; Greiner, Monroe, NC) by a Multidrop Combi Reagent Dispenser (Thermo Scientific, Waltham, MA). The plate was sealed by a PlateLoc plate sealer (cat. no. G5402-90001; Velocity 11, Santa Clara, CA) and shaken on a Big Bear plate shaker (HT-91002-1.0; Big Bear Automation, Santa Clara, CA) at 2000 rpm for 10 min at room temperature (20–25°C). Wash buffer was removed by a BioTek 406 magnetic washer (ref. no. 406PSUB3SB; BioTek, Winooski, VT). Each standard or control was mixed with an equal volume of serum matrix in an intermediate 384-well clear round-bottom Matrix plate (cat. no. 4312; Thermo Scientific). Samples were mixed with an equal volume of assay buffer on the intermediate plate. Then, 12 µL of each premixed standard, control, or test sample was transferred to the assay plate by a Matrix PlateMate Plus Automated Pipetting System (Thermo Scientific). The working bead mix was vortexed immediately before use. Then, 6 µL of the mixed beads was added to each well by a Tempest V.2 Liquid Handler (part no. 7368; Formulatrix, Bedford, MA). The plate was sealed with a PlateLoc plate sealer and shaken on a Big Bear plate shaker at 2000 rpm for 2 h at room temperature. After incubation, the content was washed by a BioTek 406 magnetic plate washer twice with 50 µL of wash buffer each time. The magnetic beads were allowed to settle for 1 min before aspiration. The dead volume after aspiration in each well was ~3 µL. After the second wash, 6 µL of detection antibodies was added into each well by the Tempest V.2 Liquid Handler. The plate was sealed with a PlateLoc plate sealer and shaken on a Big Bear plate shaker at 2000 rpm for 1 h at room temperature. Next, 6 µL of streptavidin-phycoerythrin (PE) was added to each well containing the detection antibodies. The plate was shaken for an additional 30 min at room temperature. The plate was then washed by the BioTek washer twice with 50 µL of wash buffer each time. At the end of the second wash, 80 µL of sheath fluid (cat. no. 40-50000; Luminex) was added to each well. The beads were resuspended on a plate shaker for 5 min and read on a Bio-Plex 3D instrument (Bio-Rad). The instrument was set to collect at least 50 beads per analyte. The raw data were measured as MFI.
Whole-Blood Assay
In total, 100 nL of serially diluted test compound in DMSO was dosed to each well of a 384-well clear round-bottom plate (REMP) by the Labcyte (Sunnyvale, CA) Echo Liquid Handling Platform. The same volume of DMSO was added in control wells. Human whole blood was collected from healthy individuals in sodium heparin–containing plasma collection tubes (367874; BD Bioscience, San Jose, CA). Then, 45 µL of whole blood was added to each well. The plate was shaken on a plate shaker for 1 min to mix the compound and the blood and incubated in a humidified incubator with 5% CO2 at 37 °C for 1 h. Next, 5 µL of RPMI medium containing 10 µg/mL of anti-CD3 and anti-CD28 antibodies (in house) was added to each well, to give a final concentration of 1 µg/mL of each antibody. The reaction was incubated in a humidified incubator with 5% CO2 at 37 °C for 48 h. The assay plate was spun at 2000 rpm for 5 min. Then, 15 µL of plasma from each well was transferred to a new 384-well clear round-bottom plate for cytokine measurement as described above.
Data Analysis
Raw data were converted by a Bio-Plex Results Generator and analyzed in Bio-Plex Manager 6.1 (Bio-Rad). Standard curves were generated by five parameter logistic regression. Limits of quantification were determined using the lowest or highest standard point with a recovery of 70% to 130% and a percent CV (%CV= 100 × standard deviation/average) of less than 25%.
Inhibition of cytokine production by compounds in whole blood was plotted as percent inhibition of the average positive control signal, which is DMSO control plus anti-CD3/CD28, over average negative control signal, which is DMSO plus medium. The IC50 was defined as the concentration of test compound corresponding to 50% inhibition derived from the 11-point fitted curve as determined using a four-parameter logistic regression model.
Results and Discussion
Evaluation of Assay Sensitivity and Dynamic Range
To develop and validate a miniaturized Luminex multiplexed cytokine assay in a 384-well format, a 4-plex kit containing reagents that can detect interleukin (IL)−17A, IL-5, IL-10, and interferon (IFN)−γ was used. The Bio-Plex Manager 6.1 software was used to plot standard curves and calculate observed concentrations. A 6-point standard curve was generated with a detection range of 10,000 to 3.2 pg/mL for each analyte (
Assay Dynamic Range and Sensitivity.
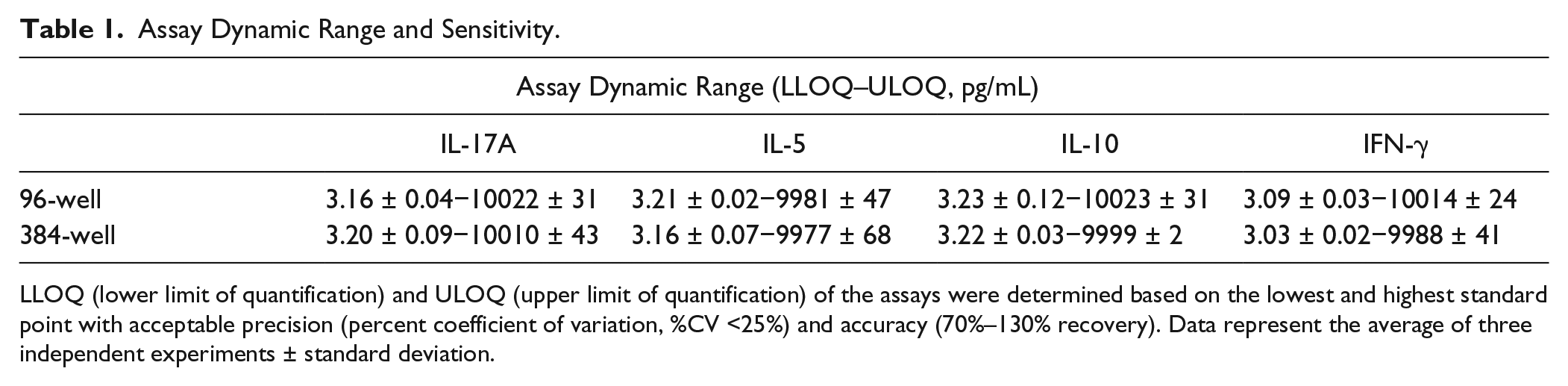
LLOQ (lower limit of quantification) and ULOQ (upper limit of quantification) of the assays were determined based on the lowest and highest standard point with acceptable precision (percent coefficient of variation, %CV <25%) and accuracy (70%–130% recovery). Data represent the average of three independent experiments ± standard deviation.
Evaluation of Assay Accuracy
Quality controls were supplied by the manufacturer in the kit and were prepared as described in the manual. Quality controls were thawed out and freshly prepared immediately before each experiment. The concentrations of the low-quality controls for IL-17A, IL-5, IL-10, and IFN-γ were 154, 165, 146, and 160 pg/mL, respectively, for the lot used. The concentrations of the high-quality controls for IL-17A, IL-5, IL-10, and IFN-γ were 825, 840, 776, and 871 pg/mL, respectively, for the lot used. All the quality control samples were well within the assay working ranges (
The concentrations of the low- and high-quality controls were determined from the standard curves (
Fig. 1A
,
B
and
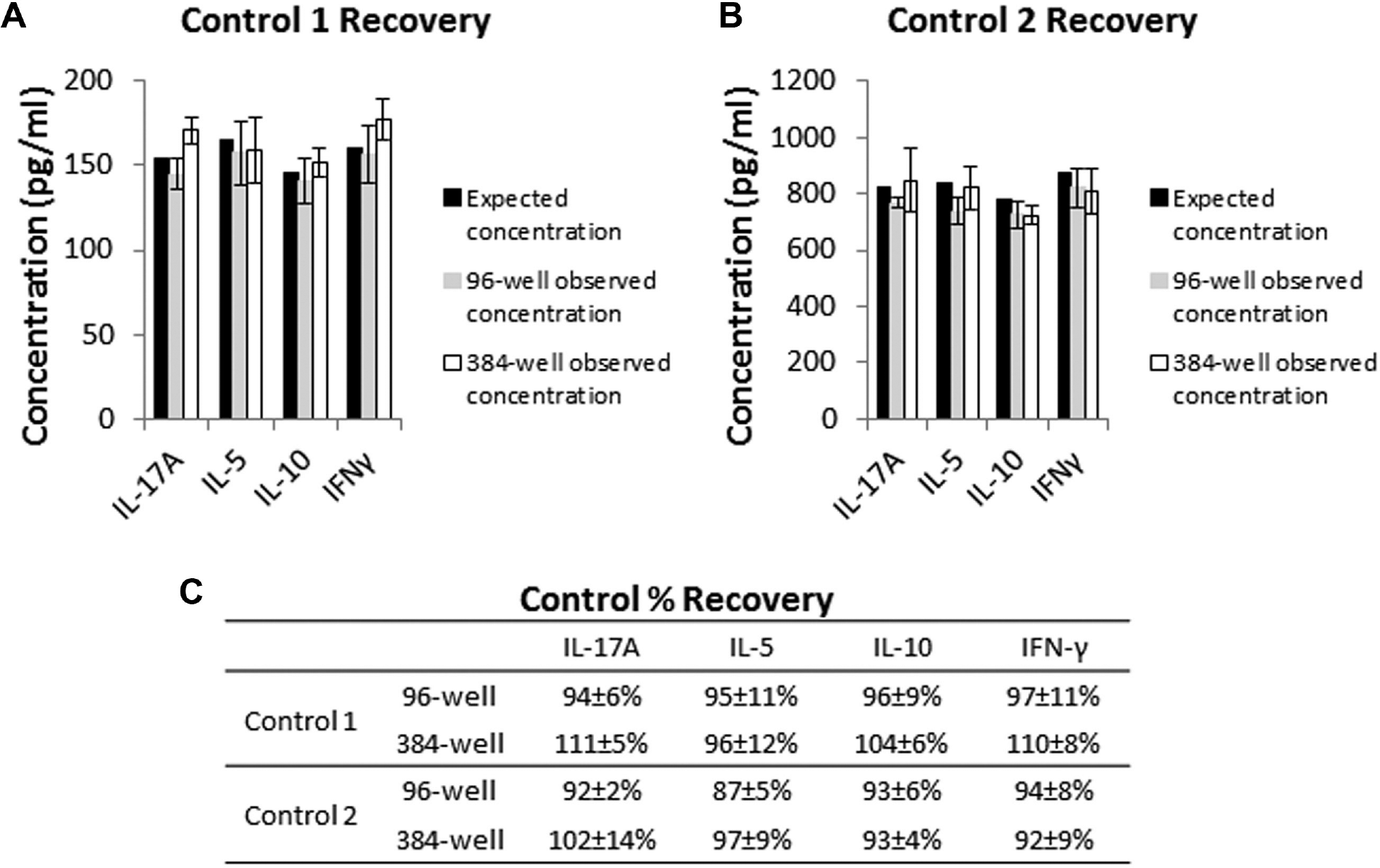
Comparison of assay accuracy in 96-well and 384-well formats. (
Evaluation of Assay Precision
Assay miniaturization may increase assay variation. Thus, assay precision (% CV of controls) was carefully monitored during the development of a 384-well Luminex multiplexed cytokine assay method. Several measures were taken to enhance the precision of the 384-well assay to similar levels as 96-well assays (
Intra-assay precision was calculated as the %CV among mean fluorescent intensity (MFI) values of six replicate wells of the quality controls. Interassay precision was calculated as the %CV of the observed concentration of spike controls from three independent assays. In all three independent experiments, the intra-assay %CV for all analytes in both the 96-well and 384-well formats was well below the 25% acceptance criteria (
Fig. 2A
,
B
). When the %CV of the same analyte from the same experiment was compared between 96-well and 384-well formats, the variations were both within acceptable limits (
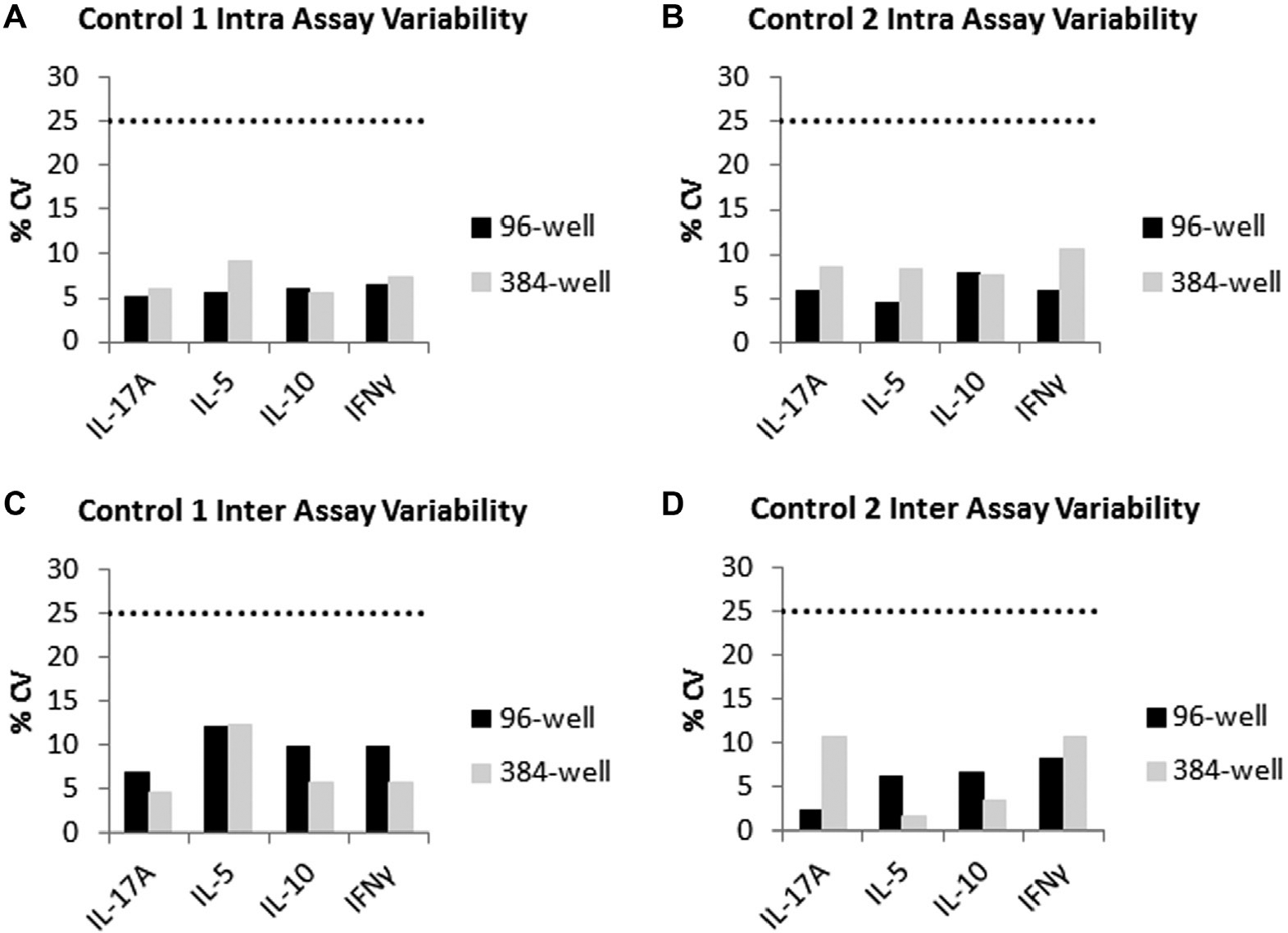
Comparison of assay precision in 96-well and 384-well formats. (
Evaluation of the Performance of the 384-Well Assay with Human Plasma in an Expanded 41-Plex Panel
The validity of the modified protocol should also be determined by its performance in measuring biomarkers in biological specimens. To confirm the 384-well method is valid, a head-to-head comparison of the modified 384-well protocol and the standard 96-well protocol was conducted using human plasma. Human whole blood was stimulated with anti-CD3 and anti-CD28 antibodies. Plasma samples were collected after 48 h of incubation and split into two parts, one for the 384-well assay and one for the 96-well assay. To test the 384-well format with larger multiplexed panels, a panel consisting of a total of 41 analytes was used.
Samples from five donors were run in both the 96-well and 384-well formats. When concentrations of the same analyte in the same sample were compared with each other, the correlation between the 96-well format and the 384-well format was very good. In a linear regression model of the log values of the back-calculated concentrations, the R2 value was >0.98 (
Fig. 3
and
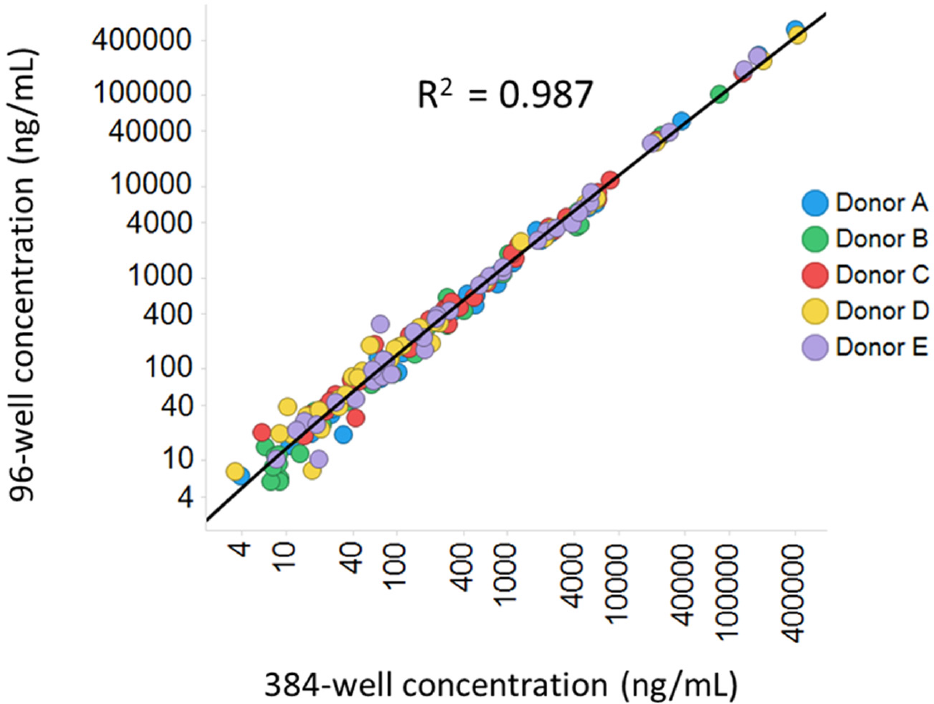
Comparison of 96-well and 384-well assay performance in an expanded panel using human plasma samples. Human whole blood from five donors was stimulated with 1 µg/mL of anti-CD3 and anti-CD28 antibodies for 48 h. Plasma was collected and run through a 41-plex panel in either a 96-well or a 384-well format. Samples were run at two dilutions, neat or 1:100. Only dilution-adjusted concentrations in range were used for comparison. When both dilutions were in range, data from neat samples were used. Each dot represents one analyte in one sample.
Application of the Automated and Miniaturized Luminex Multiplexed Assays for the Analysis of Plasma Samples from Whole Blood for Lead Optimization
Whole-blood assays are physiologically relevant systems that can provide a better clinical comparison than cell lines. Whole-blood assays have been widely used for assessing compound efficacy and/or safety in drug discovery. Cytokine/chemokine levels in plasma derived from whole blood are important end points. However, the high cost and low throughput of traditional cytokine measurement methods such as ELISA have prevented the extensive use of whole-blood assays for high-throughput SAR screening. The miniaturized Luminex assays have good sensitivity, precision, and accuracy, giving us an opportunity to screen a relatively large number of compounds in the format of dose-response curves in whole-blood derived samples using secreted cytokines as readouts. The multiplexed format enabled measuring multiple analytes in the same well at the same time, which is ideal for evaluating selectivity.
To test whether the miniaturized assays can be used to support high-throughput lead optimization screening, plasma samples derived from human whole blood stimulated with 1 µg/mL of anti-CD3 and CD-28 antibodies were used to measure the four Th cytokines. Three independent experiments with blood from three different donors were conducted. All four cytokines showed significant induction after 48 h of stimulation in all three donors. Among the three donors, IL-17A and IL-5 were induced from below LLOQ (<3.2 pg/mL) to roughly 20 to 40 pg/mL. The baseline concentration of IL-10 was close to the LLOQ. Assay donor 1 was below LLOQ (<3.2 pg/mL). Donors 2 and 3 had levels just above the LLOQ (4.3 and 8.4 pg/mL, respectively). After 48 h of anti-CD3 and anti-CD28 induction, IL-10 levels were induced to 130 to 160 pg/mL in the three donors. Background levels of IFN-γ were higher than the other analytes. The three donors had background levels of IFN-γ from 4 to 260 pg/mL. IFN-γ was also very responsive to anti-CD3/CD28 stimulation and was induced to 1 to 5 ng/mL. Overall, all four analytes showed robust and consistent induction ( Fig. 4A ).
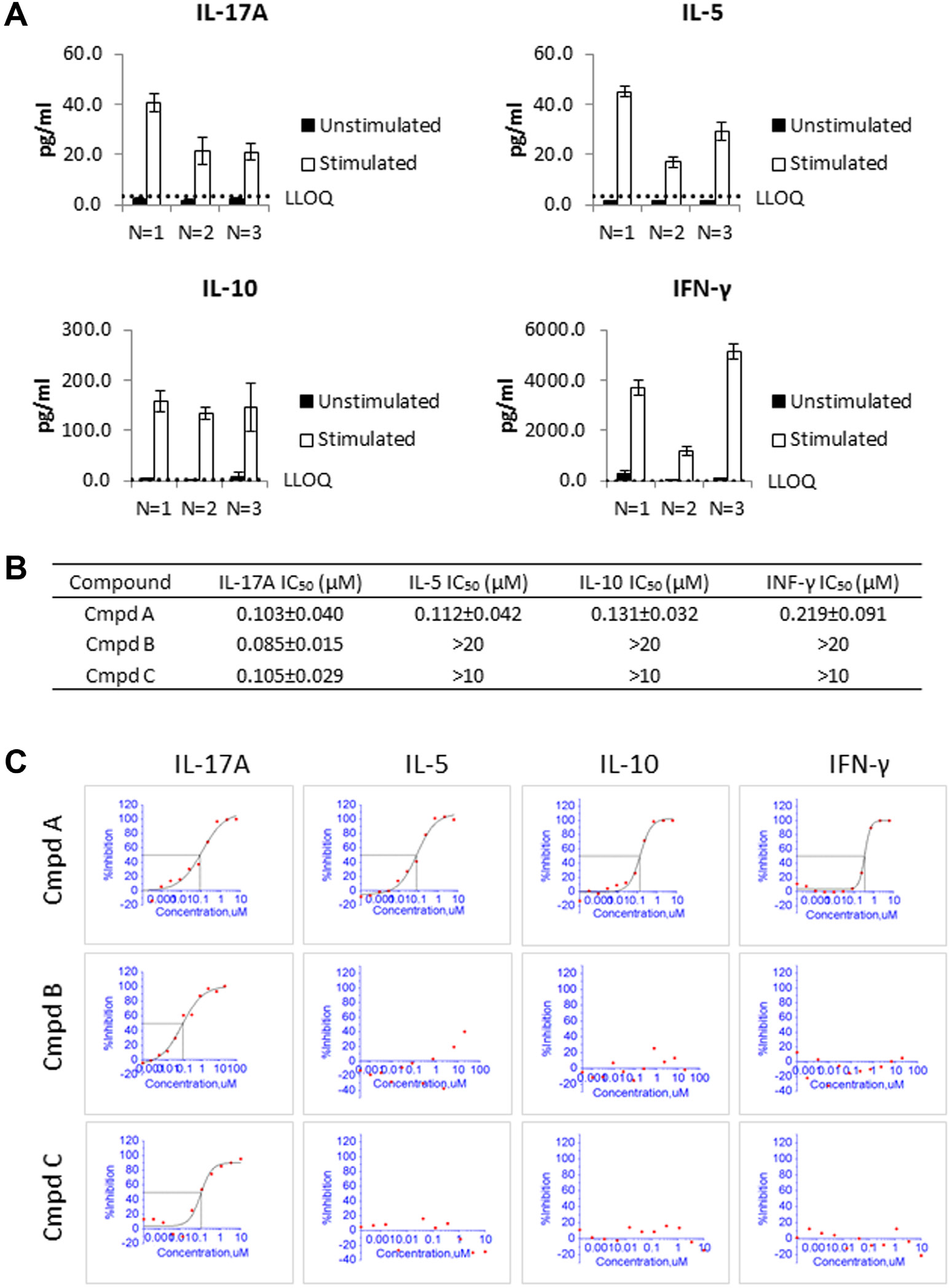
Application of the 384-well format Luminex assay to screen compounds for potency and selectivity. (
Miniaturization reduced the cost to run multiple analytes and automation increased the throughput of the assay by at least 10-fold. This miniaturized multiplex capability allows for analysis of compound potency against a target as well as selectivity against other related targets. For example, the IL-17A, IL-5, IL-10, and IFN-γ 4-plex assay panel contains four cytokines representing four major lineages of T helper cells, Th17, Th2, Treg, and Th1, respectively. 1 This assay could therefore be used to evaluate compounds that can inhibit the activation of some or all of these four T helper lineages. To test the utility of the assay panel, three reference compounds were tested to inhibit the secretion of the four cytokines induced by anti-CD3 and anti-CD28 stimulation. One compound (compound A) was an inhibitor of a major node in the signaling pathway downstream of the T-cell receptor, while the other two compounds (compounds B and C) were inhibitors of a regulator of the Th17 pathway. When the compounds were serially diluted and added to whole blood before stimulation, it was observed that compound A had similar potency across the four readouts, while the other two compounds were potent against only IL-17 secretion without affecting the secretion of IL-5, IL-10, and IFN-γ ( Fig. 4B , C ). These observations suggest that this assay panel provides a tool to evaluate compound potency and selectivity against four major T helper lineages in whole blood.
In summary, a miniaturized and automated Luminex platform with robust sensitivity, accuracy, and precision was established. The 384-well Luminex multiplexed cytokine assay platform enabled us to establish a 4-plex T helper panel using cytokines as readouts, which made screening compounds for potency and selectivity in the same assay well possible. The method can also be used to develop assays using other cytokines as physiologically relevant readouts in primary cells to drive SAR analysis. Furthermore, the miniaturized Luminex assays can be used for applications other than cell-based SAR analysis. The low cost of the miniaturized 384-well format Luminex assay makes it amenable to large-scale cytokine profiling of samples from animal in vivo studies and from human patients for systemic translational research and high-throughput biomarker profiling.
Footnotes
Acknowledgements
The authors would like to thank Jonathan Lippy, Peter Chase, Mark Daywalt, and Garrett Kolodin for automation support. We are appreciative of Murali Dhar for providing compounds for these experiments and Qihong Zhao for whole blood data discussion. We are sincerely grateful to Zhuyin Li for continuous improvement of this Luminex platform.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.