
Abstract
We previously developed an assay of cytotoxic T-lymphocyte lytic granule exocytosis based on externalization of LAMP-1/CD107A using nonphysiological stimuli to generate maximal levels of exocytosis. Here, we used polystyrene beads coated with anti-CD3 antibodies to stimulate cells. Light scatter let us distinguish cells that contacted beads from cells that had not, allowing comparison of signaling events and exocytosis from stimulated and unstimulated cells in one sample. Bead stimulation resulted in submaximal exocytosis, making it possible to detect compounds that either augment or inhibit lytic granule exocytosis. Coupled with the assay’s ability to distinguish responses in cells that have and have not contacted a stimulatory bead, it is possible to detect three kinds of compounds: inhibitors, stimulators, which cause exocytosis, and augmenters, which enhance receptor-stimulated exocytosis. To validate the assay, we screened a set of synthetic compounds identified using our previous assay and a library of 320 extracts prepared from tunicate-associated bacteria. One of the extracts augmented exocytosis threefold. Activity-guided fractionation and structure elucidation revealed that this compound is the known PKC activator teleocidin A-1. We conclude that our modified assay is suitable for screening synthetic compound plates and natural product collections, and will be useful for identifying immunologically active small molecules.
Introduction
Immunomodulation, including immune suppression and/or immune enhancement, has become an important target for drug discovery for conditions ranging from autoimmune diseases and organ transplants to vaccine adjuvants and immunodeficiency disorders. 1 Assays capable of simultaneously measuring immune suppression and enhancement would be of great value in the search for new small-molecule immunomodulatory agents. Previously, we designed a phenotypic assay based on cytotoxic T-lymphocyte (CTL) lytic granule exocytosis 2 and used it in a high-throughput screening (HTS) campaign to look for immune inhibitors using the Molecular Library Small Molecule Repository (MLSMR). 3 In that assay, we detected exocytosis via binding to CTLs of a fluorescently labeled antibody against lysosome-associated membrane protein 1 (LAMP-1; also known as CD107a4–6) and used soluble small-molecule thapsigargin (TG) and phorbol myristate acetate (PMA) that bypass the T-cell receptor (TCR) to achieve maximal cell stimulation. Our screen of the MLSMR identified 48 confirmed previously unknown immunosuppressive compounds acting via a variety of molecular mechanisms.
While our previous assay was clearly useful, it had two inherent weaknesses. First, it relied on TCR-independent nonphysiological stimuli. That meant that it could not detect inhibitors that act downstream of the TCR and upstream of phospholipase C activation, limiting the kinds of immunosuppressive compounds that it can detect. Second, because responses triggered by TG and PMA are essentially maximal, the assay can only detect inhibitors. It is not able to identify compounds that augment immune responses. However, there is reason to believe that compounds that augment immune function could be useful therapeutically. For example, immune modulatory drugs (IMiDs) such as lenalidomide7,8 and imiquimod9,10 have proven useful in the treatment of multiple myeloma and skin cancer.
Physiologically, CTLs are stimulated by contact with target cells. 11 In previous work, we have used polystyrene beads coated with anti-CD3 beads as a surrogate for target cells to stimulate CTL responses (e.g., Grybko et al. 12 ). The beads are commercially available and commonly used to stimulate mouse and human lymphocytes, including primary cells. We therefore decided to explore the use of anti-CD3 beads to stimulate TALL-104 cells in a modified version of our assay, reasoning that it would be easier to incorporate beads into the assay workflow than to use target cells. In the present communication, we show that it is possible to identify cells that have bound to a bead by virtue of the beads’ light scatter properties. This allows comparison of responses within a single sample from cells that have or have not contacted a bead. Importantly, responses to anti-CD3 beads are significantly smaller than responses triggered by TG and PMA, but still sufficiently robust that the assay can be used for screening. This gives the new assay format the ability to detect both exocytosis-inhibiting and exocytosis-enhancing compounds. When coupled with the ability to analyze signals from cells that have and have not contacted a stimulatory bead, the assay has the ability to distinguish compounds that stimulate lytic granule exocytosis on their own from compounds that enhance lytic granule exocytosis stimulated via TCR engagement, making the assay capable of detecting three kinds of compound activity. The assay can be used to screen both synthetic compound collections and crude natural product extracts, and can be used in activity-guided isolation efforts. Altering lytic granule exocytosis may result directly in immune-modulating effects. Furthermore, because key elements of signaling underlying activation of immune cells like CTLs are relatively conserved,13,14 compounds that affect lytic granule exocytosis might also affect the activity of other immune cell types. Thus, screening based on lytic granule exocytosis might provide a means of identifying multiple kinds of novel immunomodulatory compounds from a variety of sources.
Materials and Methods
Chemicals, Cells, and Solutions
TALL-104 human leukemic CTLs were maintained as described previously. 3 Experimental saline (ES) was composed of 155 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM HEPES, and 10 mM glucose. The pH was adjusted to 7.4 with NaOH, and 2% bovine serum albumin (BSA) added, except when noted. Dynabeads coated with anti-CD3 or anti-CD8 monoclonal antibody (Thermo Fisher, Waltham, MA) were washed in ES according to the manufacturer’s protocol, and then resuspended beads were added to cell suspension at a ratio of one bead per cell. Alexa647-conjugated anti-LAMP antibody (0.5 µg/mL final concentration) was purchased from BD Pharmingen (San Jose, CA). Phorbol 12-myristate 13-acetate (PMA) was from Alexis Biochemicals (San Diego, CA). Thapsigargin (TG) was from AdipoGen (San Diego, CA). Paraformaldehyde 16% (PFA) was from Alfa Aesar (Ward Hill, MA) and was diluted to 2% in phosphate-buffered saline (PBS). 1-(4-Methoxyphenyl)-3-[(5-phenyl-1,3,4-oxadiazol-2-yl)sulfanyl]pyrrolidine-2,5-dione was obtained from Chembridge (San Diego, CA). 2-N-[(2-Methoxyphenyl)methyl]-4-N-[(4-propan-2-ylphenyl)methyl]thieno[3,2-d]pyrimidine-2,4-diamine was synthesized by Dr. Jeff Aubé, then at the University of Kansas.
Flow Cytometry
All flow cytometry experiments were conducted on a two-laser BD FACSCalibur at the University of Connecticut’s Flow Cytometry Facility. Data were analyzed offline using FlowJo software (Tree Star, Inc., Ashland, OR)
Intracellular Calcium Measurements
TALL-104 cells were loaded with 1 µM Fluo-4 AM (Molecular Probes, Eugene, OR) in culture medium at room temperature for 25 min, washed to remove excess dye, and resuspended in ES without 2% BSA. Anti-CD3 or anti-CD8 beads were added, or cells were left untreated, and samples were analyzed cytometrically at selected time points over 1 h.
Intracellular p-ERK Staining
ES-washed TALL-104 cells were treated with anti-CD3 or anti-CD8 beads and incubated for 50 min at room temperature with constant rotation. After incubation, cells were fixed with 2% PFA and permeabilized by the addition of 1 mL of ice-cold methanol. Immunocytochemistry was carried out as previously described. 3 The primary antibody was rabbit anti-p44/p42 MAPK (Cell Signaling, Danvers, MA; 4370), and the secondary antibody was Cy5-conjugated goat anti-rabbit (Jackson ImmunoResearch, West Grove, PA; 111-175-045). After staining, cells were washed in FACS buffer and samples were analyzed on FACSCalibur.
Plate Format Assay and Screening Condition
For screening, 1 µL of bacterial extracts (stock concentration of 1 mg/mL in DMSO) or DMSO was added to test plates (Corning 3603) prior to cell addition. One hundred microliters of ES-washed TALL-104 cells (2.5 × 106/mL) was added to each well, mixed with DMSO or bacterial extracts, and incubated at 37 °C for 30 min. Ten microliters of stimulation solution (ES supplemented with 20 µM TG and/or 1 µM PMA, anti-CD3 beads, anti-LAMP) or control solution (ES + 4% DMSO, anti-CD8 beads, anti-LAMP) was added and mixed, and the plate was incubated for an additional 50 min in the dark at room temperature with constant rotation. One hundred microliters of 2% PFA was added to each well, and samples were transferred to flow tubes for cytometric analysis.
PKC Activity Assay by Immunocytochemistry
TALL-104 cells were resuspended in culture medium and treated with PMA or compound E1 at 9 concentrations ranging in a threefold dilution series from 0.01 nM to 200 nM for 30 min at 37 °C, followed by 50 min at room temperature with constant rotation. Cells were fixed with 2% PFA and permeabilized with ice-cold methanol. Immunocytochemistry was carried out as previously described. 3 Primary antibody was rabbit anti-active PKC substrate (Cell Signaling 2261), and secondary antibody was Alexa488-conjugated donkey anti-rabbit (Jackson 711-545-152). Values of geometric mean of fluorescence at different concentrations were fitted to the Hill equation.
LAMP/BLT Esterase Assays
Combined assays of LAMP and serine esterase release were performed as described previously. 3
Natural Product Library Generation
Bacteria were isolated from marine tunicate specimens collected in Long Island Sound, CT, the Bahamas, and the Republic of Panama. Tunicates were collected by scuba diving or from shallow water dock lines and rinsed or sterilized prior to homogenation. Homogenated biomass was then streaked onto plates made from one of several bacterial isolation media and cultured at room temperature. Morphologically distinct colonies were isolated on agar plates, grown in liquid culture for extraction, and cryogenically stored suspended in glycerol solution. Large-scale cultures for extraction were obtained via two methods, with early cultures being fermented for 2 weeks in sterile marine broth (Difco, Detroit, MI) in a shaking incubator (200 rpm; 30 °C; New Brunswick Scientific, Edison, NJ). Later cultures were fermented in sterile marine broth using a three-step process involving inoculating small cultures, growing to confluency in a shaking incubator over 3 days, followed by inoculation of medium cultures grown for 3 days and ultimately resulting in inoculation of large-scale cultures grown for 3 days prior to chemical extraction. Once fermentation was completed, 75 mL of prewashed Diaion HP-20 beads (Supelco, Bellefonte, PA) was added to each culture flask and allowed to shake at room temperature for 18 h (135 rpm). The resulting culture and beads were vacuum filtered with a vacuum frit filter. Beads and cells were extracted with consecutive washes of methanol and acetone, and the resulting eluate was concentrated in vacuo to dryness. A portion of the extract was suspended in DMSO and plated in deep 96-well plates for storage prior to screening.
Isolation and Identification of Teleocidin A-1
Teleocidin A-1 was isolated from Streptomyces sp. strain AVP053U2, isolated from the tunicate Styela clava, collected from shallow water dock lines at Avery Point, CT, in May 2011 (41° 18.975 N, 72° 3.647 W). The bacterium was isolated from ISP4 agar, replacing water with Instant Ocean (United Pet Group, Blacksburg, VA), and cultured at room temperature. The morphology of AVP053U2 resembled an actinobacterium, with a yellow base and green spores. AVP053U2 was subjected to whole genome sequencing (deMayo, J. A., Maas, K. R., Klassen, J. L., Balunas, M. J., unpublished) and determined to be a new strain from the genus Streptomyces. Streptomyces sp. AVP053U2 was cultured for 14 days in 500 mL of marine broth and extracted as described above. The organic extract was initially fractionated via C18 reversed-phase (RP) solid-phase extraction chromatography (Supelco) using a stepwise MeOH:H2O gradient as follows: 10% MeOH:H2O (fraction A), 25% MeOH:H2O (fraction B), 50% MeOH:H2O (fraction C), 75% MeOH:H2O (fraction D), and 100% MeOH (fraction E). A final wash of 100% acetone (fraction F) was used to clean any remaining material from the column. All fractions were concentrated to dryness in vacuo. Fractions were then subjected to gradient liquid chromatography–mass spectrometry (LC-MS) characterization on an Agilent ESI single quadrupole mass spectrometer coupled to an Agilent high-performance liquid chromatography (HPLC) system with a G1311 quaternary pump, G1322 degasser, and G1315 diode array detector (Agilent Eclipse XDB-C18; 5 µm; 4.6 × 150 mm column) using a 40%–100% ACN:H2O gradient (0.1% formic acid) solvent system for 30 min. During secondary screening, fraction E exhibited the most potent activity and was further fractionated. Teleocidin A-1 was isolated by C18 RP-HPLC using C18 RP-HPLC with an isocratic method of 35% H2O and 65% ACN (Agilent Eclipse XDB-C18; 5 µm; 4.6 × 150 mm column; 1 mL/min flow rate). Nuclear magnetic resonance (NMR) spectra of teleocidin A-1 were collected in DMSO-d6 on a Varian INOVA at 600 MHz for 1H, COSY, 1H-13C HSQC, and 1H-13C HMBC. NMR spectra for 13C experiments were collected on a Bruker AVANCE at 125 MHz. Chemical shifts were reported in δ units (ppm) using DMSO-d6 as the internal standard for signals. High-resolution mass spectrometry (HRMS) was performed at the University of Connecticut Mass Spectrometry Facility on an AccuTOF (JEOL) using a DART (IonSense) ionization source. Optical rotation experiments were collected on a JASCO P-2000 polarimeter (c 0.13, MeOH, 26 °C). CD spectra was collected on an Applied Photophysics Pi-star 180 circular dichroism spectrapolarimeter (Leatherhead, Surrey, UK) (c 0.5 mg/mL, MeOH, 21 °C).
Results
Identifying Cells Bound to Beads via Light Scatter and Fluorescence
To modify our previous assay, we replaced the synthetic small-molecule stimulants of the original assay with polystyrene dynabeads coated with anti-CD3 monoclonal antibody (referred to as anti-CD3 beads from here on) to stimulate lytic granule exocytosis in a more physiological manner. Thus, in the modified assay, CD3 bead binding mimics the target cell contact, yet CD3 beads are much easier to integrate into the screening process than a target cell line. Conveniently, anti-CD8-coated beads, which bind to cells but do not stimulate them, are available to serve as negative controls.
When we added anti-CD3 beads to TALL-104 human leukemic CTL cells and examined them using flow cytometry, we observed the appearance of a population with increased side scatter ( Fig. 1A ). Adding more beads further increased scatter. We suspected that the new population with increased side scatter represented cells bound by beads. To confirm this, we examined the side scatter and fluorescence profiles of beads, which fluoresce in all channels (not shown), but fluoresce most in the FL3 channel (Ex488/Em670LP on FACSCalibur). When we plotted the side scatter and fluorescence of anti-CD3 beads and of cells treated with anti-CD3 beads, we found that in the new population, both side scatter and FL3 fluorescence increased an amount consistent with that expected from the contribution of one bead ( Fig. 1B ). Analyzing populations of cells with more added anti-CD3 beads shows that side scatter and FL3 fluorescence increase linearly ( Fig. 1B ). Because anti-CD3 beads are fluorescent in all channels, measuring fluorescence signals corresponding to antibody staining from bead-bound cells requires compensation to eliminate bead fluorescence. We were able to compensate signals by treating beads as if they were a fluorophore with an emission maximum in the Fl-3 channel. This allowed us to measure signals in the FL-1 (Ex488/Em530) and Fl-4 (Ex635/Em661) channels. All histograms and calculations used in this paper are based on compensated fluorescence.
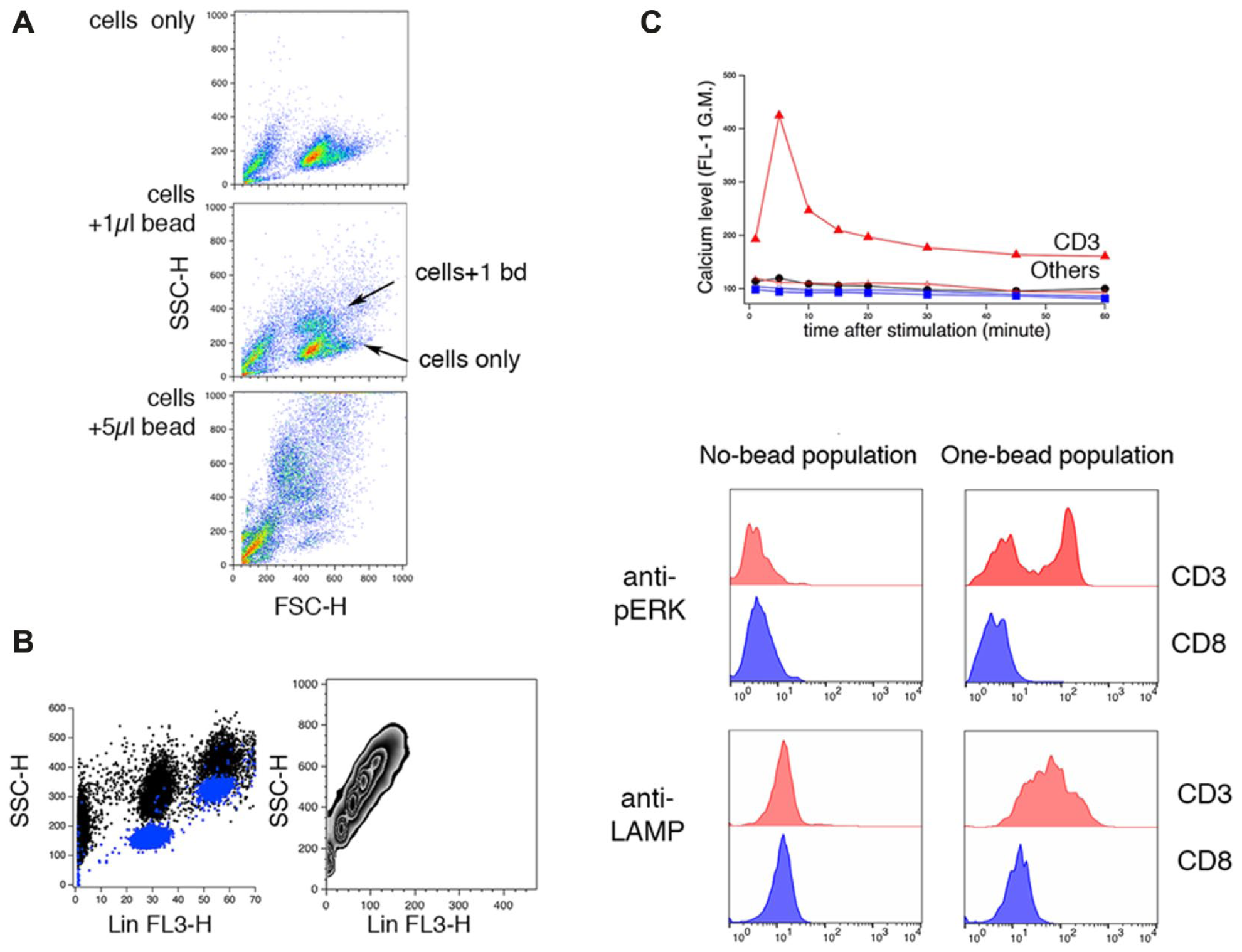
Changes in scatter allow detection of cells bound to antibody-coated beads. (
To confirm that we can identify cells bound to beads via scatter, we measured intracellular calcium levels, ERK activation, and lytic granule exocytosis, all of which are correlates of activation, 13 in samples of cells to which anti-CD3 beads or anti-CD8 beads were added. As stated above, anti-CD8 beads are used as a type of control since these bind to cells but do not activate them. We reasoned that if scatter was accurately identifying cells bound to beads, we should see correlates of activation only in the cells we suspected were bound to CD3 beads.
To assess calcium influx, we loaded cells with calcium-sensitive dye fluo-4 AM, washed them in dye-free solution, and treated them with anti-CD3 beads or anti-CD8 beads. We gated on two subsets in the forward-side scatter plot: cells with no beads bound, and cells with a single bead bound. This gating strategy was applied to all samples in Figure 1C . The top panel shows the level of intracellular calcium indicated by fluo-4 fluorescence (FL1) in two cell subsets over time after addition of anti-CD3 or anti-CD8 beads. Cells bound to anti-CD3 beads showed an immediate increase in intracellular calcium levels that declined to a level that was sustained above baseline. The no-bead population from the anti-CD3 sample did not show any changes in intracellular calcium levels. Cells bound to anti-CD8 beads also did not respond with increased fluo-4 fluorescence.
To assess PKC activation, we treated cells with beads, fixed them, and stained them with antibodies against p-ERK, 15 a signaling event that is downstream of PKC activation ( Fig. 1C , middle). In cells to which anti-CD3 beads were added, cells from the no-bead population did not show enhanced p-ERK staining, whereas some cells from the one-bead population showed an increased level of p-ERK. In cells treated with anti-CD8 beads, there was no increase in p-ERK staining when cells bound to beads.
Finally, we assessed exocytosis. Cells were mixed with beads in the presence of fluorescent anti-LAMP antibody, before they were fixed and read on the cytometer ( Fig. 1C , bottom). In CD3 bead-treated cells, the no-bead population did not show enhanced anti-LAMP staining, whereas the one-bead population displayed higher anti-LAMP fluorescence. In anti-CD8 bead-treated cells, there was no change in anti-LAMP staining. When we analyzed signals from cells with more than one anti-CD3 bead bound, we found that levels of anti-LAMP fluorescence increased with the number of beads (data not shown). We conclude that scatter allows us to identify cells bound to beads. As expected, anti-CD3 but not anti-CD8 beads can trigger calcium influx and kinase signaling, leading to granule exocytosis in TALL-104 cells. For all data presented, we selected the population of cells bound to a single bead for analysis.
Contact with Anti-CD3 Beads Stimulates Submaximal Exocytosis That Can Serve as the Basis of a Plate-Based Assay for Enhancers or Inhibitors
Inspection of the anti-LAMP responses to anti-CD3 beads suggests that exocytosis triggered via the TCR is less robust than exocytosis stimulated by TG + PMA. We compared levels of exocytosis as measured using anti-LAMP staining for a number of conditions ( Fig. 2A ), and confirmed that responses to anti-CD3 beads are smaller than responses to TG + PMA. Furthermore, we found that exocytosis stimulated by anti-CD3 beads can be augmented when cells are stimulated with the soluble agents TG, PMA, or a combination of the two. Comparison of the responses of bead-bound and unbound cells indicates that treatment with TG or TG + PMA triggers exocytosis in cells whether or not they are bound to beads. The ability of TG to trigger granule exocytosis is consistent with our previous results, and is probably due to calcium activation of conventional PKC isoforms. In contrast, treating cells with PMA enhances exocytosis in the population of cells bound to a bead, but is without effect on bead-free cells. These results suggest that comparison of the responses of cells that are or are not bound to beads could be used in an assay based on anti-CD3 bead–stimulated exocytosis to distinguish agents that trigger exocytosis from those that enhance exocytosis in receptor-stimulated cells.
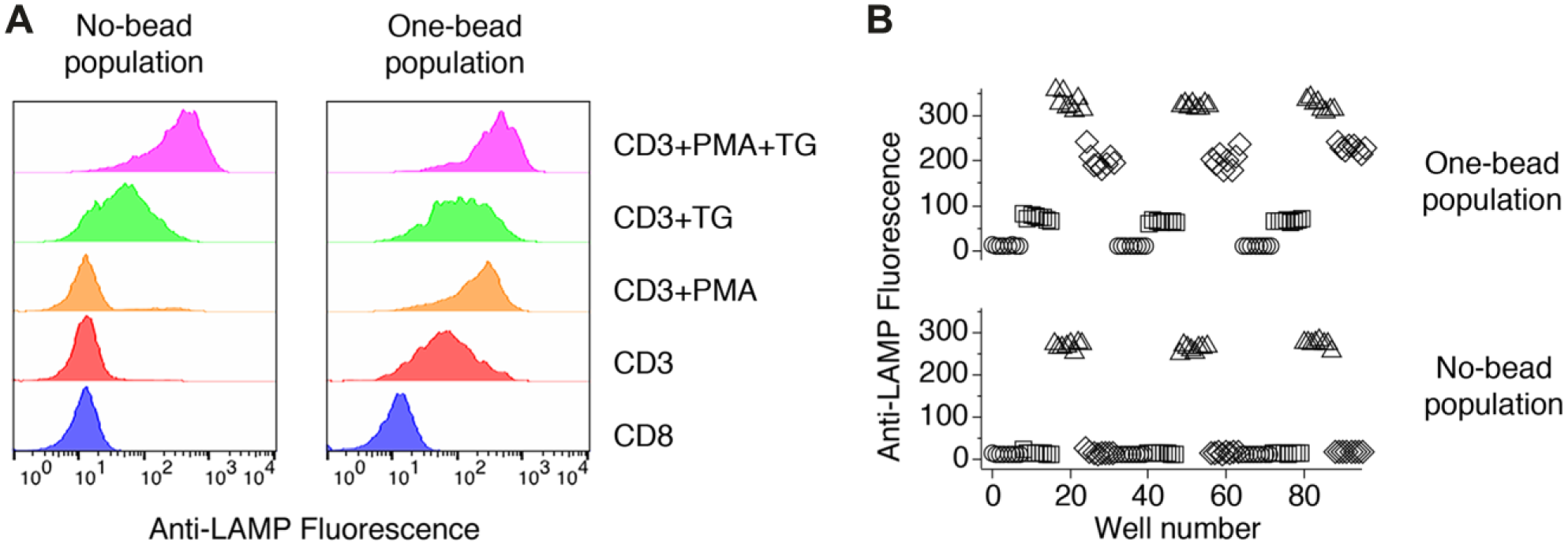
Anti-CD3 beads trigger submaximal exocytosis facilitating a robust plate-based assay format. (
To verify that an assay based on anti-LAMP staining in responses to anti-CD3 beads is sufficiently robust that it can be performed in plate format, we examined the effects of treating cells with different conditions in 96-well plates ( Fig. 2B ). Individual treatments were laid out in an interleaved pattern. After 50 min of incubation with constant rotation at room temperature, cells were fixed with PFA and transferred to tubes for cytometric analysis. They were not washed prior to analysis. We ran a total of three plates, each on a different day. We computed Z′, a statistical parameter used to assess assay robustness, 16 comparing (1) cells in contact with anti-CD3 beads to cells in contact with anti-CD8 beads (Z′ = 0.54 ± 0.13), and (2) cells in contact with anti-CD3 beads to cells in contact with anti-CD3 beads that were also treated with TG + PMA (Z′ = 0.78 ± 0.13). The first condition mimics the maximum separation expected for an inhibitor of lytic granule exocytosis, while the second mimics the maximum separation in signals expected for an agent that generates maximal exocytosis. That Z′ for both comparisons was >0.5 suggests the assay is sufficiently robust that it can detect inhibitors or enhancers of exocytosis.
Confirming the Assay’s Ability to Detect Exocytosis-Inhibiting Small Molecules
As a means of confirming that the new version of the assay can detect inhibitory compounds, we screened a plate containing 75 compounds that were identified as hits in our initial screen of the MLSMR. 3 Forty-eight of those compounds were confirmed to inhibit TG + PMA–stimulated lytic granule exocytosis by >50% when they were retested, and our subsequent analysis of the compounds’ molecular mechanism of action revealed that active compounds worked by a variety of mechanisms, including inhibition of calcium influx, ERK activation, and calcineurin activity. When we tested the plate using the new version of the assay, 36 compounds blocked exocytosis by >50%. The assay was conducted so that we could perform simultaneous BLT esterase measurements 17 from the same samples in which anti-LAMP fluorescence was measured. BLT esterase assays measure release of granzymes from lytic granules, and have served for decades as the gold standard for measuring lytic granule exocytosis. The data in Figure 3A plot percent inhibition of anti-LAMP fluorescence increases versus percent inhibition of granule exocytosis assessed using BLT esterase assays for cells stimulated with anti-CD3 beads. There was a good correlation (r = 0.88) between the two measurements. When reanalyzed the same way, our previously published data using TG + PMA stimulation yielded a similar r value (0.91, not shown). To determine how the modified assay performs compared to the original assay, we plotted the percent inhibition of lytic granule exocytosis measured using anti-LAMP fluorescence from CD3 bead–stimulated cells versus the percent inhibition of lytic granule exocytosis measured previously using anti-LAMP fluorescence from cells stimulated with TG + PMA ( Fig. 3B ). There was a positive correlation between the measurements, with r = 0.77, although it was not as strong as that observed comparing anti-LAMP responses and BLT esterase data from cells stimulated using the same means. However, when we examined the correlation between responses measured from CD3 bead–stimulated cells and TG + PMA–stimulated cells using BLT esterase assays ( Fig. 3C ), the correlation between the two modes of stimulation was very similar to what we observed using anti-LAMP staining, with r = 0.72. These data suggest that measuring exocytosis via anti-LAMP antibody binding provides similar estimates to BLT esterase assays. As a final means of assessing the performance of the modified assay, we investigated the dose dependence of inhibition for two compounds (1-(4-methoxyphenyl)-3-[(5-phenyl-1,3,4-oxadiazol-2-yl)sulfanyl]pyrrolidine-2,5-dione and 2-N-[(2-methoxyphenyl)methyl]-4-N-[(4-propan-2-ylphenyl)methyl]thieno[3,2-d]pyrimidine-2,4-diamine) that were of particular interest to us because they blocked granule exocytosis without affecting known signaling pathways ( Fig. 3D ). Dose-dependent inhibition of responses stimulated by TG + PMA or by anti-CD3 beads was similar, with 1-(4-methoxyphenyl)-3-[(5-phenyl-1,3,4-oxadiazol-2-yl)sulfanyl]pyrrolidine-2,5-dione exhibiting higher potency in response to both kinds of stimulation than 2-N-[(2-methoxyphenyl)methyl]-4-N-[(4-propan-2-ylphenyl)methyl]thieno[3,2-d]pyrimidine-2,4-diamine. Taken together, these results suggest that the new version of the assay has an ability to detect inhibitory compounds that is similar to that of the original assay format.
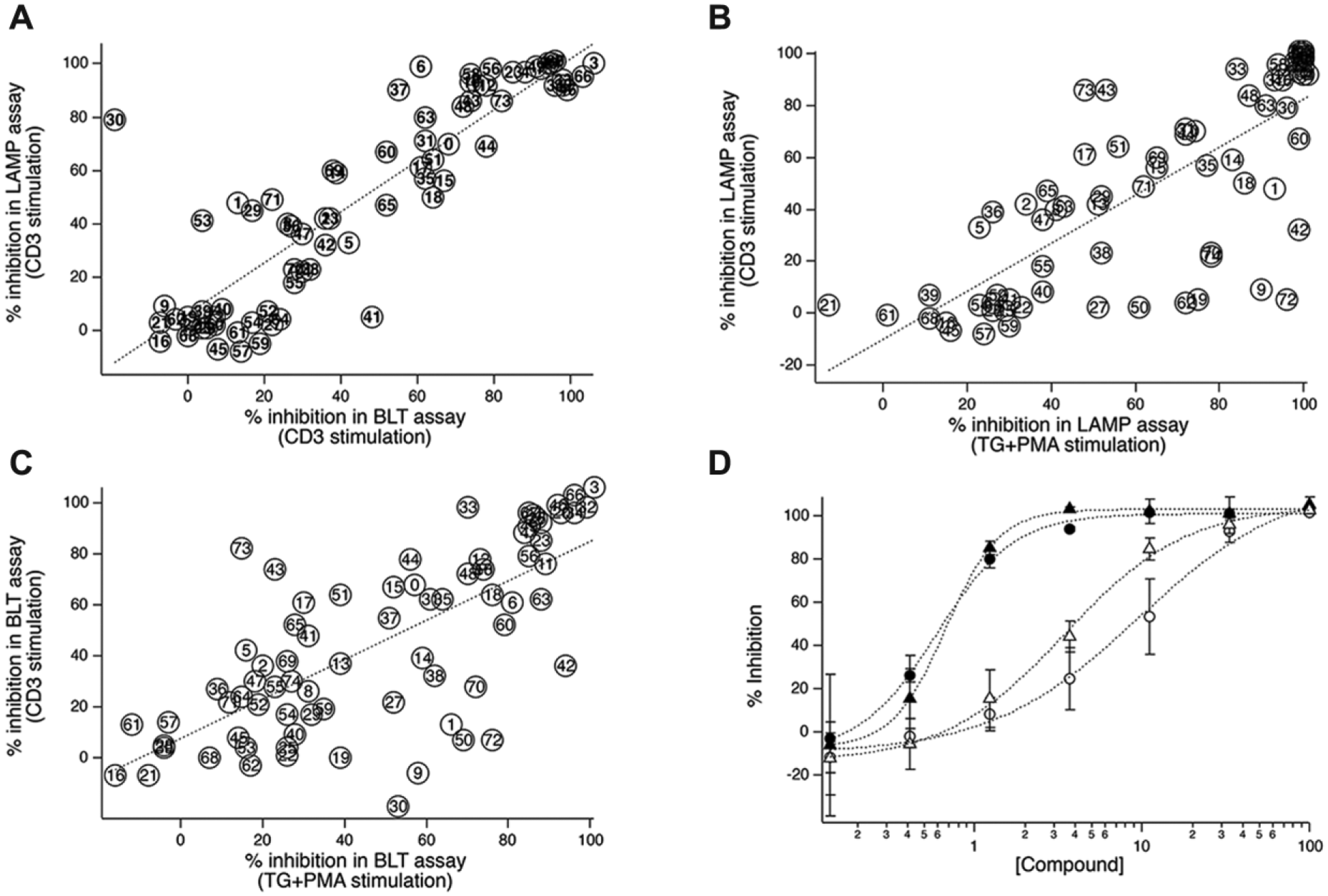
Detection of exocytosis-inhibiting compounds. (
Screening of a Tunicate-Associated Bacterial Natural Product Library Revealed an Immune-Enhancing Compound
An assay that can be used to detect small molecules that inhibit exocytosis stimulated via the TCR represents an incremental advance over our previous assay. However, as described above, our assay makes it possible to screen for compounds that enhance lytic granule exocytosis, which would represent a substantially more significant enhancement to the method. Relatively few immune-augmenting molecules have been identified to date, likely because there has been no convenient way to screen for them. Reasoning that bacteria living with hosts, such as marine tunicates, that have an immune system might produce immune-modifying secondary metabolites, we screened a collection of 320 extracts in four plates prepared from tunicate-associated bacteria. We identified two extracts that enhanced lytic granule exocytosis. Figure 4A shows data from one of the plates that yielded an augmenting hit. Each black circle represents the geometric mean of anti-LAMP fluorescence of the cell sample treated with an extract in a well, and the arrow indicates the extract-enhanced cell exocytosis in the bead-bound population. The extract had a marginal stimulatory effect on cells that were not bound to beads. The hit extract A2, named after its well location in the screening plate, was the most active enhancing extract from the library, with an approximately threefold enhancement of the physiological level of lytic granule exocytosis. The enhancing effect was confirmed upon retesting (data not shown). As mentioned, cells that were not in contact with anti-CD3 beads showed a slight enhancement of exocytosis. However, treating cells with only A2 in the presence of anti-LAMP confirmed that A2 did not stimulate lytic granule exocytosis on its own (data not shown).
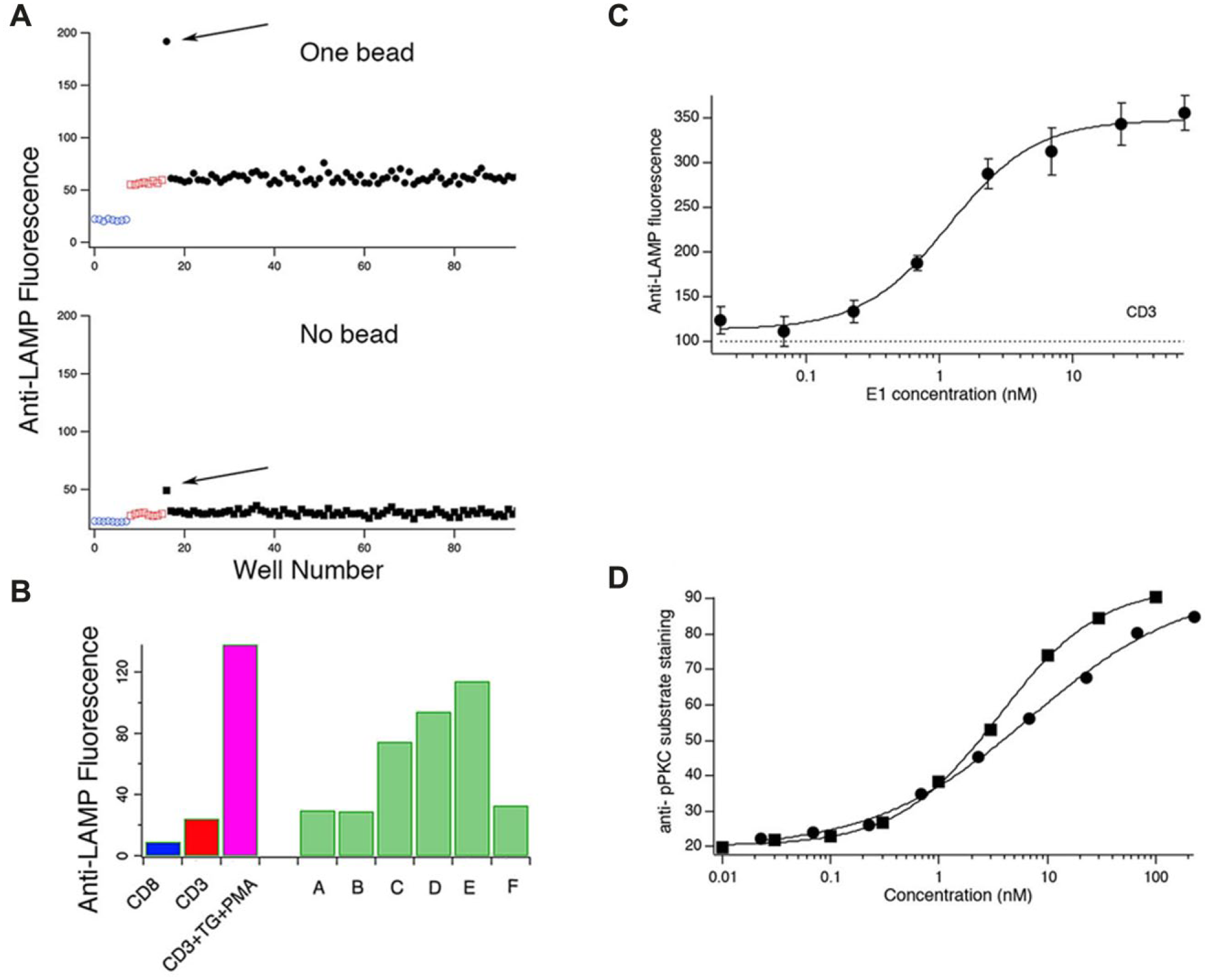
Detection and activity-guided isolation of an exocytosis-enhancing PKC-activating compound. (
Because of its potency, we pursued extract A2 further. We fractionated the extract using a reversed-phase solid-phase extraction column, and examined the activity of the individual fractions ( Fig. 4B ). Fraction E had a fivefold enhancing effect on cells stimulated by CD3 beads, almost as potent as the combination of TG + PMA. Fraction E was further separated using HPLC as described below, and the peak eluting at a retention time of 7 min was isolated as a pure compound, coded as E1. E1 was found to enhance lytic granule exocytosis and was pursued in further studies. In dose–response studies, E1 enhanced lytic granule exocytosis with an EC50 of approximately 1 nM ( Fig. 4C ).
We next sought to determine the molecular mechanism of action (MMOA) by which E1 enhances lytic granule exocytosis. As mentioned earlier, both calcium influx and activation of PKC and ERK are required for granule exocytosis. We felt it unlikely that E1 triggered calcium influx, since it did not trigger significant exocytosis in the absence of stimulation with beads. We confirmed this using live cell imaging of cells loaded with the calcium-sensitive probe Fura-2 (data not shown). We tested the effect of E1 on PKC activity using immunocytochemistry. We treated cells with E1 or PMA for 90 min before fixing and permeabilizing them. The cells were then incubated with primary antibodies raised against serine-phosphorylated PKC substrates, 18 followed by an Alexa488-conjugated secondary antibody. To assess E1’s potency relative to PMA, dose–response measurements of PKC activity were carried out at various concentrations of E1 and PMA from 0.01 to 200 nM in threefold dilution series. Figure 4D shows dose–response curves for the activation of PKC by E1 and PMA. Fits to the Hill equation indicate that E1 activated PKC with an EC50 of 6.9 nM, almost as potent as PMA (EC50 = 3.6 nM).
E1 Is Teleocidin A-1
As described above, activity-guided fractionation of the active extract from well A2 led to the isolation of E1, found to exhibit potent stimulatory activity on the PKC pathway, independent of calcium influx activity. Whole genome sequencing identified the bacterium as a novel strain of Streptomyces sp. From 16.3 mg of fraction E, compound E1 (1.4 mg) was isolated via RP-HPLC using an isocratic system of 35:65 acetonitrile–water with a retention time of approximately 7 min.
Structure elucidation of E1 utilized a series of one-dimensional (1D) and two-dimensional (2D) NMR experiments in addition to mass spectrometric analysis (
Discussion
We have shown that we can identify TALL-104 cells bound to anti-CD3 or anti-CD8 beads in cytometry via changes in light scatter. This makes it possible to study events associated with quasi-physiological activation specifically in cells that have received the activating stimulus. Importantly, it also makes it possible to conduct two-way screens in which compounds that either inhibit or enhance lytic granule exocytosis can be detected. When using the assay, comparison of responses in cells that have bound a bead to responses in cells that have not bound a bead makes it possible to distinguish between compounds that stimulate exocytosis on their own and compounds that enhance receptor-stimulated exocytosis ( Fig. 2A ). Thus, the modified version of the assay is capable of identifying three classes of compounds: (1) inhibitors; (2) stimulators, compounds that trigger responses; and (3) augmenters, compounds that enhance receptor-stimulated responses but are not themselves capable of producing responses. We believe that the strategy could facilitate studies of signal transduction and, although we have not tested this here, suspect the strategy could likely also be applied to other cell types, including primary T lymphocytes and other cell types that respond to stimuli presented on a solid phase. Similar beads are routinely used to stimulate cells for functional experiments, and so we think it highly likely that the method will be more generally applicable. Note that the requirement that cells respond to solid-phase stimulation can also be seen as a limitation of the method, as some cells may respond only to soluble stimuli. Also, some types of bead may not produce optical scatter properties that allow their identification. The assay is suitable for screening natural compound collections, and can be used as part of an activity-guided isolation scheme. In preliminary experiments, we found that further miniaturization to the 384-well format is possible (B. E. Edwards, A. Zweifach, unpublished observations.) If, as we strongly anticipate should be possible, the assay can be translated to the 384-well format, it will be possible to conduct HTS for immunologically active compounds. As it is, a 96-well format enables screening of moderately sized collections.
Our assay has both similarities to and differences from an assay that used anti-LAMP antibodies to assess exocytic responses in NK cells stimulated with plate-bound activating antibodies 21 Both assays use flow cytometry to assess exocytosis via binding of anti-LAMP antibodies. However, that assay was not able to distinguish responses from cells that did or did not receive the activating stimulus. Thus, while they were able to identify a number of stimulatory compounds in the Prestwick compound library—along with a number of inhibitors—they were unable to distinguish whether those compounds cause exocytosis on their own or augment receptor-stimulated responses. One powerful feature of their assay was that they multiplexed detection of exocytosis with measurements of protein synthesis and inside-out changes in cell adhesion. It is possible that we could enhance our assay by adding additional antibodies able to detect other cellular events. However, the fluorescence of the beads would limit the additional channels that could be used.
The assay’s ability to identify inhibitors of lytic granule exocytosis was confirmed by the fact that we were able to detect a number of inhibitory compounds with different MMOA that we originally identified with the prior version of the assay ( Fig. 3 ). Just as important, many compounds that ultimately proved not to be inhibitory using the prior assay also did not inhibit using the new format. Agreement between the results with the old and new assays was not perfect, but that might be explained by degradation of compounds in storage over time or by differences in the dependence of granule exocytosis on strength of signals when cells were stimulated with soluble TCR-independent chemicals compared to stimulation through their TCRs. Little is known about how the strength of signals is translated to lytic granule exocytosis, and so it is not clear that perfect correspondence in the degree to which exocytosis is inhibited by compounds should always be expected. Supporting the ability of the new assay to identify compounds that enhance lytic granule exocytosis, the compound we identified from bacterial extracts, teleocidin A-1, is a known PKC activator, 19 and has been shown to enhance granule-dependent cytotoxicity 22 in primary human lymphocyte preparations.
The assay we developed was successfully utilized to screen 320 natural product extracts from tunicate-associated bacteria. Compared to screening synthetic compound collections, screening of natural product extract libraries presents several difficulties in the search for novel compounds with biomedically relevant activities. 23 Extracts are complex mixtures of compounds that often range in polarity and solubility. Active natural product compounds may be present in only minute quantities, and their activity may be overshadowed by other bioactive components or interfering nuisance compounds within the mixture. Crude extracts often contain pigmented compounds and may also contain autofluorescent metabolites. In addition, compounds may be less soluble when isolated and/or be unstable when not in the extract matrix.
That our assay is suitable for screening both synthetic compound collections and complicated natural product mixtures for exocytosis-suppressing and exocytosis-enhancing compounds suggests it might be a powerful means of discovering immunologically active small molecules from diverse sources. Using short compound treatment times, as we did here, we suspect that the assay is most likely to find compounds that, like teleocidin A-1, enhance signals leading to lytic granule exocytosis. However, we could likely detect compounds with other mechanisms of action with longer treatment times. For example, we found that treating cells with the known immunomodulator imiquimod increased anti-LAMP staining by ~75% in cells bound to anti-CD3 beads after a 6 h incubation (data not shown). To facilitate identification of compounds with a wide variety of MOAs, it may be possible to devise an assay format in which fluorescent cellular bar coding is used to assess the effects of treating cells with compounds for varying lengths of time. Compounds that affect lytic granule exocytosis would affect immune functions that involve lytic granule exocytosis, and on this basis might be expected to modulate antiviral and antitumor immunity. Furthermore, we would anticipate that many compounds identified as active in a screen based on lytic granule exocytosis would also affect the activity of other immune cells. Thus, the assay we describe here may provide a general means of screening compound collections for immunologically active small molecules. We surmise that one reason there are so few immunomodulatory small molecules is that to date, there has not been a convenient way to screen for them. With our assay, the ease of screening both synthetic and natural extract libraries should make for more rapid discovery of new small-molecule immunomodulators.
Footnotes
Acknowledgements
We thank Drs. Carolyn Teschke and Tina Motwani (UConn Department of Molecular and Cell Biology) for help in collecting CD spectra data, Divya Chennamadhavuni (UConn Department of Chemistry) for assistance in collecting polarimetry data, and Dr. You-Jun Fu (UConn Mass Spectrometry Facility) for obtaining HRMS data. We thank Dr. Carol Norris and the UConn Flow Cytometry Facility for invaluable assistance. We thank Dr. Jeff Aubé, currently at the University of North Carolina, for synthesizing 2-N-[(2-methoxyphenyl)methyl]-4-N-[(4-propan-2-ylphenyl)methyl]thieno[3,2-d]pyrimidine-2,4-diamine.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
We acknowledge funding from the Robert F. Kaiko and Lucy T. Li Pharmacy Honors Research Scholarship (to A.M.W.) and the University of Connecticut Summer Undergraduate Research Fellowship (to A.M.W.). Funding was also received from the University of Connecticut Multicultural Scholars Program (to J.A.D.)
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.