
Abstract
Soluble epoxide hydrolase (sEH) is a bifunctional enzyme that possesses an epoxide hydrolase and lipid phosphatase activity (sEH-P) at two distinct catalytic domains. While the physiological role of the epoxide hydrolase domain is well understood, the consequences of the phosphatase activity remain unclear. Herein we describe the bacterial expression of the recombinant N-terminal domain of sEH-P and the development of a high-throughput screening protocol using a sensitive and commercially available substrate fluorescein diphosphate. The usability of the assay system was demonstrated and novel inhibitors of sEH-P were identified.
Introduction
Soluble epoxide hydrolase (sEH) is a cytosolic enzyme ubiquitous in mammals. It is mainly expressed in the liver and kidneys but also in many other tissues, including red blood cells, smooth muscle cells, adipocytes, proximal tubules, leucocytes, and endothelial cells. sEH usually occurs as a homodimer arranged from two antiparallel monomers. Every monomer of 62.5 kDa contains two distinct domains connected by a proline-rich linker. The eponymous epoxide hydrolase domain (sEH-H, EC 3.3.2.10) is located at the C-terminus and catalyzes the conversion of epoxidated fatty acids such as epoxyeicosatrienoic acids (EETs) to the corresponding diols. Murine knockout and pharmacological inhibition of sEH showed anti-inflammatory, antihypertensive, neuroprotective, cardioprotective, and antidiabetic effects mediated by the accumulation of EETs.1,2 The N-terminal domain was found out to have phosphatase activity (sEH-P, EC 3.1.3.76).3,4
The sEH-P has structural and sequential similarity to the haloacid dehalogenase family and cleaves phosphate esters in a magnesium-dependent nucleophilic attack by Asp9. 5 Endogenic substrates of sEH-P are isoprenoid phosphates 6 and lysophosphatidic acids.7,8 Phenotypic differences between mice treated with sEH-H inhibitors and sEH knockout mice, which lack both domains, indicate a role of sEH-P in hypercholesterolemia 9 and pulmonary hypertension. 10 A knockdown experiment in cells showed an influence of sEH-P on vascular endothelial growth factor, 11 therefore regulating endothelial nitric oxide synthase activity and nitric oxide–mediated endothelial cell functions. 12 Recent studies by Hou et al. 13 could demonstrate that sEH-P activity downregulated eNOS activated by simvastatin. Studies on human polymorphism showed that subjects with Arg287Gln or Lys55Arg have lowered sEH-P activity but higher risks for type 2 diabetes or coronary heart diseases. 14 R103C and R287Q variants of sEH exhibited impaired phosphatase activity in vitro. 15 Despite these findings, the physiological role of sEH-P is far from being completely understood. A chemical probe, defined as “a selective small-molecule modulator of a protein’s function that allows the user to ask mechanistic and phenotypic questions about its molecular target in biochemical, cell-based or animal studies” 16 would be useful to investigate the physiological and pathophysiological roles of sEH-P.
The development of a chemical probe for sEH-P has not advanced so far, as typical phosphatase inhibitors do not modulate sEH-P activity. 17 Lipid sulfates 18 are inhibitors of sEH-P, but intrinsic toxicity makes them incompatible with investigation in cells or in vivo studies. Recently, ebselen was found to inhibit sEH-P, but this compound reacts irreversibly with the enzyme assumedly in a redox mechanism. 19 Furthermore, ebselen is relatively promiscuous and has been shown to interact with a great variety of different proteins, 20 making its suitability as a chemical probe for sEH-P rather questionable. The experimental thrombolytic drug SMTP-7 and its derivatives were found to inhibit sEH-P and hydrolase activity simultaneously, which is supposed to contribute to the anti-inflammatory effect of these compounds.21,22 Therefore, there is still a need for a chemical probe to investigate the effects mediated by the sEH-P domain. In this study, we present the development of a novel sEH-P assay suitable for high-throughput screening (HTS) and the medium-throughput screen to identify new inhibitors that should further facilitate the development of a chemical probe for sEH-P.
Materials and Methods
Materials
The substrate fluorescein diphosphate (FDP) was purchased as tertaammonium salt from Biomol (Hamburg, Germany). 6,8-Difluoro-4-methylumbelliferyl phosphate (DiFMUP) was from ThermoFisher Scientific (Waltham, MA), and PHOME from Cayman Chemical (Ann Arbor, MI). Buffer reagents and chemicals were obtained from AppliChem (Darmstadt, Germany) or Sigma Aldrich (Taufkirchen, Germany) and used without further purification. The screening library containing approved drugs was obtained from Enzo (Lörrach, Germany). Medium-throughput screening was performed using the ChemBioNet library. 23
Cloning and Enzyme Preparation
The complementary DNA (cDNA) clone encoding human sEH 24 was used to separate the phosphatase domain by PCR (forward primer AAACATATGACGCTGCGCGCG and reverse primer AAACTCGAGAAGCTGGATTCCGGT). Separated sEH-P was inserted into a pET28b(+) expression vector (Novagen, Gibbstown, NJ) with kanamycin resistance and a thrombin cleavage site at the C-terminus. The final plasmid carried a hexa-His-tag at the C-terminus and a second hexa-His-tag at the N-terminus. The plasmid was transformed and overexpressed in Escherichia coli BL21(DE3) cells (Invitrogen, Darmstadt, Germany).
To yield a second plasmid coding for the inactive form of sEH-P with Asp9Ala mutation, a mutagenesis PCR was conducted using the following primers: forward, 5′-GCC GTCTTCG
Wild-type sEH-P and Asp9Ala sEH-P mutant were expressed and purified using the same protocol. Cells were grown in TB medium containing kanamycin (100 µg/mL) and sodium phosphate (90 mM) at 37 °C and 180 rpm to an OD600 of 0.8 to 1. After cooling to 21 °C, cells were induced with isopropyl β-D-1-thiogalactopyranoside (ITPG) (400 µM), and the culture was incubated overnight. Cells were harvested by centrifugation (5000 rpm, 30 min) and the pellet was resuspended in 30 mL buffer A (25 mM Bis-Tris, 500 mM NaCl, pH 7.0) with one dose of Complete EDTA free (Roche, Basel, Switzerland) and a trace amount of DNase I (Applichem, Darmstadt, Germany). Cells were passed through a cell disruptor (Constant Systems, Daventry, UK) three times at 14,500 psi. Disrupted cells were centrifuged for 60 min at 20,000 g at 4 °C to remove cells debris and subsequently passed to high-spin centrifugation at 100,000 g for 60 min at 4 °C. The supernatant was loaded on a 5-mL HisTrap HP (GE Healthcare, Solingen, Germany) column, which was washed with buffer A containing 150 mM imidazole. Purified double-hexa-His–tagged protein was eluted using a linear gradient from 150 to 500 mM imidazole at around 250 mM imidazole. Fractions containing sEH-P were concentrated using a CentriPrep concentrator (Millipore, Darmstadt, Germany) with a 5-kDa cutoff membrane. A Superdex200 column (GE Healthcare, Germany) was equilibrated with assay buffer (50 mM acetate, 10 mM MgCl2, pH 5.75), and concentrated protein was isocratically eluted using the same buffer. Protein concentration was determined by using Nanodrop spectrophotometry (Implen, Muenchen, Germany). Purity and identity of sEH-P (>95%) were determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and fingerprint mass spectrometry analysis, respectively.
sEH phosphatase activity assay using 6,8-difluoro-4-methylumbelliferyl phosphate
An alternative assay system with the substrate DiFMUP was performed similar to the procedure described by Hahn et al. 24 Different concentrations of compound (1 % DMSO final concentration) were incubated with 0.25 µM sEH phosphatase in acetate buffer (50 mM) and MgCl2 (10 mM) pH 5.8 with 0.01% Triton X-100. A negative control without protein was entrained as well as a positive control with protein and no compound. After incubation for 30 min at room temperature, 300 µM DiFMUP was added and the fluorescence of dephosphorylated DiFMU was measured at 37 °C for 45 min (λex 360 nm, λem 450 nm). The percent inhibition was evaluated, and a sigmoidal dose-response curve was fitted with GraphPad Prism (version 5.0; GraphPad Software, La Jolla, CA) to determine the IC50 value.
sEH hydrolase activity assay using 3-phenyl-cyano(6-methoxy-2-naphthalenyl)methyl ester-2-oxirane-acetic acid
Fluorescence-based assay sEH hydrolase activity was performed as published previously. 24 In brief, 1 µL of the compounds with DMSO was transferred to a black 96-well microplate. The compounds were incubated with recombinant full-length sEH (0.22 µM) and Triton-X 100 (final concentration 0.01%) for 30 min at room temperature. After 30 min of incubation, 3-phenyl-cyano(6-methoxy-2-naphthalenyl)methyl ester-2-oxirane-acetic acid (PHOME) was added to a final concentration of 50 µM. Measurement of the fluorescence of the product 6-methoxynaphtaldehyde (λex 360 nm, λem 465 nm) was performed by using an infinite F200pro plate reader (Tecan, Crailsheim, Germany) every minute at 30 time points. Experiments to identify percent inhibition values were carried out in triplicates. The percent inhibition was calculated in comparison to activities of blank (without protein) and positive wells (without inhibitor).
FDP-Assay Development
The general assay setup in a 96-well format was established as follows: for compound testing, 1 µL of compound stock solution in DMSO (or DMSO only for the positive control) and then 89 µL of the protein solution (0.1 µM final concentration) in assay buffer (50 mM acetate, 10 mM MgCl2 [pH 5.75], 0.01% Triton X-100) were added to each well. Assay buffer without protein was added to the wells for measuring blank fluorescence. After 30 min, the reaction was started by addition of 10 µL FDP solution (10 µM final concentration) in assay buffer. Fluorescence changes (λex 485 nm, λem 525 nm) were detected in 30 time points using an infinite F200pro plate reader (Tecan, Crailsheim, Germany). Data were fitted using Microsoft Excel (Microsoft Corp., Redmond, WA) to a linear model, and the slope was used as a relative measure of enzyme activity. Data were normalized relative to the blank wells without protein (0%) and wells without inhibitor (100%). Each substance was measured in triplicate in three independent experiments. IC50 values were calculated using data obtained from measurements with at least six different inhibitor concentrations, applying a sigmoidal dose-response (variable slope with four parameters) equation using GraphPad Prism (version 5; GraphPad Software). For assay development, different concentrations of sEH-P (0.01–0.5 µM) and FDP (1–50 µM) were tested in assay buffer (50 mM acetate, 10 mM MgCl2 [pH 5.75], containing 0.01% Triton X-100 and 1% DMSO) in a checkerboard assay to determine the optimal conditions.
Assay Miniaturization and Library Screening
For medium-throughput screening of the Screenwell Food and Drug Administration (FDA)–approved drug library of 780 compounds (Enzo, Lörrach, Germany), 20 nL of compound (10 mM in DMSO) was transferred to assay plates by an Echo 550 (Labcyte, Dublin, Ireland) acoustic dispenser. Then, 10 µL sEH-P (30 nM final concentration) was added to each well, and the mixture was incubated at 25 °C for 15 min. After the addition of 10 µL FDP (5 µM final concentration), a second incubation step of 60 min at 25 °C occurred before the reaction was stopped using 5 µL NaOH (1 M). In a second screening, the ChemBioNet library of 14,000 diverse compounds 23 was screened using the same protocol.
Results and Discussion
Protein Expression
Recombinant sEH-P domain was expressed in E. coli BL21 (DE3) cells in TB medium. For purification, nickel-affinity chromatography was used with an imidazole gradient. Double His-tagged sEH-P was eluted with 250 mM imidazole. Further purification was done with gel filtration to reach pure sEH-P (>95 %). Of a 1-L culture, 15 to 20 mg sEH-P was obtained. To ensure the specificity of the substrate turnover, the negative mutant Asp9Ala sEH-P was expressed using the same protocol.
Assay Development
The colorless nonfluorescent FDP was shown to be a good substrate for many phosphatases by Huang because of its amphoteric solubilty. 25 After the two-step cleavage, fluorescein is readily detected at wavelengths compatible with compound screening (λex 485 nm, λem 525 nm). FDP has not been reported as a sEH-P substrate to date, and therefore we first performed a checkerboard assay to optimize conditions (see Table 1 ), with the goal to develop a sensitive 96-well format with linear enzyme kinetics over a reaction time of at least 1 h. Key derived parameters were percent turnover rate of the substrate after 1 h (%TO), the signal-to-background ratio (S/B), and Z′. 26
Checkerboard Assay to Obtain Suitable Relation between Substrate FDP and sEH-P.
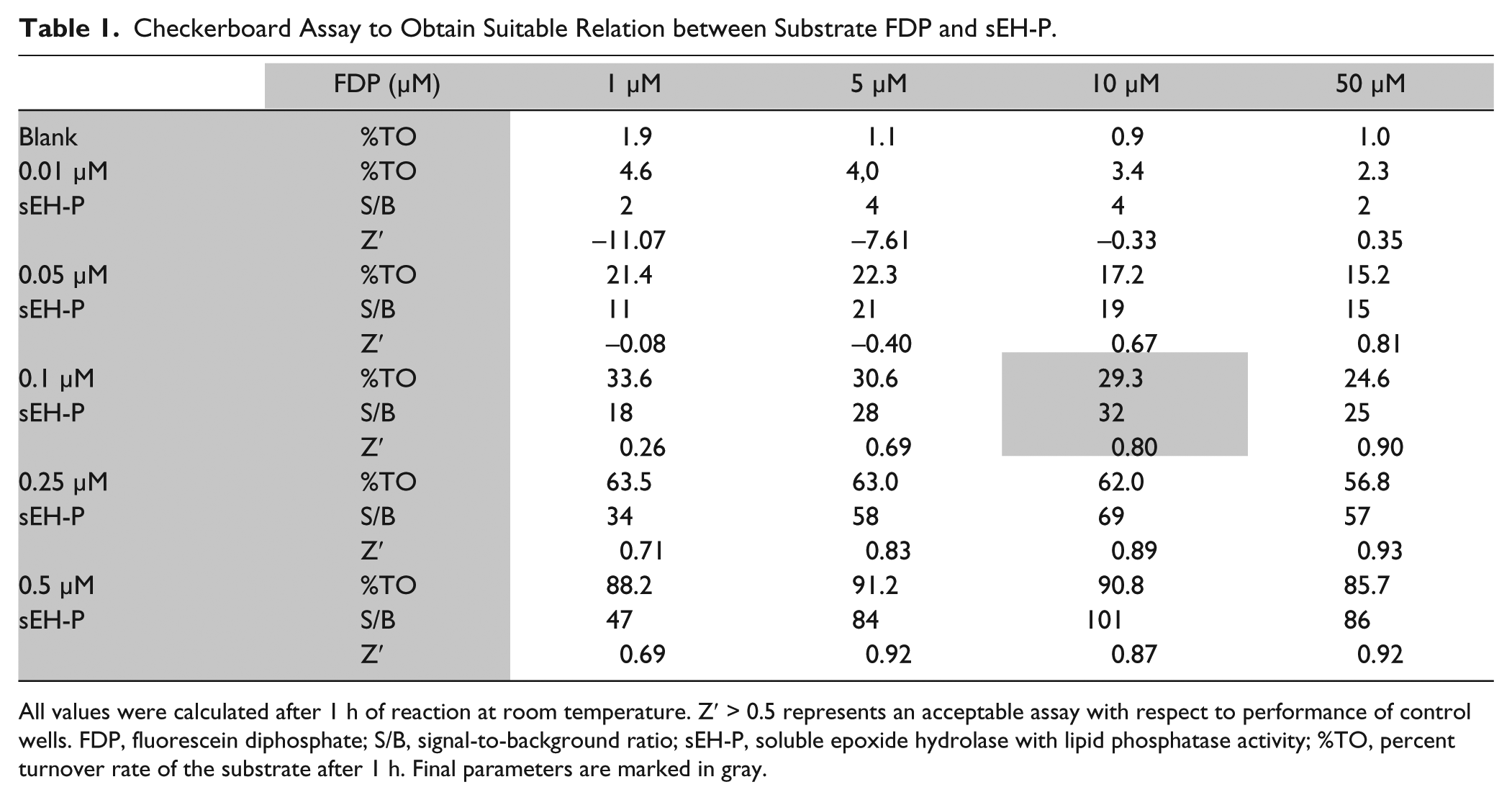
All values were calculated after 1 h of reaction at room temperature. Z′ > 0.5 represents an acceptable assay with respect to performance of control wells. FDP, fluorescein diphosphate; S/B, signal-to-background ratio; sEH-P, soluble epoxide hydrolase with lipid phosphatase activity; %TO, percent turnover rate of the substrate after 1 h. Final parameters are marked in gray.
The optimal configuration (marked in gray in the table), which fulfilled all desired parameters (approximately 20% TO, >25-fold S/B, Z′ > 0.6) contained 0.1 µM sEH-P and 10 µM FDP. The assay was performed with the Asp9Ala mutant, under normal conditions (0.1 µM protein) and elevated protein (1 mM). Substrate turnover could not be observed with the Asp9Ala mutant. As part of assay validation, the influence of DMSO was examined. DMSO showed no influence on the assay up to 4% (data not shown). Triton X-100 was used to prevent compound aggregation,27,28 but addition of more than 0.02% Triton X-100 reduced sEH-P activity (data not shown). Therefore, a constant level of 1% DMSO and 0.01% Triton X-100 was kept in the final assay setup. In a pharmacological validation step, the IC50 values of sodium dodecyl sulfate (SDS), a known inhibitor, was determined at different plate positions and pipetting schemes. The IC50 of SDS was 3.0 ± 0.6 µM, which aligns with the reported value of 5.2 µM. 18
To allow for screening of a higher number of compounds, the assay was adapted to a 384-well format with 20 µL total volume per well. To eliminate cross-fluorescence and adapt to the sensitivity of the EnVision Reader (Perkin Elmer, Hamburg, Germany), the FDP concentration was reduced to 5 µM in a 384-well format. A range of protein concentrations (5–50 nM) was tested to readjust the linear turnover rate of 20% after 1 h. This parameter was fulfilled by a concentration of 30 nM sEH-P. The kinetic parameters under these conditions were determined in 384-well plates ( Fig. 1 ). In this adapted format, the IC50 determined for ebselen was 483 ± 10 nM, which is comparable to the literature. 19 To cater for the throughput needed for high-throughput screens, the assay was reconfigured to an end-point format. Sodium hydroxide was used because sEH-P is inactive at alkaline pH. 19 Furthermore, deprotonation of the fluorescent product increases the signal window to a minimum 80-fold S/B ratio. Z′ > 0.7 was determined for all plates. To validate the change from kinetic cycles to an end-point format, the IC50 of ebselen was measured ( Fig. 2 ). In end-point format, the IC50 of ebselen was 377 ± 11 nM, which is in the range measured in kinetic cycles.
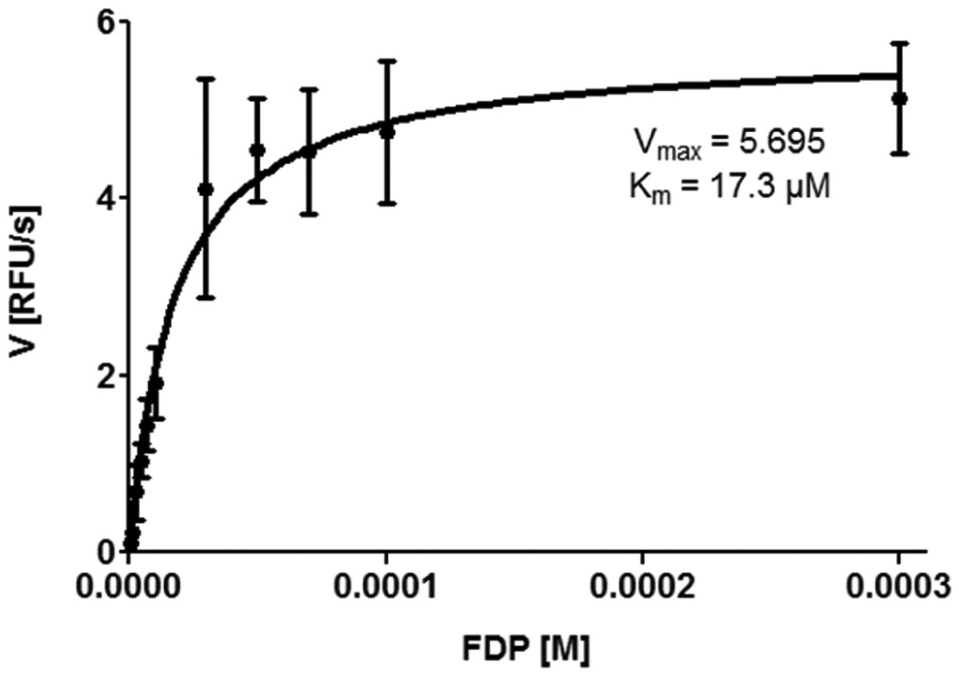
Kinetic parameters of soluble epoxide hydrolase with lipid phosphatase activity (sEH-P) using a fluorescein diphosphate (FDP) substrate. RFU, relative fluorescence units.
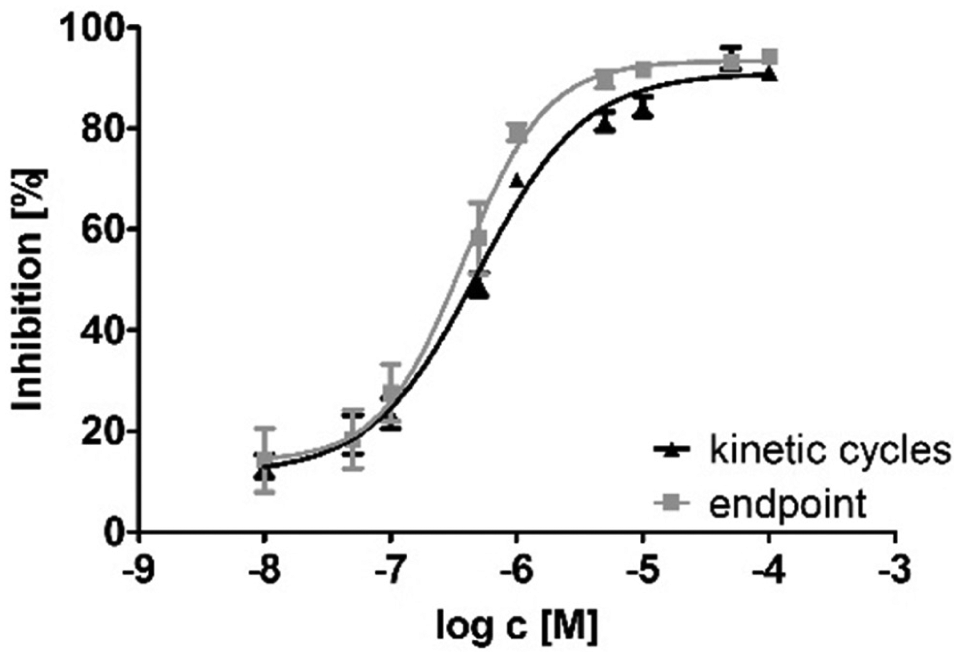
IC50 curves of ebselen measured in end-point format (black triangles) and in 60 kinetic cycles (gray squares).
From the Enzo library–approved drug library screen, nine compounds showed more than 50% inhibition at a 10-µM concentration: auranofin, carboplatin, cefotetan, cisplatin, entacapone, tolacapone, oxaprozin, silver sulfadiazine, and tiludronic acid. From these, five were selected for IC50 evaluation (
Table 2
). The silver ion of silver sulfadiazine disturbed the assay, so the compound was reordered as sodium salt but showed no activity and was eliminated from the hit list. Furthermore, we could not reproduce the inhibitory activity of tiludronic acid using the compound bought from Sigma-Aldrich. We validated entacapone, tolacapone, and oxaprozin for quenching activity (
Hit Compounds with IC50s on sEH-P and Their Original Targets as Defined within the ChEMBL Database.
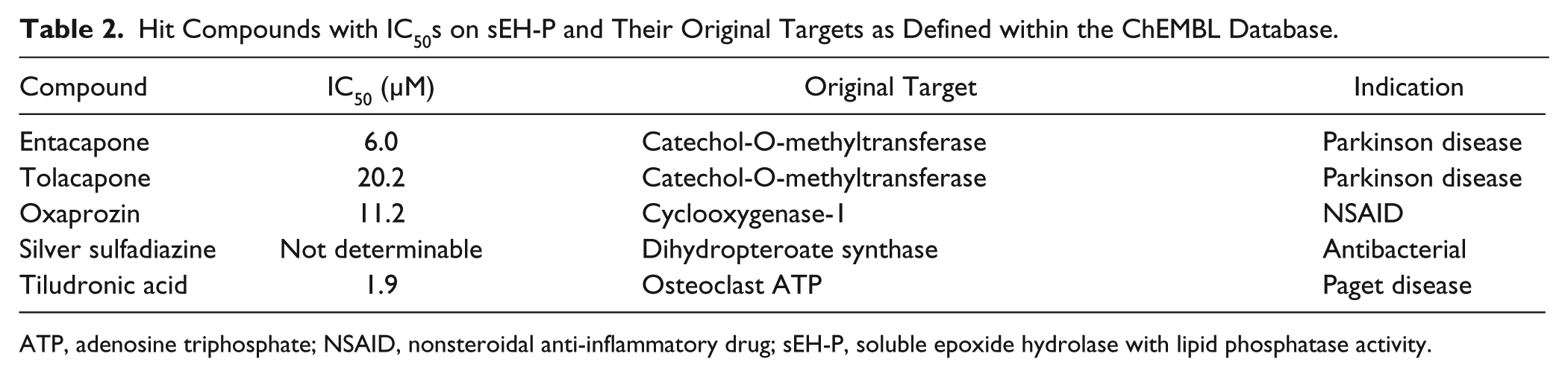
ATP, adenosine triphosphate; NSAID, nonsteroidal anti-inflammatory drug; sEH-P, soluble epoxide hydrolase with lipid phosphatase activity.
In the second screening round, the ChemBioNet library (~16,000 compounds) was screened at a 10-µM concentration, of which 400 substances showed significant inhibition over 3σ ( Fig. 3 ). After visual inspection of these substances, many were discarded because of interference compounds (PAINS, pan-assay interfering substances), 29 or they represented unwanted inhibition mechanisms of the hydrolase function (amides or ureas). Subsequently, 59 compounds were chosen for determination of dose-response curves, of which 12 gave reasonable curves.
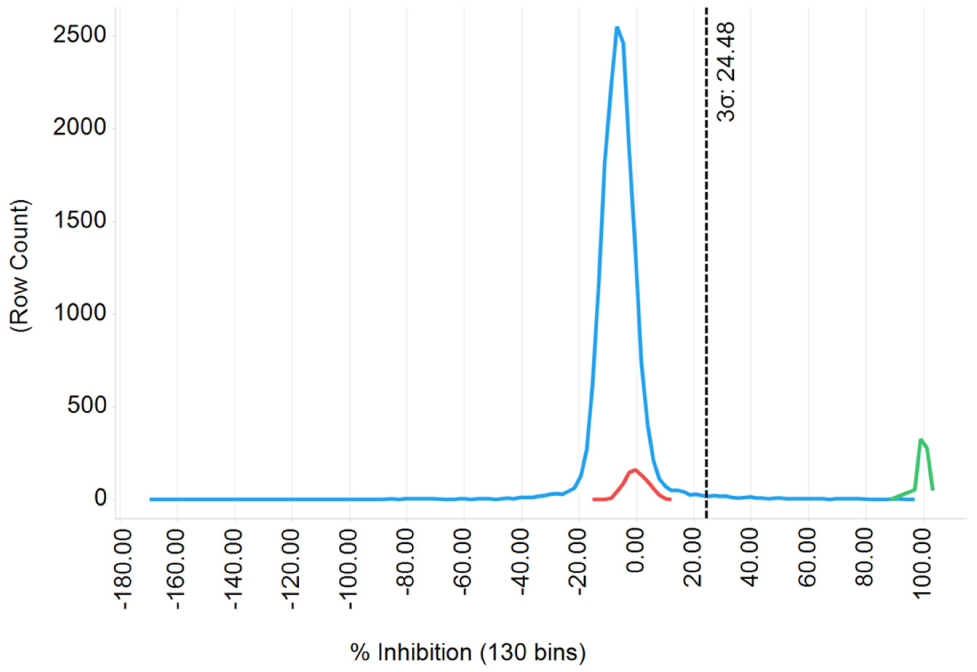
Frequency distribution graph derived from ChemBioNet library (blue line) screening. Blank samples are represented in red, with positive control (ebselen) in green.
In this study, we developed a robust assay system for identification of novel inhibitors of the phosphatase activity of soluble epoxide hydrolase. Previously, we and other groups reported the use of different substrates for quantification of sEH-P activity. P-Nitrophenolphosphate was the first reported sEH-P substrate, 3 which is chromogenic and unfortunately not suitable for HTS due to low sensitivity. The use of DiFMUP is limited to the excitation and emission wavelengths (λex 358 nm, λem 450 nm), which interfere with several heterocyclic compounds that are broadly present in HTS libraries. Tran et al. 18 developed an HTS assay using AttoPhos as a substrate. It exhibits excellent sensitivity and a large stoke shift. However, AttoPhos is suitable for alkaline phosphatases, while the highest activity of sEH-P is observed under acidic conditions. 3 Thus, FDP is suitable for kinetic observations of sEH-P activity at low pH.
Furthermore, it is proposed that the hydrolase domain of sEH regulates the activity of sEH-P 30 and vice versa. Thus, it was important to develop an assay system with an isolated N-terminal sEH-P domain to exclude the influence of the C-terminal domain. The N-terminal sEH-P domain can be easily expressed in a convenient E. coli expression system and purified in two steps in high yield. We could show that the mutated sEH-P domain does not interfere with the FDP substrate. The FDP assay was subsequently transferred to the 384-well format, and two compound libraries were successfully screened. We were able to identify oxaprozin as a promising starting point for the development of a chemical probe to investigate the physiological function of the phosphatase activity of soluble epoxide hydrolase.
Footnotes
Acknowledgements
We thank Paul Schmanke for the preparation of sEH-P Asp9Ala mutant and Steffen Hahn for purification of sEH-P.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the German Research Foundation (DFG; SFB 1039 Teilprojekt A07), the research funding programe Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz (LOEWE) of the State of Hessen, Research Center for Translational Medicine and Pharmakology TMP, and Fraunhofer Institute for Molecular Biology and Environmental Ecology.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.