
Abstract
Kinases are attractive drug targets because of the central roles they play in signal transduction pathways and human diseases. Their well-formed adenosine triphosphate (ATP)–binding pockets make ideal targets for small-molecule inhibitors. For drug discovery purposes, many peptide-based kinase assays have been developed that measure substrate phosphorylation using fluorescence-based readouts. However, for some kinases these assays may not be appropriate. In the case of the LIM kinases (LIMK), an inability to phosphorylate peptide substrates resulted in previous high-throughput screens (HTS) using radioactive labeling of recombinant cofilin protein as the readout. We describe the development of an HTS-compatible assay that measures relative ATP levels using luciferase-generated luminescence as a function of LIMK activity. The assay was inexpensive to perform, and proof-of-principle screening of kinase inhibitors demonstrated that compound potency against LIMK could be determined; ultimately, the assay was used for successful prosecution of automated HTS. Following HTS, the secondary assay format was changed to obtain more accurate measures of potency and mechanism of action using more complex (and expensive) assays. The luciferase assay nonetheless provides an inexpensive and reliable primary assay for HTS that allowed for the identification of LIMK inhibitors to initiate discovery programs for the eventual treatment of human diseases.
Introduction
LIM kinases play central roles in Rho GTPase regulation of the actin cytoskeleton by phosphorylating cofilin proteins (cofilin1, cofilin2, destrin/ADF) on Serine3 and inactivating their F-actin severing activity. 1 The LIM kinase family consists of two members; LIM kinase 1 (LIMK1) and LIM kinase 2 (LIMK2), which have 50% overall identity and 70% identity in their kinase domains. 1 LIMK activation results from activation loop phosphorylation by Rho GTPase–regulated kinases, including ROCK, PAK, and MRCK. 1 There are numerous ways that Rho GTPase activation has been associated with human cancers, particularly with progression to invasive and metastatic stages. 2 Therefore, LIMK makes an interesting prospective target for antimetastatic drug discovery. 3 The association of LIMK with a number of additional diseases, including primary pulmonary hypertension and glaucoma, highlights LIMK as an attractive drug target. 4
Convenient methods for kinase inhibitor high-throughput screens (HTS) often make use of short synthetic peptide substrates. However, the two published methods for screening chemical libraries to identify LIMK inhibitors used radioactive phosphate incorporation into recombinant cofilin 5 or the related cofilin family protein destrin. 6 Radiation-based screening presents numerous challenges for HTS, including radioactive contamination of the work environment and waste disposal. Therefore, we sought to develop a nonradioactive screening method that could be easily and cheaply transferred to HTS platforms in an academic screening environment.
One nonradioactive screening method uses luminescence generated by firefly luciferase, which uses ATP to catalyze the mono-oxygenation of luciferin to generate light over a broad linear range. 7 In this report, we demonstrate that a reliable and robust nonradioactive LIMK assay was developed using recombinant cofilin as substrate. After optimization of enzyme, substrate, and adenosine triphosphate (ATP) levels, the assay presented Z′ values compatible with transferability to HTS. A pilot screen identified several initial hits, thereby providing proof of concept. Therefore, this luminescence-based HTS assay is an attractive alternative to the radiation-based methods previously used to identify LIMK inhibitors or indeed any kinase where peptide substrates are not available. The assay proved reliable and inexpensive to operate, suffering only from a requirement for high amounts of relatively pure recombinant protein substrate and limited ability to perform mechanism-of-action studies and routine robust IC50 determinations.
Material and Methods
Cofilin Expression
Overnight cultures of pGEX-KG cofilin or pGEX-KG S3A cofilin in Escherichia coli BL21 (DE3) pLyss were grown in 200 mL of L-Broth containing 200 µg/mL ampicillin and 20 µg/mL chloramphenicol at 37 °C. Each culture was diluted 1:10 into 2 L of L-Broth with ampicillin and grown to OD600 0.6 to 1.0 at 37 °C before induction with 100 µM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 3 h at 37 °C. Cells were pelleted by centrifugation at 4500 g for 20 min at 4 °C, resuspended in 5 mL of Tris-buffered saline (TBS; pH 7.4) containing 3 mM dithiothreitol (DTT) and 1× Complete protease inhibitor cocktail (Roche, Basel, Switzerland), and then disrupted by three 1-min rounds of sonication at 20% intensity using a Branson Digital Sonifier. (Thistle Scientific, Glasgow, UK) Debris was removed by centrifugation at 12 000 g for 30 min at 4 °C, and clarified supernatants were incubated with 10 mL bed-volume of TBS/DTT-washed glutathione-Sepharose (GE, Piscataway, NJ) bead slurry in BioRad (Hercules, CA) 14-cm Econo-Pac Chromatography Columns overnight at 4 °C. Beads were washed with >50 bed-volumes of TBS/DTT and cofilin released from the glutathione S-transferase (GST) moiety by incubation with 250 units of bovine thrombin (Sigma, St. Louis, MO) overnight at 4 °C. Supernatant was removed and incubated with 30 µL of washed p-aminobenzamidine beads (Sigma) for 1 h at room temperature to remove thrombin, before snap-freezing aliquots and storing at –80 °C. Protein concentration and purity were determined by running on a 15% polyacrylamide gel against bovine serum albumin (BSA) standards, followed by staining with SimplyBlue Safestain (Invitrogen, Carlsbad, CA) and quantification using a LiCor (Lincoln, NE) Odyssey infrared scanner.
Radioactive Kinase Assays
Recombinant cofilin or synthetic peptides were evaluated as LIMK1 substrates in an assay carried out at 30 °C for 10 min in a total volume of 50 µL containing 62.5 mM Tris-HCl (pH 7.5), 1.25% BSA (w/v), 0.1% (v/v) 2-mercaptoethanol, 0.5 mM EGTA, 0.01% Brij35 (v/v), 20 mM MgCl2, 0.2 mM ATP + [γ-32P] ATP (2 × 106 cpm/nmol) with 0.02 µg LIMK1 active kinase domain (Upstate Biotechnology, Millipore, Dundee, UK), and 500 µM peptide substrate or 0.2 µg cofilin (200 nM) as indicated. Reactions were stopped by spotting onto 4-cm2 squares of p81 paper and submerging in 0.5% (v/v) orthophosphoric acid, followed by 3 × 15-min orthophosphoric acid washes and a final 5-min acetone rinse. Once dried, phosphate incorporation was determined by Cerenkov counting.
Phosphorylation of Peptide Array
PepChip (Pepscan, Lelystad, the Netherlands) kinase slides were used to identify LIMK1 peptide substrates. On each glass slide was an array of 1176 peptides in duplicate. Phosphorylation was performed according to the manufacturer’s instructions. A 60-µL reaction mix containing 62.5 mM Tris-HCl (pH 7.5), 1.25% BSA (w/v), 0.1% (v/v) 2-mercaptoethanol, 0.5 mM EGTA, 0.01% Brij35 (v/v), 20 mM MgCl2 with 0.03 µg LIMK1, and 10 µM ATP containing 10 µCi [γ-32P] ATP was made for each slide; 50 µL of this reaction mixture was then pipetted into the center of a glass coverslip and the peptide array gently lowered onto it. The slide was then turned over and incubated in a humidified chamber at 30 °C for 2 h before washing off the coverslip and reaction mix in phosphate-buffered saline (PBS) with 1% Triton X-100 (v/v). The slide was transferred to 2 M NaCl, 1% Triton X-100 for 2 × 5-min washes at room temperature, followed by 3 × 5-min H2O washes before the slides were air-dried and exposed to film overnight. The resulting films were aligned with the provided grid image in Adobe Photoshop (Adobe Systems Inc., San Jose, CA).
Luciferase-Based Kinase Assay
Kinase-Glo (Promega, Madison, WI) is a luminescence-based assay in which kinase inhibition may be determined by changes in luciferase activity as a function of ATP depletion. 8 LIMK assays were carried out in 96-well white plates in a total volume of 40 µL of 40 mM MOPS (pH 7.0) and 1 mM EDTA in the presence of 25 µM cofilin. The reaction was incubated for 1 h at room temperature, an equal volume of Kinase-Glo reagent was added to each well, the plate was further incubated for 30 min, and luminescence was read in Tecan (Männedorf, Switzerland) Safire 2 microplate reader.
Kinase Inhibitors
The standard inhibitor plate purchased from Biomol (BML-2832; San Diego, CA) contained 80 known kinase inhibitors supplied as 10-mM DMSO solutions in a 96-well plate format. Compounds were tested in duplicate at 1 and 10 µM in the Kinase-Glo assay. As a counterscreen, the Kinase-Glo assay was also run in the absence of LIMK1 to eliminate compounds that interfere with the luciferase enzyme. Inhibitors that result in >50% inhibition at 1 µM compound and did not interfere with luciferase activity were resupplied from Calbiochem (San Diego, CA).
HTS and IC50 Determinations
HTS was performed in 384-well white plates. Compounds were tested singly at 30 µM. The Kinase-Glo assay was carried out in a total volume of 10 µL of 20 mM HEPES (pH 7), 10 mM MgCl2, 0.25 mM EGTA, 0.01% (v/v) Triton X-100, and 1 mM DTT in the presence of 7.5 ng LIMK1, 8 µM cofilin, and 20 µM ATP. The reaction was incubated for 1 h at room temperature, and then an equal volume of Kinase-Glo reagent was added to each well. The plates were sealed and shaken on a Denley Minimix (Thermo Scientific, Epsom, UK) for 20 s and then incubated for a further 20 min, protected from light before reading on an Analyst HT microplate reader. Resupply of confirmed hits and analogue by catalogue (ABC) compounds were from ChemBridge (San Diego, CA), Ambinter (Paris, France), ChemDiv (San Diego, CA), Key Organics (Camelford, UK), Biofocus (Saffron Walden, UK), and InterBioScreen (Moscow, Russia). Ten-point IC50 curves were generated for all resupplied and ABC compounds.
Alternative Assay Format
The Homogeneous Time-Resolved Fluorescence (HTRF) Transcreener adenosine diphosphate (ADP) assay (Cisbio, Bedford, MA) is a competitive immunoassay that directly measures the competition of native ADP generated from the hydrolysis of ATP with d2-labeled ADP for a monoclonal antibody labeled with Eu3+ cryptate. LIMK1 assays were carried out in 384-well low-volume white plates in a total volume of 10 µL of 20 mM HEPES (pH 6.8), 10 mM MgCl2, 0.25 mM EGTA, 0.01% v/v Triton X-100, and 1 mM DTT in the presence of 0.5 ng LIMK1, 2.8 µM cofilin, and 20 µM ATP. The reaction was incubated for 1 h at room temperature, then 2.5 µL of 1X ADP-d2 followed by 2.5 µL of 1X anti-ADP cryptate was added to each well, the plate was further incubated for 60 min, and fluorescence was read at ex 320 nm/(em 620 nm) (donor) and at ex 320 nm/(em 665 nm) (acceptor) on a Tecan Ultra microplate reader. For each plate blank controls, cryptate was added in quadruplicate to monitor the cryptate signal at 620 nm and define the lower limit of the assay window. The fluorescence ratio (acceptor/donor) was calculated and used to determine the Delta F%, that is, the relative energy transfer rate for each data point (Delta F% = (ratio – ratioblank)/ratioblank × 100). Percentage inhibition of LIMK1 activity was then calculated from Delta F%.
Results
The major LIMK substrates are the cofilin family proteins (cofilin1, cofilin2, and destrin/ADF), which are phosphorylated on a conserved serine residue at position 3 (S3). Recombinant cofilin1 can be efficiently phosphorylated in vitro ( Fig. 1A ) but not if S3 was mutated to alanine (S3A). Since NMR studies of cofilin (PDB ID 1Q8G and 1Q8X) revealed an unstructured and flexible amino-terminus, 9 it seemed likely that LIMK would phosphorylate corresponding peptides in solution. The first 11 amino acids of cofilin1 (Cfl1) and cofilin2 (Cfl2) were assayed for phosphorylation at equimolar concentration to recombinant cofilin. Surprisingly, neither peptide was phosphorylated above background ( Fig. 1B ). A peptide corresponding to the first 20 amino acids of cofilin1 was also not phosphorylated by LIMK1 (data not shown).
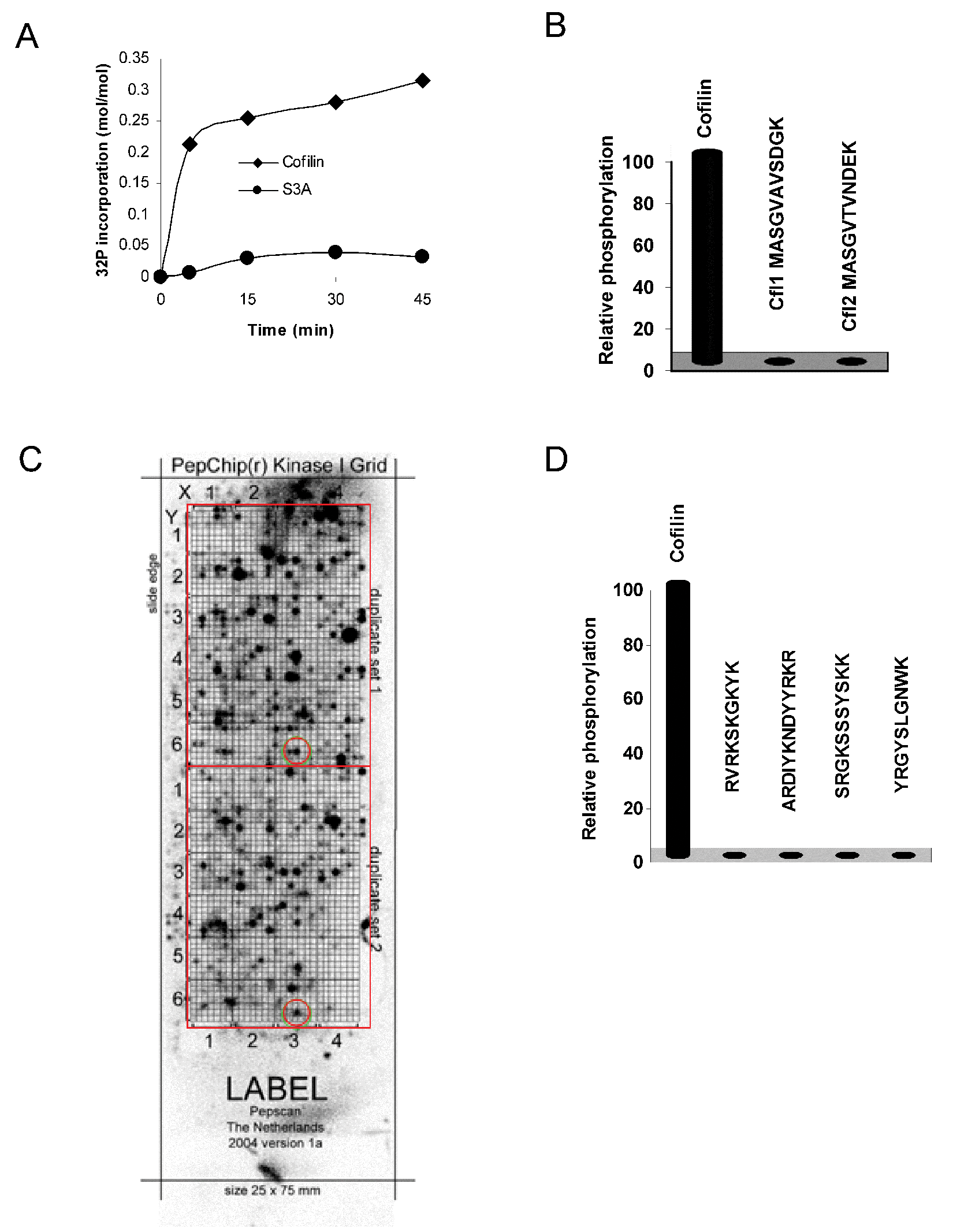
LIMK phosphorylation is restricted to cofilin protein. (
Any peptide phosphorylated by LIMK1, regardless of whether it was a physiological substrate, could potentially be used for assay development. To identify peptide substrates, we used a PepChip slide that displays 1176 peptides in duplicate on a glass slide. After incubation with LIMK1 and [γ-32P]-labeled ATP ( Fig. 1C ), individual spots comparably labeled in each duplicate array were identified and ranked. The four most reproducible and radioactively labeled peptides were synthesized and assayed in solution for phosphorylation. Despite LIMK1 phosphorylating cofilin on a serine residue, it actually belongs to the tyrosine kinase-like family, raising the possibility that tyrosine-containing peptides might be substrates. However, none of the four peptides were phosphorylated in solution ( Fig. 1D ). Although an immobilized peptide array makes screening candidate peptide substrates convenient, it has been reported that immobilization of peptides on solid supports may result in artifact, possibly due to high local concentrations of substrate. 10 Because of these negative results, the lack of reported LIMK peptide substrates, and published methods for assaying LIMK activity that have used the entire cofilin protein, we next aimed to develop a nonradioactive LIMK HTS-suitable assay that used recombinant protein as substrate.
Luciferase-Based Assay for Determination of LIM Kinase Activity
The best LIMK substrate was full-length recombinant cofilin, which, given the ~20-kDa size, meant that fluorescence polarization methods were not viable assay formats. Instead, we examined a luciferase-based assay that reflects kinase activity as a function of ATP depletion as a candidate assay.
To optimize kinase conditions, we evaluated activity as a function of various parameters. A full ATP-dependence determination was carried out, which resulted in a Kmapp ATP extrapolated to be in the region of 10 to 20 µM ( Fig. 2A ). Next, the Km for protein substrate was determined as 8 µM by performing a cofilin dose-response assay at 10 ng/well enzyme concentration and 50 µM ATP ( Fig. 2B ). LIMK1 enzyme titrations were carried out at saturating substrate concentrations (25 µM), 50 µM ATP, and one in two serial dilutions of the kinase ( Fig. 2C ). By incubating luciferase with increasing LIMK1 concentrations without cofilin, we determined that an LIMK1-induced decrease in luciferase activity was substrate dependent ( Fig. 2D ). Cofilin alone yielded no apparent ATP hydrolysis, indicating that no contaminating ATPases were co-purified (data not shown).
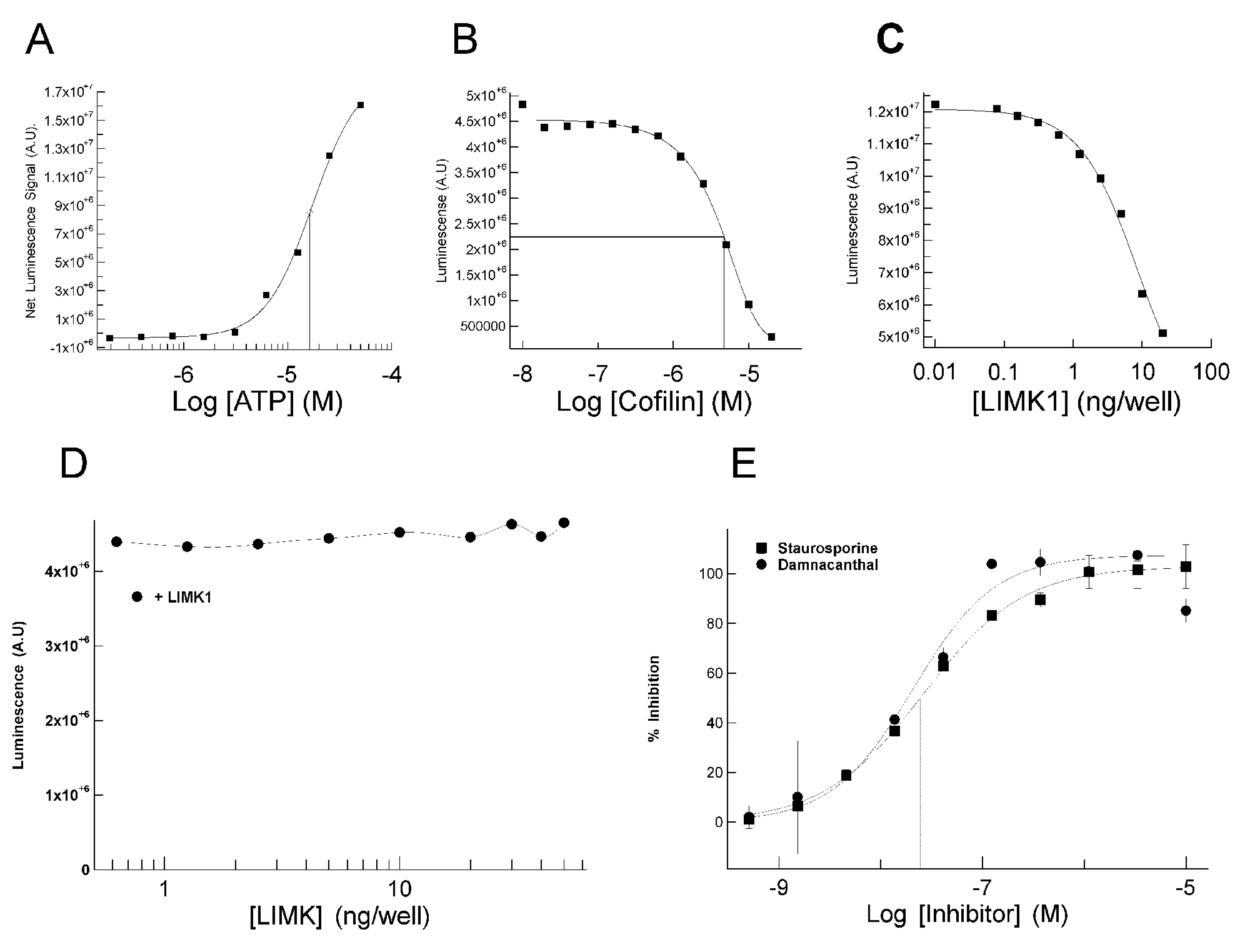
Characterization and optimization of luciferase-based LIMK assay. (
Prior to initiating HTS, a panel of kinase inhibitors was screened for proof of concept and to identify LIMK inhibitor(s) for use as a standard. From this panel, staurosporine and damnacanthal were identified and subsequently determined to inhibit LIMK1 with IC50 values of 28 and 19 nM, respectively ( Fig. 2E ). Staurosporine was previously reported to bind LIMK1. 11 LIMK inhibition by the phytochemical anthraquinone damnacanthal, previously identified as an Lck inhibitor, 12 was a novel observation. Staurosporine was subsequently used at a fixed concentration on each HTS assay plate to provide an internal control.
Summary of Chemical Library HTS
Using the luciferase-based LIMK1 assay, an HTS of kinase-focused (9459 entities) and diverse compound libraries (48 209 entities) was prosecuted. LIMK was used at 7.5 ng/well with 30 µM of each inhibitor and ATP at the calculated Km (20 M). It was estimated that in each well, hydrolysis of approximately 30% (6 µM) of the ATP was required to give a robust signal. Plate-by-plate values ( Fig. 3A ) for negative (blank) and positive (enzyme) controls and calculated Z′ factors, calculated according to the method of Zhang et al., 13 showed good assay reproducibility during the HTS with a global mean Z′ of 0.5. Plates with initial Z′ factors <0.5 were rescreened and subsequently had acceptable Z′ factors >0.5. Staurosporine was used in quadruplicate at a calculated 50% inhibitory concentration (actual inhibition = 59% ± 16%). Histogram analysis of the raw data set revealed a typical skew normal distribution centered on 0% to 10% inhibition ( Fig. 3B ). The compound hit rates with ≥50% inhibition from the diverse and kinase-focused libraries were 0.5% and 3.4%, respectively. Retesting of 320 initial hits from both libraries in the primary assay and against the luciferase reaction alone confirmed 258 as having greater than 50% inhibition, yielding final hit rates from the diverse and kinase-focused libraries of 0.08% and 2.3%, respectively. Preliminary analysis of the mode of inhibition with respect to ATP was performed by determining inhibition by each compound at 30 µM with ATP concentrations of 2 or 20 µM ( Fig. 3C ). A diagonal line was simulated for ATP noncompetitive inhibitors, whereas the curved line was simulated for ATP competitive inhibitors. 14 The compounds tested were largely ATP competitive, clustered around a curved simulated fit, indicating lower inhibitory activity with increased ATP.
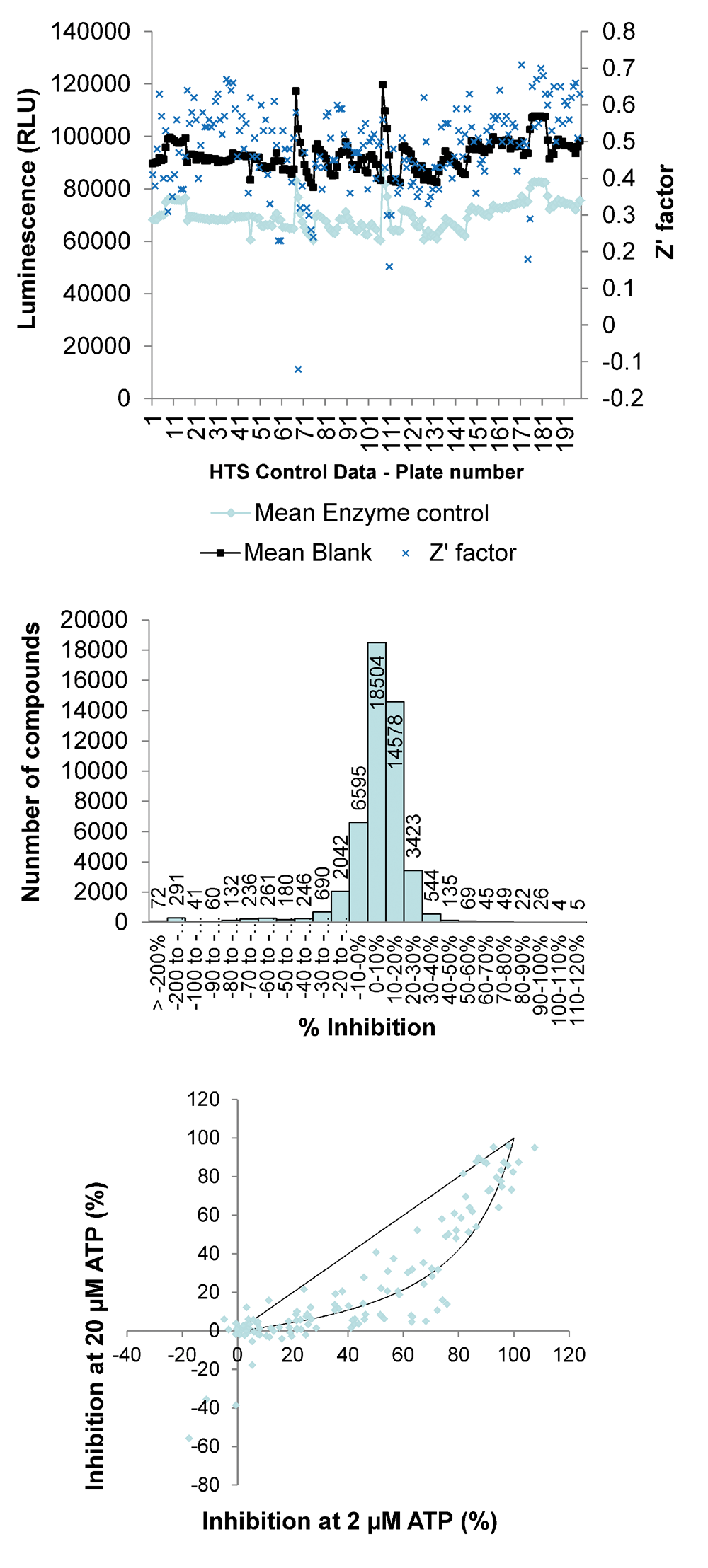
Results of a high-throughput screen (HTS) using luciferase-LIMK assay. (
Comparison of the Luciferase ATP Depletion Assay with an ADP Accumulation Assay
The luciferase-based LIMK assay has shown itself as a robust and economical assay for HTS. However, a second kinase assay was established to expand capability in mechanism and IC50 studies. The HTRF Transcreener assay is a competitive immunoassay in which ADP labeled with the acceptor fluorophore d2 and unlabeled ADP compete for binding to a monoclonal anti-ADP antibody conjugated with Europium cryptate (Eu3+ cryptate) that acts as an energy donor. 15 This assay is more specific than the luciferase-based assay as it measures cofactor product accumulation rather than loss of ATP, which could be susceptible to any ATP-depleting mechanism (e.g., nonspecific degradation to AMP or adenosine). A wider range of concentrations of ATP can be used in this assay by modifying the concentration of ADP-d2 tracer used. At 20 µM ATP, as little as 20 to 50 nM ADP production could be accurately measured ( Fig. 4A ). A good signal window could be achieved (>75% of max signal) at a low conversion of ATP to ADP (~11%) using this assay, in contrast to the luciferase-based assay in which a much higher percentage of ATP consumption was necessary to get a good signal window (>30%). During development of this more sensitive format, it was determined that LIMK1 hydrolyzed ATP somewhat even in the absence of cofilin in a concentration-dependent manner ( Fig. 4B ; 0 ng/well cofilin data points). This effect was not observed in the Kinase-Glo assay due to its lower sensitivity than the HTRF Transcreener assay. As a result, background-contaminating ATPase activity fell within the noise of the control signal. The superior sensitivity of the Transcreener method allowed the amounts of LIMK1 and cofilin to be reduced while maintaining a good signal window and low product (ADP) formation. Final assay concentrations of 0.5 ng/well LIMK1 and 2.8 µM cofilin were used to generate IC50 curves for subsequent hit-to-lead studies. IC50 values for staurosporine from the Kinase-Glo and HTRF Transcreener ( Fig. 4C ) assays were comparable at 29 nM and 17 nM, respectively. Comparison of pIC50 values for 81 of the HTS hit compounds using the Kinase-Glo assay were next compared with data obtained using the HTRF Transcreener assay ( Fig. 4D ). Overall, the IC50 values were lower with the HTRF Transcreener assay than for the Kinase-Glo, but the rank orders of potency were similar between the two assay formats with a Pearson r = 0.59 and a slope fitted by linear regression approaching 1 (0.97).
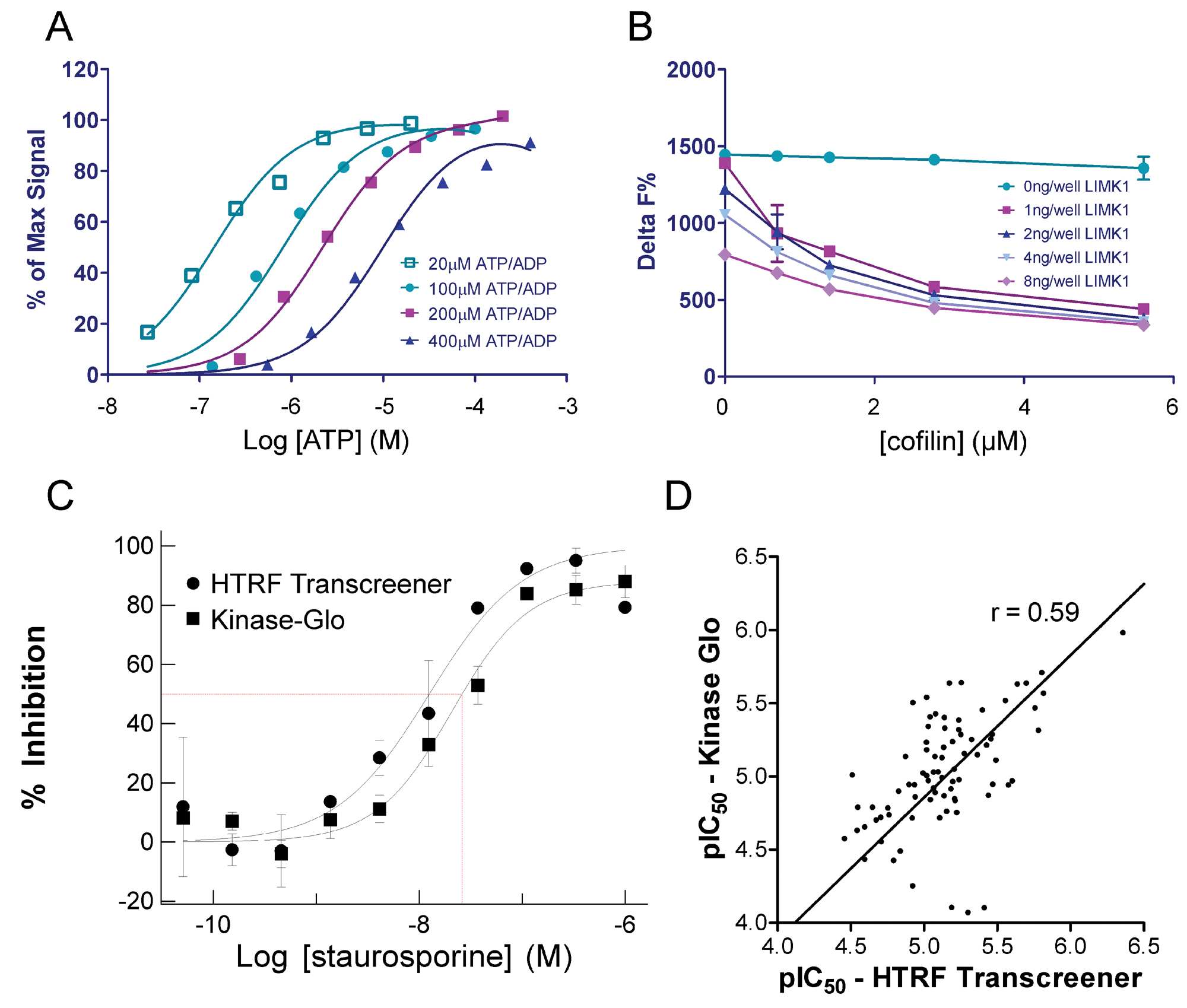
(
Discussion
A large number and variety of kinase assays are currently used as drug screening platforms. Although robustness and reproducibility are key factors for such assays to be implemented in HTS labs, additional factors including convenience, cost, and safety are routinely considered. The majority of these assays measure phosphate incorporation into a peptide substrate either directly or indirectly. However, in some cases where it is not possible to follow phosphate incorporation, alternative approaches that indirectly determine kinase function have been developed that measure ATP depletion or ADP accumulation as a function of kinase activity.
In the present work, we sought to develop a rapid, nonradioactive, robust, reproducible, and inexpensive HTS-compatible assay for LIMK1. Based on the current literature, the best characterized substrate for these enzymes is cofilin, which is phosphorylated on a single serine residue located at position 3. With the availability of anti-phospho-cofilin antibodies that recognize phosphorylated S3, we attempted to develop a fluorescence polarization-competition assay in which a fluorescently labeled phospho-S3-containing peptide complexed to antibody is competed off with nonfluorescent peptide or protein that has been phosphorylated in an in vitro kinase reaction. This approach failed for two reasons: Although validated for phospho-selectivity and adequate for Western blotting or immunohistochemistry of phosphorylated cofilin, 16 commercial phospho-selective antibodies had insufficient avidity for phosphopeptides (data not shown) or for use in an enzyme-linked immunosorbent assay (ELISA) format against phosphorylated cofilin protein (data not shown), and peptides were not phosphorylated by LIMK ( Fig. 1B ). The latter deficiency proved to be an insurmountable hurdle for the development of a peptide-based assay format.
Given the somewhat surprising inability of LIMK1 to phosphorylate peptides corresponding to the amino-terminus of cofilin, an attempt was made to identify any peptide that could be phosphorylated in vitro by screening an immobilized peptide array. Despite seemingly encouraging results from the array experiment ( Fig. 1C ), none of the identified peptides could be validated as substrates in solution ( Fig. 1D and data not shown). The fact that recombinant protein was also used in the two published radioactive screens for LIMK inhibitors5,6 and the absence of commercially available peptide-based LIMK assays suggest that LIM kinases do not readily phosphorylate anything other than full-length proteins. This is probably a rare property among protein kinases, as many have had peptide substrates identified for use in assays, but it nonetheless raises the question as to how well peptide substrates (especially those from nonnatural kinase targets) represent the in vivo activity of a kinase and whether HTS using these substrates disadvantages the chemistry output as a consequence. An unanswered question was whether full-length LIMK would phosphorylate peptides, but given that the kinase domain does phosphorylate cofilin suggests that an absence of substrate docking sequences does not account for the lack of peptide phosphorylating activity.
Because of the inability of LIMK to phosphorylate peptides, we sought to develop a nonradioactive LIMK assay that used recombinant full-length cofilin protein as substrate. Fortunately, cofilin expression as a GST fusion protein in E. coli works very well, with high levels of expression, solubility, purity, and recovery in the range of 10 to 20 mg per liter of bacterial culture (data not shown). Cleavage of the GST moiety was found to be essential for phosphorylation by LIMK1. Optimization of kinase reaction variables (e.g., substrate, ATP, enzyme) resulted in the establishment of parameters that yielded satisfactory Z′ values for HTS and an assay that was capable of generating reasonably robust IC50 measurements. The luciferase assay format was extremely easy to work with and automate, and as such was a suitable assay for HTS with certain limitations. Care should be taken with any ATP-based assay to have extremely clean protein starting materials, especially if the specific activity of the kinase in question is low. Bacteria and baculovirus used in most protein expression systems contain a plethora of ATP-using enzymes that have very high activity; as a result, even a trace of contamination from one of these highly active enzymes would register a false-positive signal and potentially mislead data analysis. Specificity of reaction can be proven easily if, as in our case, there is limited ATP hydrolysis in the presence of either enzyme or substrate individually and a high rate in the presence of both. Proof of concept was also demonstrated through the dose-dependent inhibition of LIMK1 activity by staurosporine ( Fig. 2E ), which, although promiscuous among kinases, had previously been shown to bind LIMK1. 11 The inhibition of LIMK1 activity by only two inhibitors suggests that it might be possible to develop potent inhibitors with good selectivity profiles. Once HTS was complete, the limitation of the ATP depletion assay of most concern was the lack of scope to change the ATP concentration to determine inhibitor mechanism of action. The relatively high substrate usage (up to 40%) required to gain a robust signal also resulted in an unacceptable degree of variability in subsequent structure-activity test cycles within the LIMK project. For these reasons, the more expensive HTRF Transcreener assay was developed and became the assay of choice for hit-to-lead work on this project. Using the HTRF Transcreener assay, we were able to provide further post hoc validation of the less expensive luciferase-based assay ( Fig. 4C , D ). In conclusion, a rapid and robust nonradioactive HTS-compatible assay for LIMK has been developed based on the relative determination of ATP levels by luciferase-mediated luminescence.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported by Cancer Research UK.