
Abstract
Members of the ether-à-go-go (EAG) family of voltage-gated K+ channels are involved in several pathophysiological diseases, and there has been a great interest in screening for drugs that modulate the activity of these channels. Many drugs have been shown to bind in the pore of these channels, blocking ion flux and causing disease pathology. In this report, we present two independent screening campaigns in which we wanted to identify small molecules that bind to either the intracellular cytoplasmic amino terminal Per-Arnt-Sim (PAS) domain from the human EAG-related gene (ERG) channel or the amino or carboxy terminal globular domains from the mouse EAG1 channel, affecting their interaction. We report that in both cases, compounds were identified that showed weak, nonspecific binding. We suggest alternative routes should be pursued in future efforts to identify specific, high-affinity binders to these cytoplasmic domains.
Introduction
The EAG family of voltage-gated K+ channels clusters by sequence similarity into three subfamilies: ether-à-go-go (EAG), EAG-related gene (ERG), and EAG-like K+ channels (ELK). They are encoded by the KCNH1 to 8 genes. 1 These channels are involved in several human pathophysiological diseases. Loss of human ERG (hERG) K+ channel function either by inherited mutations or by secondary pharmacological effects is associated with long QT syndrome (LQTS), which can lead to ventricular arrhythmias and sudden cardiac death. In addition, expression of both hERG and human EAG1 is increased in a number of cancer cell types, where they are associated with proliferation and invasiveness phenotypes.2,3 Over the years, most studies on potassium channels have focused on the inhibitory effects of various compounds on channel gating. However, in an effort to understand the molecular mechanisms underlying channel dysfunction and associated rescue strategies, an increasing number of studies are shifting the focus from channel blockage to other aspects of channel regulation. 4
Channels in the EAG family share a characteristic architecture with four subunits arranged to form a central K+ conducting pore. Each subunit has large cytoplasmic amino and carboxy terminal regions ( Fig. 1 ). The amino terminal region contains the Per-Arnt-Sim (PAS) domain that in other proteins has been shown to bind small molecules and modulate protein activity. 5 Crystal structures have been reported for the PAS domains from hERG and mouse EAG (mEAG) channels; they show that these domains have cavities similar to where other PAS domains bind small molecules; however, no ligands have been reported thus far. 6 The carboxy terminal region contains a cyclic nucleotide-binding homology (CNBh) domain, but the EAG family of channels is not regulated by direct binding of cyclic nucleotides. Rather, it has been shown that an intrinsic ligand occupies this pocket; it has also been suggested that flavonoids could compete within this binding pocket to potentiate channel function.7,8 In addition, biochemical and functional data have shown that the PAS domain associates with the CNBh domain in solution. A recent X-ray crystal structure of a complex of the mEAG1 PAS domain and CNBh domain shows that this protein interface is a hotspot for many of the known mutations that are associated with human disease pathology. 9 It is therefore likely that the interaction between the PAS domain and the CNBh domain plays a key role in modulation of channel function.
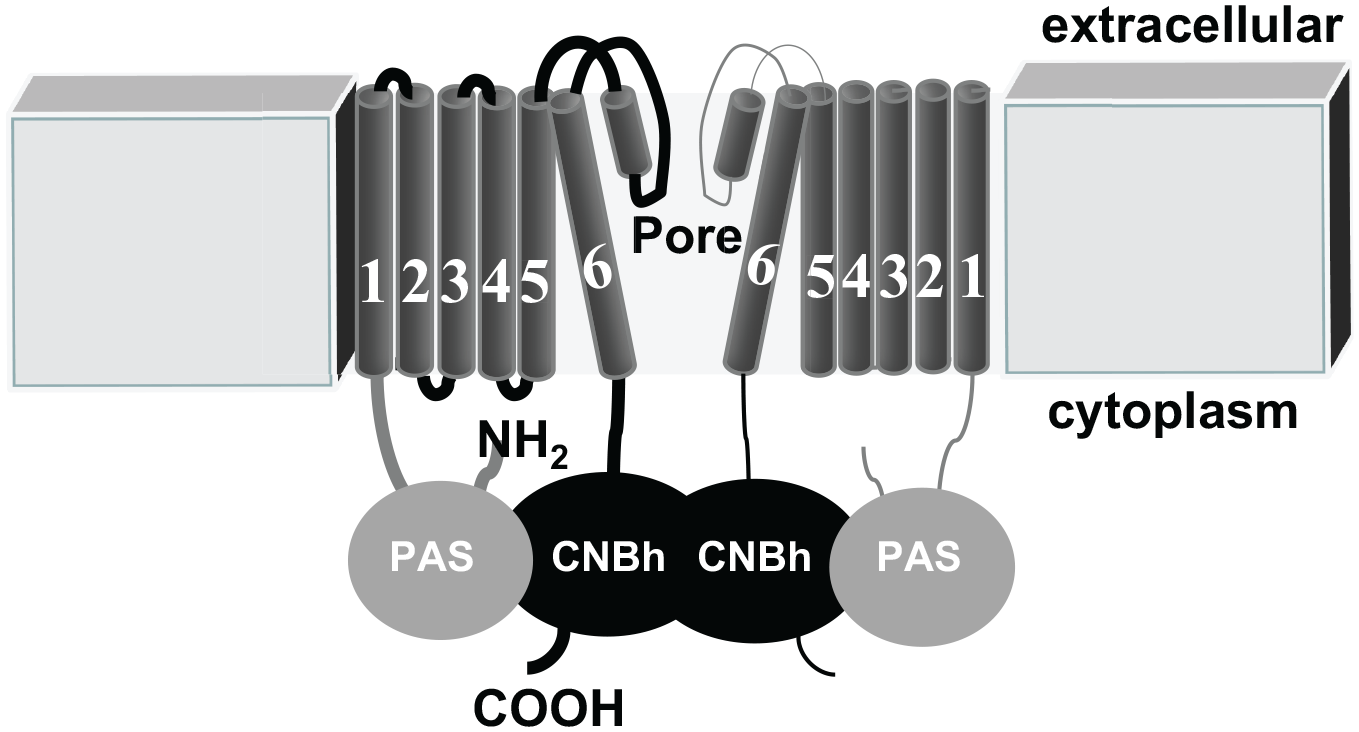
Schematic diagram showing two subunits of a tetrameric ether-à-go-go (EAG) K+ channel. Each subunit consists of six transmembrane-spanning domains. S1 to S4 represent the voltage-sensing domain and S5-helix-S6 represent the pore domain. Each subunit contains a Per-Arnt-Sim (PAS) domain in the amino terminus and a cyclic nucleotide-binding homology (CNBh) domain in the carboxy terminus.
In this study, we present our conclusions from two independent screening campaigns, each of ~5000 compounds, to identify small molecules that bind specifically and with high affinity to the cytoplasmic domains of EAG channels. These high-affinity binding molecules could potentially be used as novel modulators of channel activity. First, we screened for small molecules that affect the stability of the hERG PAS domain using a fluorescence-based thermal shift assay, and concurrently we screened for small molecules that could modulate the association between the mEAG1 PAS and CNBh domains (which are 98.5% and 100% identical to the human EAG1 proteins, respectively) using a fluorescence polarization assay.
Materials and Methods
Protein Production and Labeling
The hERG PAS wild-type domain (residues 1–135) was expressed and purified as previously described. 10 The mouse EAG1 PAS protein (residues 1–137) was cloned by using sequence-specific primers, as well as expressed and purified following the procedure described previously for a construct spanning residues 27 to 136. 6 The mouse CNBh domain (residues 552–724) was expressed and purified as described. 7 KtrA from Bacillus subtilis was purified as previously described. 11
To label mEAG1 PAS on accessible cysteine residues with fluorescein, the purified protein was loaded on a HiTrap Desalting column (GE Healthcare, Piscataway, NJ) to remove dithiothreitol (DTT) by buffer exchange with 50 mM Tris (pH 8.0) and 150 mM NaCl as the buffer. Protein was incubated with a 5- or 10-fold molar excess of fluorescein 5-maleimide (Thermo Fisher Scientific, Waltham, MA) for 1 h at room temperature protected from light. The reaction was stopped by adding DTT to 5 mM. Free fluorescein was removed by dialysis overnight at 4 °C against 20 mM Tris (pH 8.0), 150 mM NaCl, and 5 mM DTT. The fluorescein to protein molar ratio of fluorescein-labeled PAS (F-PAS) was calculated as indicated by the manufacturer and was typically found to be 3 or 4.
hERG PAS Thermal Shift Screen
A screening campaign was conducted using the ApexScreen Library from TimTec (Newark, DE). The ApexScreen is a collection of 5080 compounds that were selected to represent the diversity within the TimTec stock of over 160,000 compounds. Compounds were provided at 3 mM in 100% DMSO in a 96-well format and 100 µL/well. Columns 1 and 12 were empty and used as positive and negative controls, and therefore each plate contained 80 compounds. Then, 25 µL of stock compounds was transferred to an intermediate plate containing 275 µL of buffer (50 mM Hepes [pH 7.5], 150 mM NaCl, 5 mM DTT, and 4% DMSO) to produce an intermediate plate that contained 250 µM of compound and 12% DMSO.
A thermal shift assay that reports the stability of the hERG PAS domain in solution was essentially as previously described 10 but adapted for compound library plate-based screening. In brief, 20 µL of purified hERG PAS protein (7.5 µM) was combined with 20 µL Sypro Orange Dye (6.25×) (Sypro Orange solution from Sigma-Aldrich [St. Louis, MO] comes as a 5000× stock) in a 96-well Bio-Rad (Hercules, CA) Multiplate white PCR plate. To this assay plate, 10 µL of intermediate compound plate was added, resulting in an assay volume of 50 μL with final concentrations of 3 µM hERG PAS, 2.5× Sypro Orange Dye, and 50 µM compound in 50 mM Hepes (pH 7.5), 150 mM NaCl, 5 mM DTT, and 2% DMSO. The final protein to compound molar ratio was 0.06. Importantly, we verified that the thermal profile of the PAS protein was not affected by the presence of 2% DMSO. The plates were heated from 25 °C to 80 °C in an iQ5 Real Time Detection system (Bio-Rad). Unfolding of hERG PAS protein was monitored using the excitation and emission filters 545/30 and 585/20, respectively.
Thermal Shift Assay for Retest
Compounds from both screening assays were tested in a thermal shift format at a final concentration of 50 µM or 100 µM. Purified mEAG1 PAS, CNBh, or hERG PAS was added to a 96-well plate at 3 µM concentration in 50 mM Hepes (pH 7.5), 150 mM NaCl, 5 mM DTT, and 2% DMSO. Data were recorded as mentioned above. When thermal stability of PAS was tested against compound concentration, DMSO was kept at 2%. Change in Tm was plotted against compound concentration, and the Kd was determined from fitting the experimental data with equation (1) or with a similar equation described in Vivoli et al. 12 :
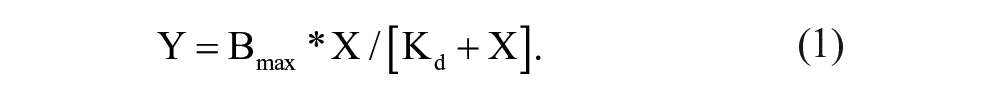
In this equation, Y represents the change in Tm on binding, X represents the concentration of compound, and Bmax represents the maximal binding.
mEAG1 PAS and CNBh Domain Fluorescence Anisotropy Screen
A screening campaign was conducted using a compound library purchased from the University of Wisconsin Small Molecule and Screening Facility. The facility possesses a compound library from the ChemBridge DIVERSet, a collection of “universally” diverse, predesigned drug-like small molecules. The compounds have been rationally selected based on 3D pharmacophore analysis to cover the broadest part of biologically relevant pharmacophore diversity space. Compounds were provided at 10 mM in 100% DMSO in a 384-well format and 50 µL/well. Using a Liquidator 96 Manual Pipetting System (Mettler Toledo, Greifensee, Switzerland), 5 µL of stock compounds (5760 compounds) was transferred to an intermediate plate containing 95 µL of buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 5 mM DTT, and 15.8% DMSO) to produce an intermediate plate that contained 500 µM of compound and 20% DMSO.
A fluorescence anisotropy assay that reports the interaction of fluorescein-labeled mEAG1 PAS protein with the CNBh domain was adapted for plate-based screening from a previously described assay. 9 The assay is based on an increase of F-PAS fluorescence anisotropy (r value) when the F-PAS and CNBh complex is formed. In brief, F-PAS (0.11 µM) and CNBh (11 µM) or F-PAS (0.11 µM) alone were added in assay buffer (90 μL) to a 96-well black, flat-bottomed microplate (Greiner Bio-One, Monroe, NC). In each fourth plate, rows A to D of column 1 contained no compound and were used as positive controls, and rows E to H of the same column contained F-PAS only and were used as negative controls. To this assay plate, 10 µL of intermediate diluted compound was added, resulting in an assay volume of 100 μL, with final concentrations of 0.1 µM F-PAS, 10 µM CNBh, and 50 µM compound in 20 mM Tris (pH 8), 150 mM NaCl, 5 mM DTT, and 2% DMSO. The final protein to compound molar ratios were 0.002 and 0.2 for F-PAS and CNBh, respectively. We verified that the interaction between F-PAS and CNBh protein is not altered by the presence of 2% DMSO. F-PAS (0.1 µM) and CNBh (10 µM) concentrations were chosen such that under these conditions, the measured r value is 60% of saturation, allowing for either an increase or decrease of r value upon compound addition. The plates were incubated for 70 min at room temperature in the dark before fluorescence anisotropy (r value) was read on a BioTek Synergy 2 plate reader (BioTek, Winooski, VT), using excitation and emission filters 485/20 and 528/20 nm, respectively.
Screening Statistics
The Z′ value for both screening campaigns was on average 0.9, showing that both assay formats were of excellent quality 13 to screen at a high final compound concentration of 50 µM. During the screening campaign, any plate that had a Z value less than 0.5 was individually analyzed for “outlying high-hitting” wells, which generally correlated with highly colored compounds. For the thermal shift screen, all compounds that gave a Tm shift of either ≥1 °C or ≤1 °C from the no-compound control wells were retest confirmed in duplicate at 50 µM. For the fluorescence anisotropy screen, all compounds that gave a change in anisotropy of 3× standard deviations from the mean value were retest confirmed in triplicate at 50 µM with the inclusion of six positive and six negative controls per plate. To eliminate false hits due to intrinsic compound fluorescence contributing to the measured r value, a control without F-PAS was included for each selected compound. Individual fluorescence intensities from the vertical polarized light (I∥) and horizontal polarized light (I⊥) of the control were subtracted from those of the complete assay, and fluorescence anisotropy was calculated using the equation r = (I∥ – I⊥)/(I∥ + 2I⊥).
Retest Confirmation
Compounds that retest confirmed at a single dose of 50 µM from both screening campaigns were reordered as powders and tested for dose response. Commercial analogues of these compounds were also obtained to evaluate structure-activity relationships.
Affinity Measurements Using Fluorescence Anisotropy
The fluorescence anisotropy assay was performed as described above but with increasing concentrations of CNBh protein, from 2 to 80 µM, in the presence or absence of compounds at 100 µM in triplicate. The dependence of r with CNBh concentration was plotted and the dissociation constant Kd was determined from the fitting of the experimental data with equation (2), as previously described 9 :
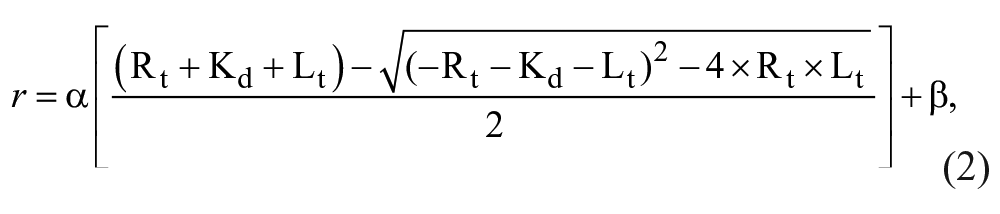
where Rt and Lt are total receptor (F-PAS) and ligand (CNBh) concentrations, and α and β are a scaling factor and an offset factor, respectively.
Affinity Measurements Using Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) was performed in a MicroCal VP-ITC instrument (Malvern Instruments, Malvern, UK) at 15 °C with a first injection of 2 µL, followed by 28 injections of 10 µL. Purified mEAG1 PAS and CNBh proteins were dialyzed overnight against 50 mM Hepes (pH 7.5), 150 mM NaCl, and 1 mM tris(2-carboxyethyl)phosphine (TCEP). DMSO was added to the protein to be injected, and DMSO or a selected compound dissolved in DMSO was added to the protein to be titrated; in all situations, the final concentration of DMSO was 2%. The concentration of protein in the syringe was between 200 μM to 300 μM, while the titrated protein was 10 times less concentrated in the cell. Titrations were performed with mEAG1 PAS into CNBh and CNBh into mEAG1 PAS, in the presence or absence of 50 or 100 µM of the selected compounds. Data were analyzed using MicroCal Origin 7 (Malvern Instruments). The dissociation constant of interaction was obtained by fitting the data to a single-site binding model.
Results and Discussion
Initial Screening and Retest Confirmation
Two independent in vitro screening campaigns for small molecules that bind specifically and with high affinity to the cytoplasmic domains of EAG channels were executed. First, to identify novel small molecules that bound to the hERG PAS protein, the ApexScreen Library from TimTec comprising 5080 chemically diverse compounds was screened at 50 µM using a thermal shift assay format. From the initial screen, 83 compounds were identified as “hits” that gave a change in Tm of hERG PAS protein of at least 1 °C with a 1.6% hit rate ( Table 1 ). Initial hits were retest confirmed in duplicate from liquid at 50 µM, resulting in 30 compounds: 18 compounds that gave a Tm shift ≥1 °C (stabilizers) and 12 compounds that gave a Tm shift ≤1 °C (destabilizers), resulting in a 36% retest confirmation rate ( Table 1 ).
Summary of High-Throughput Screening and Lead Follow-Up Strategies.
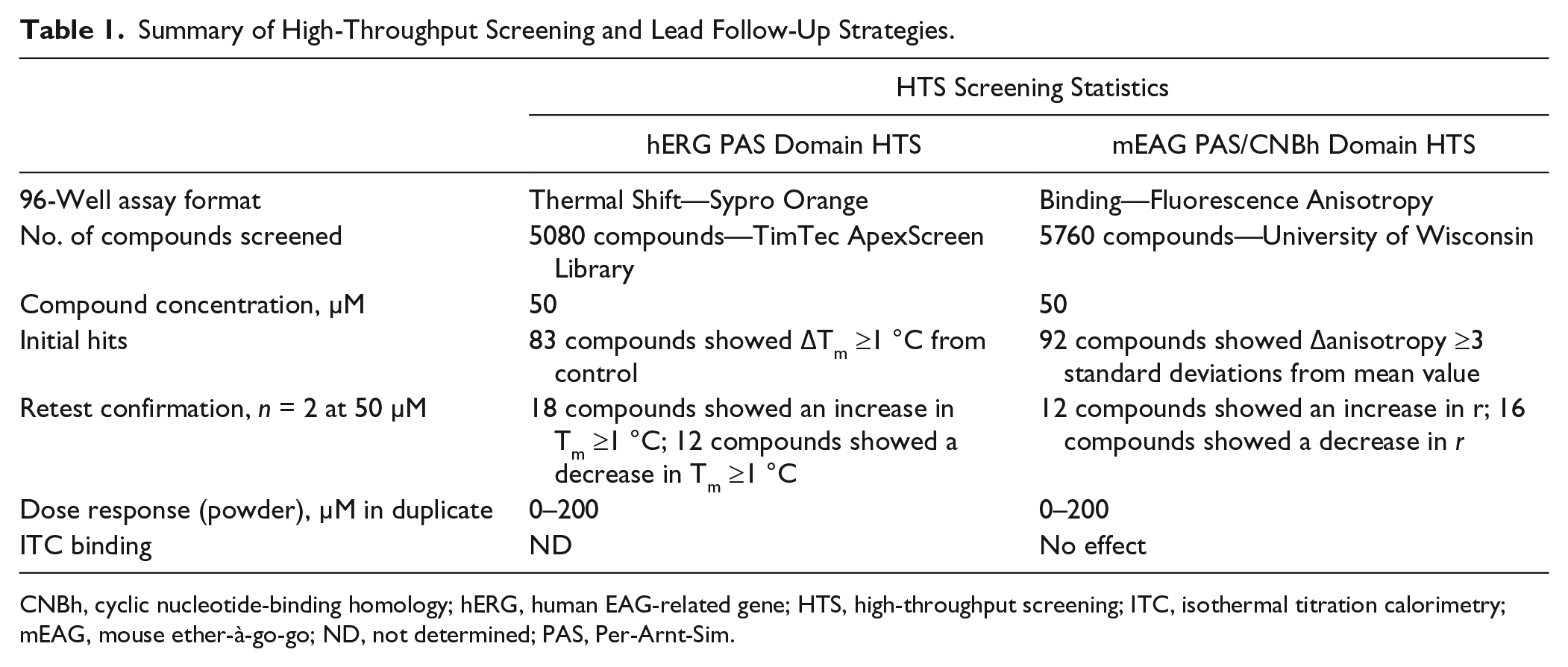
CNBh, cyclic nucleotide-binding homology; hERG, human EAG-related gene; HTS, high-throughput screening; ITC, isothermal titration calorimetry; mEAG, mouse ether-à-go-go; ND, not determined; PAS, Per-Arnt-Sim.
Second, to identify small molecules that could modulate the interaction between the mEAG1 PAS and CNBh domains 5760 compounds from the Chembridge DIVERset were screened at 50 µM using fluorescence anisotropy as an assay format. From the initial screen, 92 compounds were identified as “hits” that showed a change of fluorescence anisotropy greater than or equal to 3× standard deviations from the mean value with a 1.6% hit rate ( Table 1 ). Initial hits were retest confirmed in triplicate from liquid at 50 µM, resulting in 28 compounds: 12 compounds showed an increase in anisotropy and 16 compounds showed a decrease in anisotropy, a 30% retest confirmation rate ( Table 1 ).
Dose-Response Confirmation
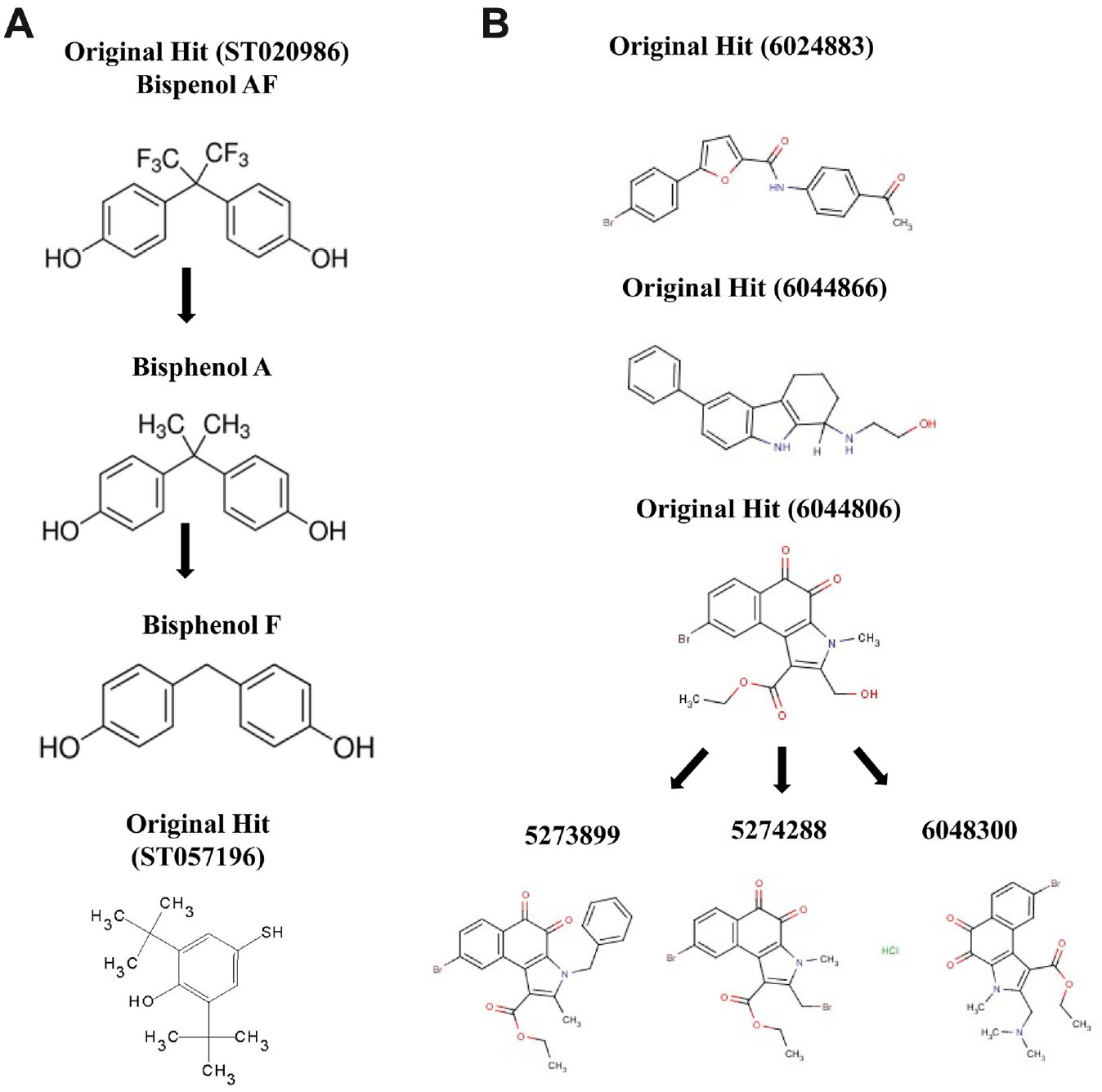
From the screening campaigns, we have identified compounds that alter either the protein stability of hERG PAS protein or the mEAG1 PAS/CNBh complex. Since both compound libraries were obtained as frozen liquid stock solutions, we reordered all compounds for retest confirmation as powders to analyze in each assay format by dose response through a range of compound concentrations (0–200 µM). For hERG PAS, all compounds that stabilized or destabilized the apparent Tm in the thermal shift assay were assessed. However, it became clear that the 18 compounds that apparently had stabilized the Tm profile all showed a decrease in maximum fluorescence response as the compound concentration increased, suggesting that the PAS protein was aggregating or precipitating in the well, resulting in a false shift in Tm. We had previously noticed this effect during assay development in that as the concentration of hERG PAS protein decreased, the apparent Tm increased. In contrast, compounds that had destabilized the Tm of hERG PAS protein at a single dose showed compound titration up to 200 µM without affecting the maximal fluorescence response, and we focused on hit compounds ST020986 for further hit expansion ( Fig. 2A ) and ST057196, a phenol-based compound.
(
From our second screen, 28 compounds were tested for dose response, of which 14 compounds were selected for retest from powders. On testing these compounds from 0 to 200 µM, seven compounds showed an increase in anisotropy, suggesting that they affected the mEAG1 PAS interaction with CNBh. From these retest compounds, we focused on three compounds: 6024883, 6044866, and 6044806 (the latter included hit expansion, as shown in Fig. 2B ).
Chemical Series Expansion and Proposed Mechanism of Action
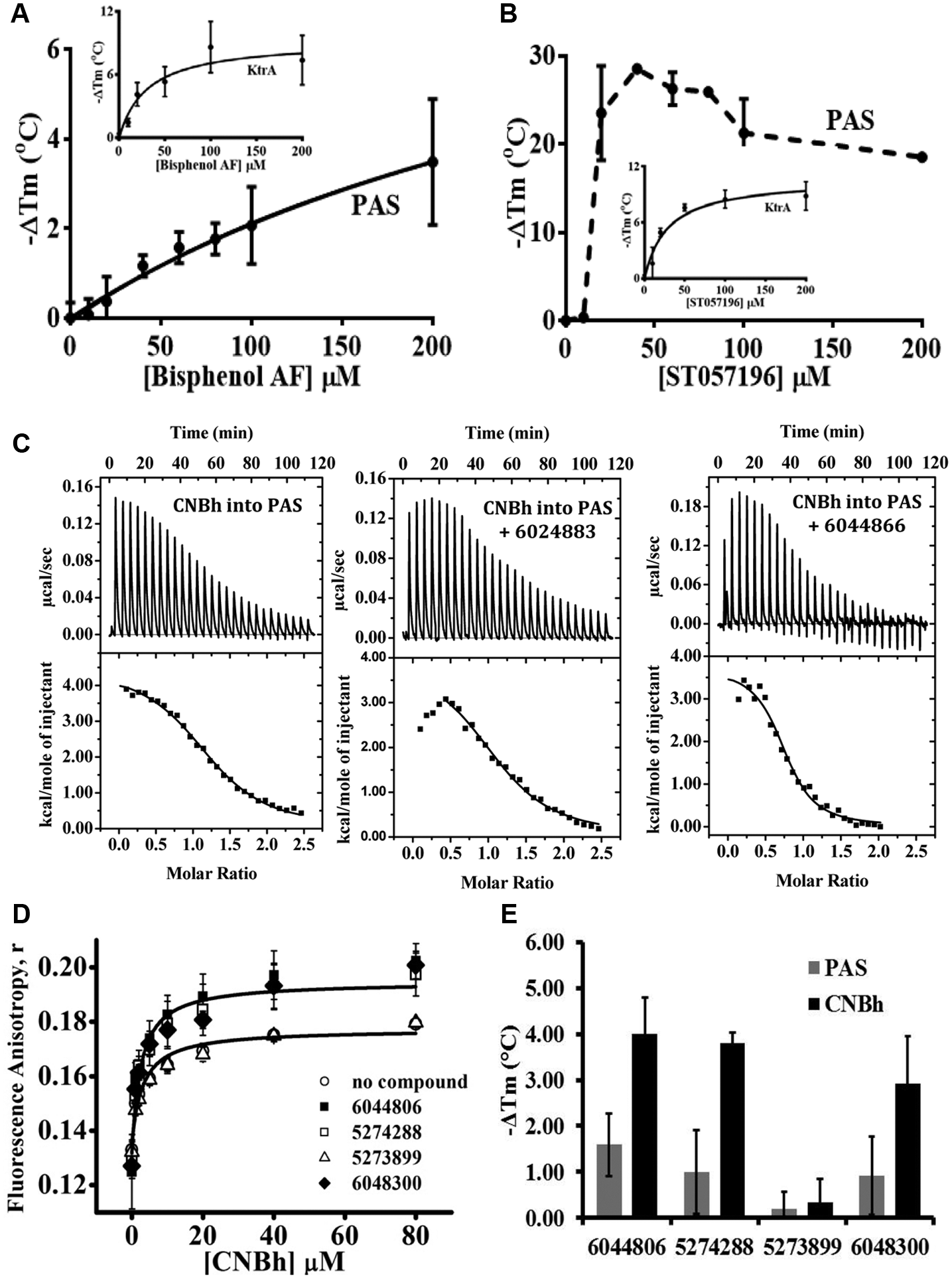
The compound ST020986 has the familiar chemical name of Bisphenol AF, and two analogues were commercially available, Bisphenol A and F ( Fig. 2A ). Interestingly, compounds within this series have previously been shown to bind to the human PAS kinase (hPASK) domain with varying affinities. 14 The structure-activity relationship of binding of this compound series and hydrophobic nature of these compounds suggested that they were ideal candidates for binding in the hydrophobic environment of the PAS protein cavity. We also noticed that ST057196, another phenol-like compound, also destabilized hERG PAS protein in the thermal shift assay, and so it was tested in dose response also. As shown in Figure 3A , titration of 3 µM PAS protein with 0 to 200 µM Bisphenol AF resulted in a significant dose-dependent decrease in Tm of the hERG PAS protein. At the addition of 200 µM compound, Tm decreased at least 4.5 °C. Saturation in the assay was not achieved due to difficulties in compound solubilization at higher concentrations. Fitting these data with two binding equations (see equation (1) in Materials and Methods and equation in Vivoli et al. 12 ) revealed a Kd of 388 µM or 310 µM, respectively, indicating a low-affinity interaction between protein and compound. No shift in Tm was observed for Bisphenol A and F, suggesting weak to no binding of these compounds; however, compound solubility issues limited this study. This rank order of potency for the hERG PAS protein of Bisphenol AF > Bisphenol A or F is consistent with that observed previously for the hPASK PAS domain. It is generally thought that the extra interactions established between a receptor and its ligand result in increased temperature stability of the complex relative to the apo-receptor. However, the decrease in temperature stability observed for the PAS protein after exposure to Bisphenol AF probably resulted from interaction of the compound with an unfolded state of the protein. 15 In addition, we verified that the destabilizing effect of this compound was nonspecific, since when titrated against an unrelated protein, KtrA from Bacillus subtilis, 11 it also decreased the Tm in a dose-dependent manner and the data could be fitted as above (Kd of 31 µM). Altogether, this led us to stop pursuing this series of compounds. Relative to the single phenol derivative ST057196, titration of PAS with 0 to 200 µM compound resulted in a steep concentration dependence of Tm, where on addition of 50 µM compound, the protein Tm already decreased by more than 18 °C ( Fig. 3B ). We noticed that ST057196 contains a sulfhydryl group that could potentially modify any of the eight cysteine residues present in the hERG PAS domain and cause destabilization of the protein. In fact, as shown in the inset in Figure 3B , titration of KtrA 11 with ST057196 also decreased the Tm in a dose-dependent manner, which could be fitted as above with a Kd of 25 µM. These results show that the effect of ST057196 is nonspecific and is consistent with a covalent modification of the protein. This led us to stop pursuing this series of compounds.
(
From our second screening campaign, we wanted to know whether compounds 6024883, 6044866, and 6044806 induced changes in the Kd of binding of mEAG1 PAS with CNBh protein. To do this, compound was present at 100 µM during ITC titrations performed between the PAS and CNBh proteins. Solubility issues prevented analysis of compound 6044806 in this assay format. The interaction between mEAG1 PAS and CNBh protein is an endothermic reaction with a Kd of 2.1 ± 0.8 µM and n = 1.3 ± 0.27. Presence of 100 µM of compounds 6044866 (Kd = 1.1 µM; n = 1.2) and 6024883 (Kd = 3.3 µM; n = 0.77) had no effect on this interaction ( Fig. 3C ). Compound 6044806 and three other compounds from this series were ordered as powders (5273899, 574288, and 6048300), and all four were tested in the fluorescence anisotropy assay at 100 µM with F-PAS at 0.1 µM and CNBh protein from 2 to 80 µM to see whether they would affect the interaction. As shown in Figure 3D , after fitting the data with equation (2), none of the compounds affected the Kd of interaction (no-compound control, Kd = 3.1 ± 1.1 µM; 6044806, Kd = 1.8 ± 0.56 µM; 5273899, Kd = 3.6 ± 1.2 µM; 6048300, Kd = 2.3 ± 0.93 µM; and 5274288, Kd = 2.4 ± 0.67 µM). However, we noticed that three of the compounds (6044806, 574288, and 6048300) increased the maximal anisotropy signal compared with the no-compound control and compound 5273899. Using the thermal shift fluorescence assay, we individually added each compound to either the mEAG1 PAS or CNBh domain alone and interestingly saw that these same three compounds had varying destabilizing effects on the CNBh protein compared with minor effects on the mEAG1 PAS protein ( Fig. 3E ). To see whether this was a true binding effect, we further tested compound 6048300 for binding to CNBh using ITC, as this compound had the best solubility profile. Unfortunately, no signal above heat of dilution could be obtained (data not shown), and therefore we cannot find evidence for a specific interaction with CNBh protein.
Future Prospects for Non–Pore Modulation of EAG K+ Channels
Although members of the EAG voltage-gated K+ channels have been under extensive study over the years, a lot is still unknown about their molecular mechanisms of modulation. Small molecules that specifically bind to the channel’s cytosolic domains and alter the functional properties of the channel would be of great utility for understanding those properties. In addition, novel small molecules that modulate channel activity would be of great interest due to the role of EAG channels in LQTS or cancer.
We have executed two independent screening campaigns to identify small-molecule ligands for EAG channel regions that have not been extensively explored in terms of their small-molecule binding potential. We searched for compounds that could either bind to the N-terminal PAS domain of the hERG K+ channel or interfere with the interaction between mEAG1 PAS and CNBh domains. We recognize that both of the screening libraries used in these campaigns were of limited size, but they contained a good chemical diversity, and both assays were robust and of excellent quality. High-resolution crystal and nuclear magnetic resonance structures of PAS domains from hERG, mEAG, and Drosophila Elk channels have been reported,6,16 and although these structures do not contain ligands, they have reasonably large hydrophobic cavities that have been suggested to bind ligands for other PAS domains. In fact, in our first screen, we focused on phenol-based compounds, of which the bisphenol series had been identified as binding to the hPASK PAS domain with varying affinities. However, we showed that in both cases, these compounds had solubility issues and caused protein destabilization, a nonspecific effect. Similarly, for our second screening effort, we identified a series of compounds in which some seemed to increase the apparent maximal anisotropy signal of mEAG1 PAS binding to the CNBh protein, but they turned out to not have an effect on the affinity of this interaction.
In conclusion, both screening campaigns resulted in compounds that have weak in vitro effects and challenging solubility issues. Our results strongly suggest that it will be challenging to identify small molecules that recognize these cytoplasmic regions (PAS domain or PAS-CNBh interface) and modulate channel function. As a consequence, it could be argued that modulation of EAG channels through these domains is not feasible. However, considering the functional impact of mutations within these channel regions, we believe that alternative strategies should be pursued. One such strategy would be the use of antibodies or antibody fragments. These molecules have the advantage of being highly specific and can be reengineered in terms of potency and toxicity. They also have great potential for interfering with protein-protein interfaces such as the one between the PAS and CNBh domains. We are aware that this strategy raises the specific problem of targeting a biological molecule to the cytoplasmic environment. However, this is a common challenge to many different therapeutic fields and is one that is being addressed by many research groups. 17
Footnotes
Acknowledgements
We thank our colleague Paula Magalhães in the CCGen service for access to the RT PCR machines for thermal shift screening assays.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the following grants: National Institutes of Health NIH-NS081320 awarded to JHMC and FCT Fellowship SFRH/BPD/105672/2015 awarded to ASF.