
Abstract
For the development of therapeutically potent anti-cancer antibody drugs, it is often important to identify antibodies that internalize into cells efficiently, rather than just binding to antigens on the cell surface. Such antibodies can mediate receptor endocytosis, resulting in receptor downregulation on the cell surface and potentially inhibiting receptor function and tumor growth. Also, efficient antibody internalization is a prerequisite for the delivery of cytotoxic drugs into target cells and is critical for the development of antibody–drug conjugates. Here we describe a novel activatable fluorescence–quencher pair to quantify the extent of antibody internalization and degradation in the target cells. In this assay, candidate antibodies were labeled with a fluorescent dye and a quencher. Fluorescence is inhibited outside and on the surface of cells, but activated upon endocytosis and degradation of the antibody. This assay enabled the development of a process for rapid characterization of candidate antibodies potentially in a high-throughput format. By employing an activatable secondary antibody, primary antibodies in purified form or in culture supernatants can be screened for internalization and degradation. Because purification of candidate antibodies is not required, this method represents a direct functional screen to identify antibodies that internalize efficiently early in the discovery process.
Keywords
Introduction
Antibodies represent an important class of therapeutic agents in the treatment of cancer. These molecules exert pharmacological activity through a variety of mechanisms.1,2 Currently, there are 16 therapeutic antibodies approved by the U.S. Food and Drug Administration (FDA) for cancer therapy, including two radio-immunoconjugates and two antibody–drug conjugates (ADCs). In addition to binding an antigen with high affinity and specificity, internalization of targets on the cell surface is often one of the critical properties that define an antibody’s functional potential as an anticancer agent. 3 Following specific binding, efficient internalization of an antibody–receptor complex can be a critical mechanism of therapeutic action. Downmodulation and subsequent degradation of receptors on the cell surface may result in inhibition of receptor-dependent signaling, which can have a significant impact on tumor growth. Tumor-specific antibodies that internalize efficiently also provide a means for delivery of various cytotoxic agents (small-molecule drugs, toxins, DNAs/RNAs, etc.) conjugated to an antibody into the cytosolic compartment of target cells. 4 In contrast, rapid internalization may not be desirable if effector function, such as antibody-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC), is critical for antitumor activity. Thus, characterization of the internalization potential of potential therapeutic candidates during the screening process may be an important selection criterion. Several reports have shown that the efficiency of antibody internalization varies greatly, and often depends on the target antigen and the epitope recognized by the antibody.5–7 However, the parameters governing this process are still unclear. For this reason, engineering of optimal internalization properties via rational design remains a challenge, and selection of optimal internalization properties during the process of screening antibody candidates can be of significant value.
Flow cytometry has been used extensively to measure internalization of antibodies and receptors. Conventional methods require the removal of a fluorescent signal from the cell surface, usually by washing with an acidic buffer to remove the labeled probe or by quenching with an anti-fluorophore antibody. These steps are labor-intensive and sometimes harmful to the cells. In addition, these assays measure internalized antibody, but are not able to directly assess degradation. The first aim of the present study was to establish a rapid and robust flow cytometric, cell-based assay to evaluate antibody binding, endocytosis, and degradation simultaneously. For this purpose, we developed a protocol using an activatable fluorescence–quencher probe, which is not fluorescent on the cell surface, but is activated within the cell after antibody degradation. Furthermore, we tested this protocol for rapid screening of antibodies early in the discovery process where typically only binding and blocking assays are evaluated. Cell-based functional assays are normally conducted using purified antibody, potentially limiting the throughput with which potential candidates can be assessed. Incorporation of a cell-based assay that relates directly to the therapeutic potential of the antibody at a point earlier in the discovery process was accomplished by employing a secondary antibody labeled with a fluorescence–quencher pair directed against a common epitope on primary candidate antibodies from crude cell culture supernatants.
Materials and Methods
Cells and Antibodies
The cancer cell lines A431 and BxPC-3 were obtained from the ATCC (Manassas, VA) and grown in Dulbecco’s modified Eagle’s medium (DMEM), 10% (v/v) fetal bovine serum (FBS) (Life Technologies, Grand Island, NY). The anti-receptor A antibody (AbA1), anti-receptor B antibodies (AbB1, B2, B3, and B4), and isotype control antibody were produced by Eli Lilly and Company (New York, NY). Human IgG and goat anti-human Fab were purchased from Jackson ImmunoResearch (West Grove, PA). Antibody transient transfection supernatant was generated by transfecting DNA constructs into a 293F cell line with Lipofectamine™ 2000 (Life Technologies) according to the manufacturer’s manual. The antibody concentration of the supernatant was measured by Octet (Pall ForteBio, Menlo Park, CA) with human IgG as standard.
Analysis of Receptor Degradation by Western Blot
BxPC-3 or A431 cells were seeded in 24-well plates at 3 × 105 cells/well and cultured overnight. A total of 10 µg/mL of anti-receptor antibody (AbA1, AbB1, AbB2, AbB3, or AbB4) or an isotype control was added, and cells were incubated at 37 °C for 2, 4, or 6 h. Cells were washed three times with PBS, lysed by M-Per with protease inhibitor (Thermo Fisher Scientific, Rockford, IL). Total protein (25 µg) was run on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to nitrocellulose membranes. After blocking with Odyssey Blocking Buffer (Li-Cor, Lincoln, NE) for 1 h at 37 °C, primary goat antibody (anti-receptor A, α and β subunits, or anti-receptor B) was added and the membrane was incubated overnight at 4 °C. Anti-β actin antibody (Sigma-Aldrich, St. Louis, MO) was used as a loading control. After washing three times with PBST (phosphate buffered saline + 0.05% Tween 20), secondary antibody (IRDye 800CW anti-goat IgG, Li-Cor) was added. After 1 h incubation, the membrane was washed with PBST and scanned by an Odyssy CLx Infrared Imaging System (Li-Cor).
Labeling of Antibody with Fluorophore and Quencher
First, test antibodies and goat anti-human Fab were labeled with DyLight650 (DL650) NHS ester (Thermo Fisher Scientific). Briefly, 0.5 mL of antibody or anti-human Fab at 1–2 mg/mL in 0.05 M borate buffer was added to 50 µg of the DL650 dye. After mixing, the reaction mixture was incubated for 2 h at room temperature, protected from light. Free dye was removed using a Zeba desalting spin column (Thermo Fisher Scientific) according to the manufacturer’s manual. The degree of labeling (dye-to-protein ratio) was calculated from absorbance at 280 nm and the Amax of the dye. Second, 1.25 µL of IRDye QC-1 NHS ester (Li-Cor) at 10 mg/mL was added to 100 µL of fluorophore-labeled antibody at 1 mg/mL. After 2 h incubation at room temperature in the dark, the dual-labeled antibodies were purified using a Zeba desalting spin column.
Estimation of Quenching and Recovery Efficiencies In Vitro
To estimate the quenching efficiency, the single- or dual-labeled antibody was serially diluted in a flat-bottom, 96-well plate with a starting concentration of 0.05 mg/mL. The fluorescent signal was scanned using an Odyssy CLx Infrared Imaging System (Li-Cor). The quenching efficiency was calculated as follows:

To estimate the recovery efficiency, 0.5 mg/mL of the single- or dual-labeled antibody was incubated in the presence of 2 mg/mL of proteinase K, 10% SDS, and 2% 2-mercaptoethanol at 50 °C for 30 min, and then 95 °C for 5 min to enable full denaturation and digestion of the antibody, separating the quencher from the fluorophore. The resulting sample was then diluted to 0.05 mg/mL and serially diluted in a flat-bottom, 96-well plate and scanned using an Odyssy CLx Infrared Imaging System. The recovery efficiency was calculated as follows:

Internalization/Degradation Assessed Using High-Throughput Flow Cytometry
For measurement of internalization and degradation of the directly labeled antibody, target cells were seeded at 3–6 × 104/well in 96-well plates and cultured overnight at 37 °C. On the next day, 0.5–10 µg/mL of DL650 single- or DL650 and QC-1 dual-labeled antibody was incubated with cells for 30 min on ice to enable specific binding of the antibody to the cell surface targets. After incubation, the cells were washed three times with medium to remove unbound antibody. The cells were then incubated at 37 °C with 5% CO2 for antibody internalization to occur. At different time points, the cells were dissociated with Trypsin (Life Technologies) and stained with 30 nM SYTOX Green Dead Cell Stain (Life Technologies) for 5 min before being analyzed using a high-throughput flow cytometry (HTFC) system (IntelliCyt, Albuquerque, NM). ForeCyt software was used to analyze the flow cytometry data. Dead cells were gated away by the SYTOX Green Dead Cell Staining plotted by FL1-H. The median fluorescence intensity of FL4-H indicated the DL650 signal. For measurement of internalization and degradation of the indirectly labeled antibody, 0.5–10 µg/mL of test antibody was incubated with a 6-fold molar excess of DL650 single- or DL650 and QC-1 dual-labeled secondary anti-human Fab for 30 min on ice in prior to the experiment. The antibody–dye mixture was added to the cells, and the assay was conducted as for the directly labeled antibody. Each experiment was done in duplicate.
Results and Discussion
Targeted probe-based fluorescence quenching was reported previously for imaging of tumors in vivo. 8 In that study, the TAMRA (fluorophore)–QSY7 (quencher) pair was conjugated to either avidin or an anti-Her2 antibody (trastuzumab), and used successfully to detect tumors in mice with a high activation ratio and low background signal through fluorophore activation upon uptake by cancer cells. In the strategy reported here, we chose DL650 as the fluorophore, and QC-1 as the quencher. 9 A test antibody was labeled with DyLight dye and the quencher, either directly or indirectly, through binding of a secondary Fab to human IgG ( Fig. 1 ). QC-1 is a nonfluorescent, dark quencher dye that is efficient at quenching fluorescence from a broad range of visible and near-infrared fluorophores (approximately 550–950 nm) by Förster resonance energy transfer (FRET). 10 Other fluorophores in this range can also be used, depending on available laser sources and detection channels of flow cytometers. When the fluorophore is in the proximity of a quencher conjugated to the same antibody ( Fig. 1 , right), the fluorescent signal is quenched by FRET. When an antibody labeled with the fluorophore and the quencher (dual labeled) is incubated with target cells, no fluoresce is detected outside the cells. Signal only appears following internalization of the antibody and subsequent degradation to separate the dye and the quencher. Antibody labeled with the fluorophore alone is used to detect total antibody on the surface and inside of the cell ( Fig. 1 , left).
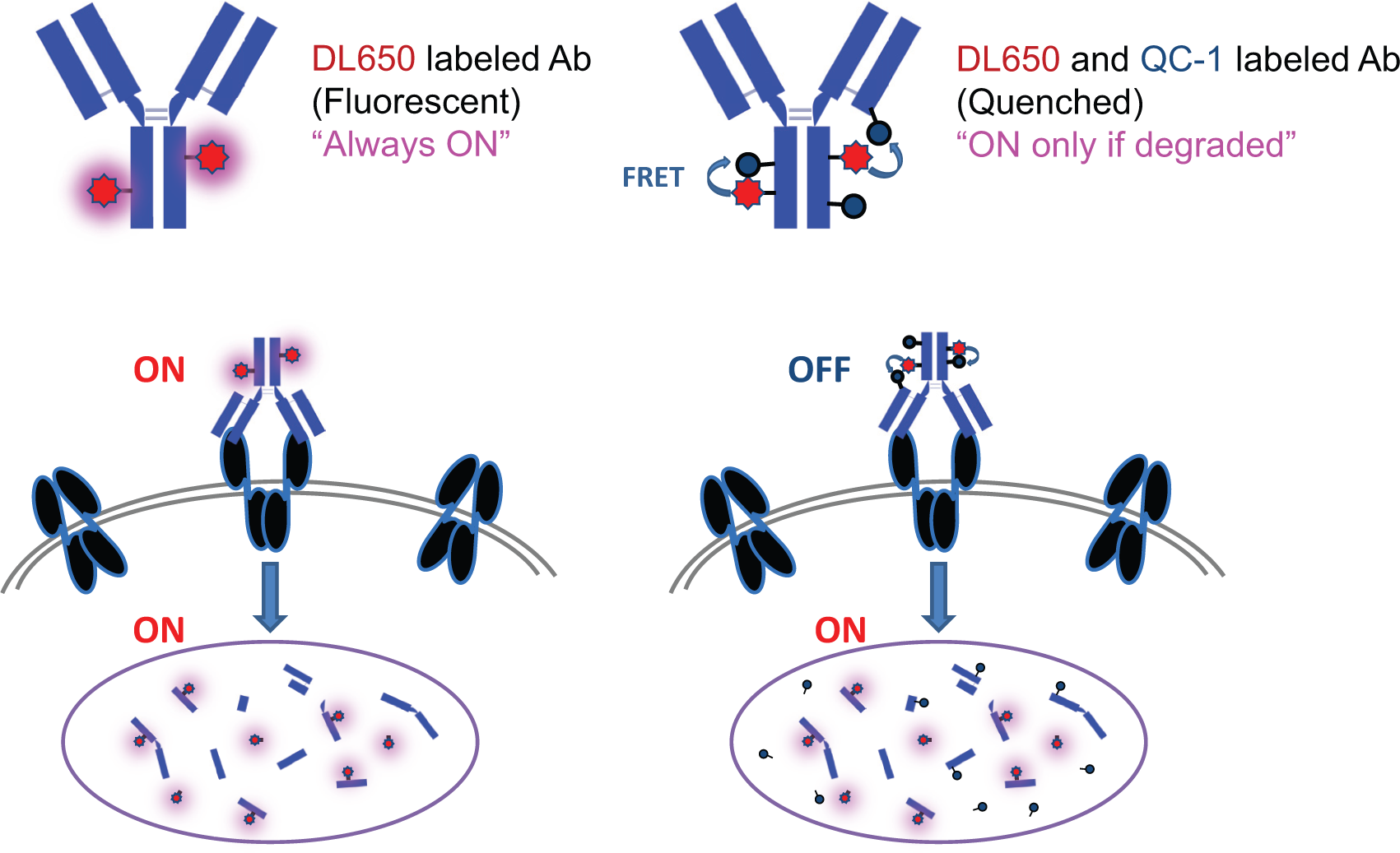
Schematic illustration of the concept of the antibody internalization and degradation assay. Left and right panels show fluorophore-labeled (single-labeled) antibody and fluorophore–quencher-labeled (dual-labeled) antibody, respectively. The labeled antibody binds to the target on the cell surface (black) and internalizes into cells. Since the fluorophore (red star) is not quenched in the single-labeled antibody (left), the fluorescent signal is “always on” and measures cell surface and internalized antibody. Using flow cytometry, the total amount of antibody is detected. The fluorophore on the dual-labeled antibody (right) is masked by the quencher (grey circle) by FRET when it is bound on the cell surface. When antibody is internalized and degraded, the fluorophore is activated by physical separation from the quencher and the resulting fluorescent signal is detected by flow cytometry. After complete internalization and degradation, the levels of fluorescence should be similar for single- and dual-labeled probes.
In addition to the direct labeling approach, a secondary Fab fragment was labeled with dye or dye and quencher and used to detect the extent of internalization and degradation for a number of primary antibodies in parallel. With a labeled secondary Fab, purification and individual labeling of each candidate are not necessary, thus facilitating the screening of antibodies for internalization and degradation in a high-throughput format.
Table 1 summarizes the efficiencies of the labeling reactions for various antibodies. Dye DL650-to-protein (D:P) ratios for IgG and Fab were typically 8–12 and 2.5–3.5, respectively. The quenching efficiency was higher than 80%, verifying effective quenching by dual labeling with QC-1. When the antibodies labeled with 125 µg/mL of QC-1 were denatured with SDS in vitro and digested by proteinase K treatment, more than 70% of the fluorescent signal was recovered, indicating that the dual-labeled probe is activatable by protein degradation.
Labeling of Antibody and Secondary Antibody with DyLight650 Fluorophore and Li-Cor QC-1 Quencher.
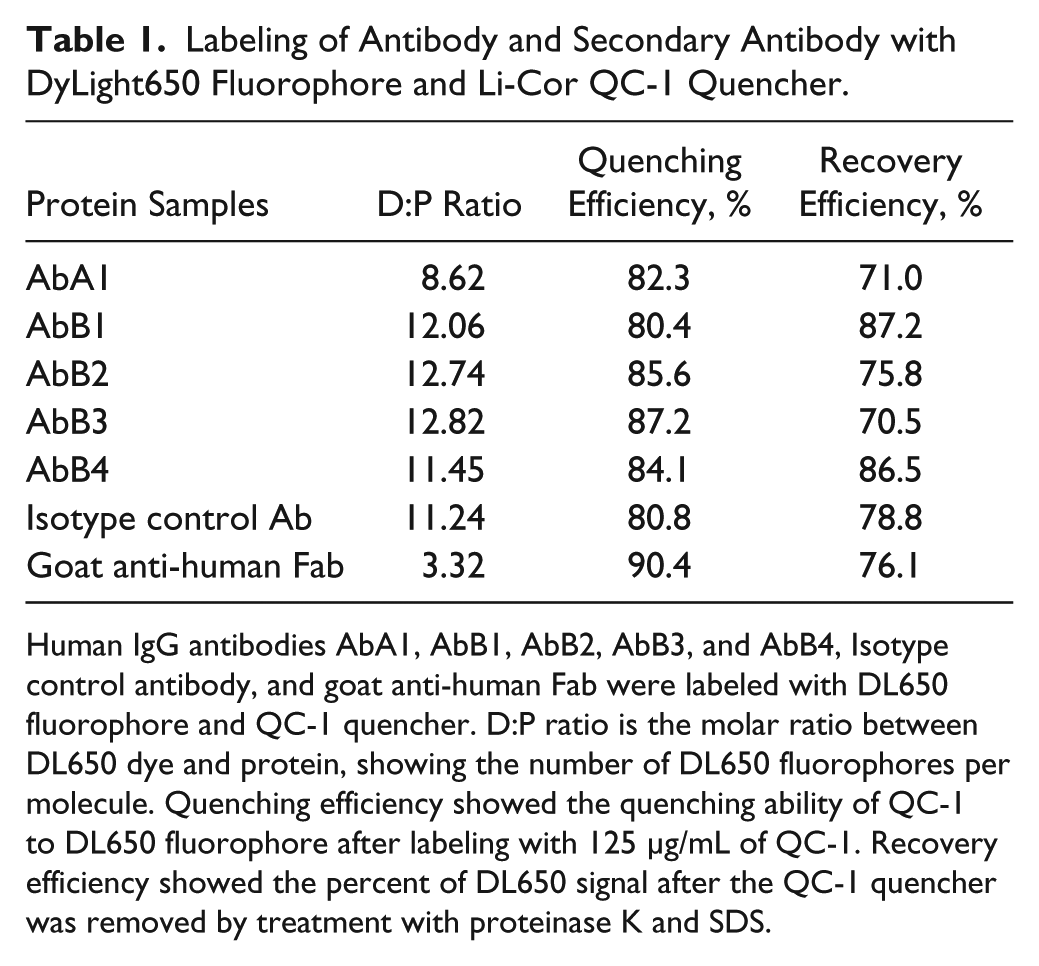
Human IgG antibodies AbA1, AbB1, AbB2, AbB3, and AbB4, Isotype control antibody, and goat anti-human Fab were labeled with DL650 fluorophore and QC-1 quencher. D:P ratio is the molar ratio between DL650 dye and protein, showing the number of DL650 fluorophores per molecule. Quenching efficiency showed the quenching ability of QC-1 to DL650 fluorophore after labeling with 125 µg/mL of QC-1. Recovery efficiency showed the percent of DL650 signal after the QC-1 quencher was removed by treatment with proteinase K and SDS.
As a proof-of-concept study, we first tested our protocol using a fully human, internalizing antibody, AbA1, directed against receptor A. As shown in Figure 2A , AbA1 induced rapid internalization and degradation of receptor A on BxPC-3 cells. The total levels of receptor A were considerably reduced after 2 h of antibody treatment. To test degradation of the antibody, AbA1 and the isotype control antibody were labeled with DL650 only, or DL650 and QC-1, followed by incubation with A431 cells overexpressing receptor A. Figure 2B showed internalization and degradation of AbA1 at 5 µg/mL measured by flow cytometry. After incubation for 30 min on ice (time 0), unlike the dual-labeled AbA1, the cells treated with single-labeled AbA1 showed strong fluorescence, indicating binding of antibody to the cell surface ( Fig. 2B ). After about 2 h incubation at 37 °C, the signal from the dual-labeled AbA1 began to increase, gradually indicating internalization and degradation of the antibody initially bound to the cell surface ( Fig. 2B ).
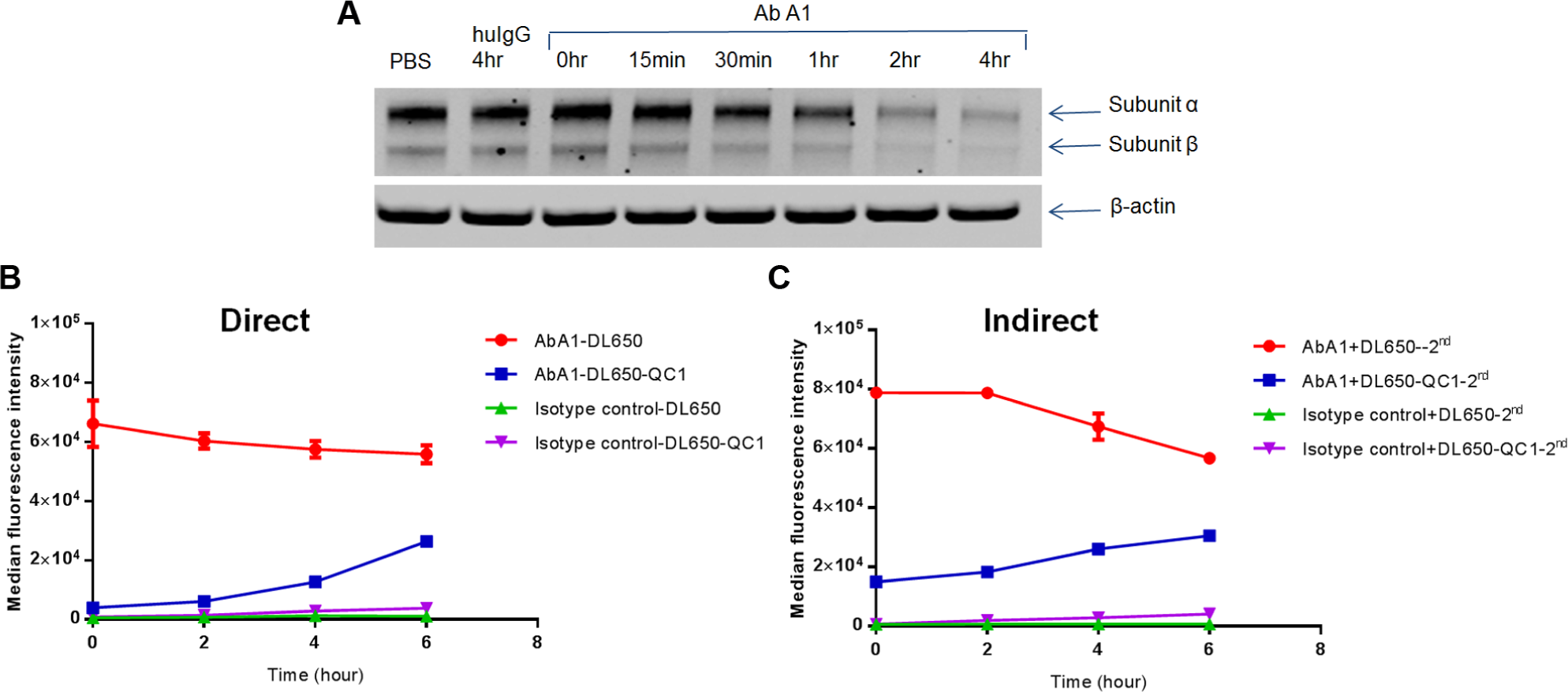
(
Next, we compared the methods using antibody labeled directly with antibody labeled indirectly through a secondary Fab. As shown in
Figure 2B,C
, the results obtained with direct and indirect labeling were similar, validating the utility of a secondary Fab in this internalization and degradation assay. Different antibodies (
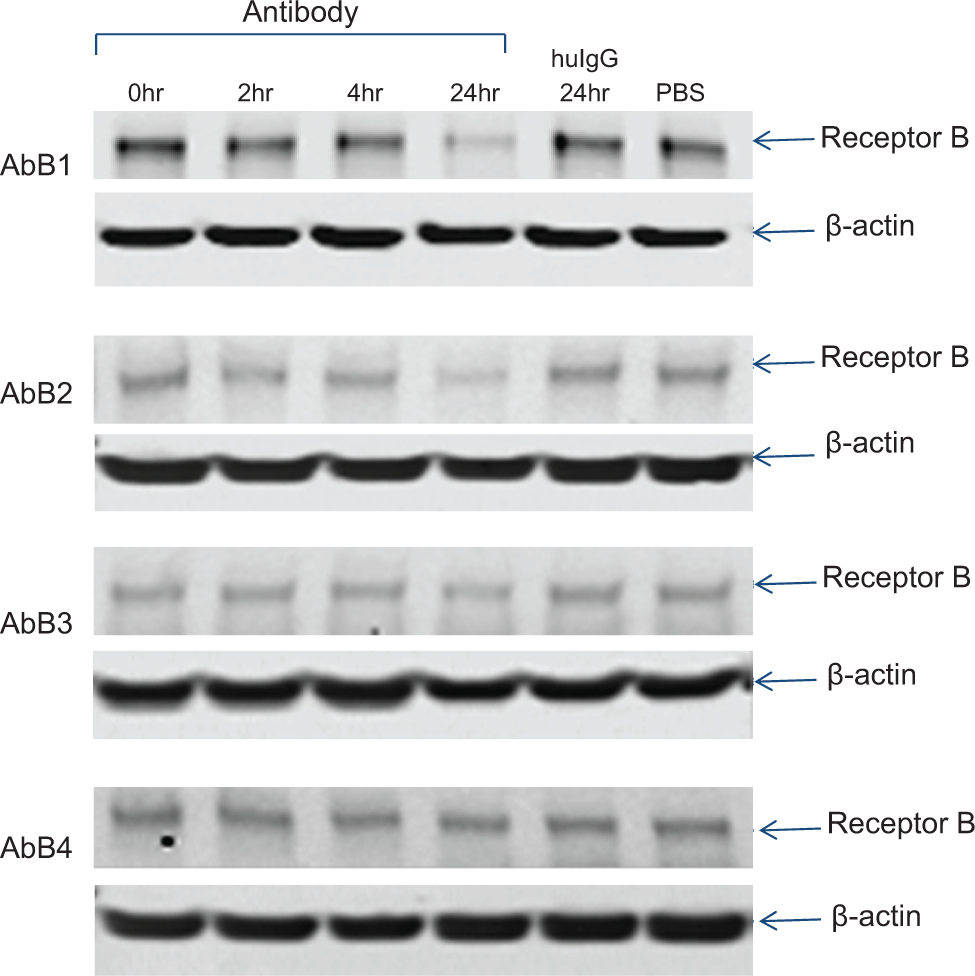
Degradation of receptor B induced by anti-receptor B antibodies. A431 cells were treated with AbB1, AbB2, AbB3, AbB4, or isotype control for the durations indicated, and total cell lysate (25 µg each) was analyzed by Western blot. Total receptor B was detected with an anti-receptor B antibody, and β-actin was used as a loading control.
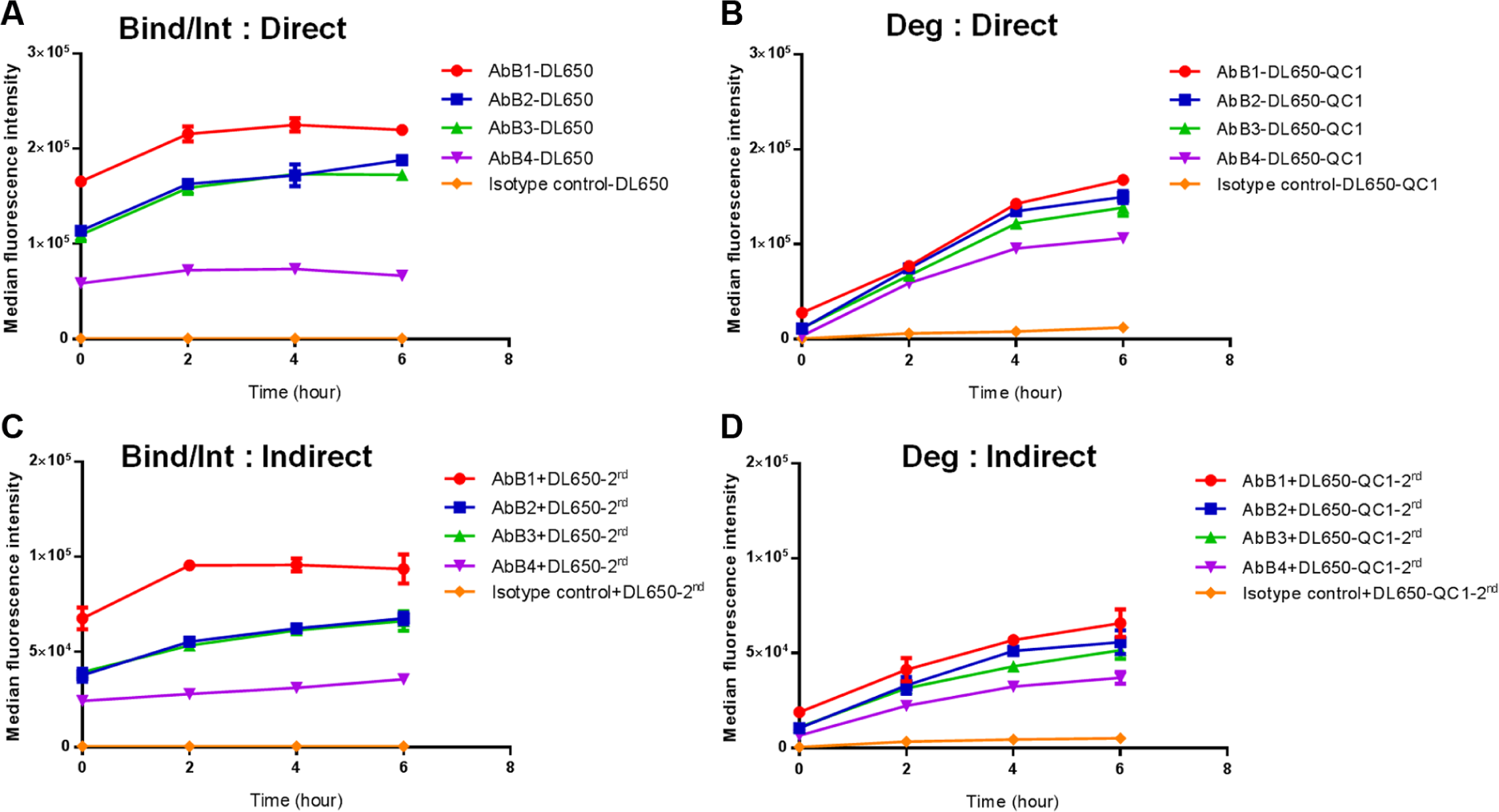
Comparison of directly labeled and indirectly labeled anti-receptor B antibodies using the internalization/degradation assay. A series of anti-receptor B antibodies (AbB1, AbB2, AbB3, and AbB4) were labeled directly with DL650 (
We also evaluated the assay using antibodies targeting antigens with potential as ADC targets. The results showed good correlation with levels of cytotoxicity measured in saporin conjugation assays (data not shown), which is dependent on the ability of antibody to internalize and be degraded. These results demonstrate that the assay is a useful tool for evaluation of antibodies for use as ADCs.
To demonstrate the utility of this method for screening antibody properties early in the discovery process, we performed a study comparing the use of purified antibodies and supernatants from antibody-expressing cells using a secondary Fab labeled with a fluorophore and a quencher. Fresh supernatants from cells expressing anti-receptor B antibodies were diluted to various concentrations, and compared with the same concentrations of purified antibodies. Figure 5 shows an example of the results obtained at 1 µg/mL. The rank order of antibodies based on assessment of internalization and degradation using the crude supernatants (AbB1 > AbB2 ≥ AbB3 > AbB4) was identical to that obtained using the purified antibodies. We further demonstrated that differences in internalization and degradation between antibodies were distinguishable at the lowest concentration tested (0.5 µg/mL), much lower than the concentration of antibody typically observed in supernatants obtained from small-scale production.
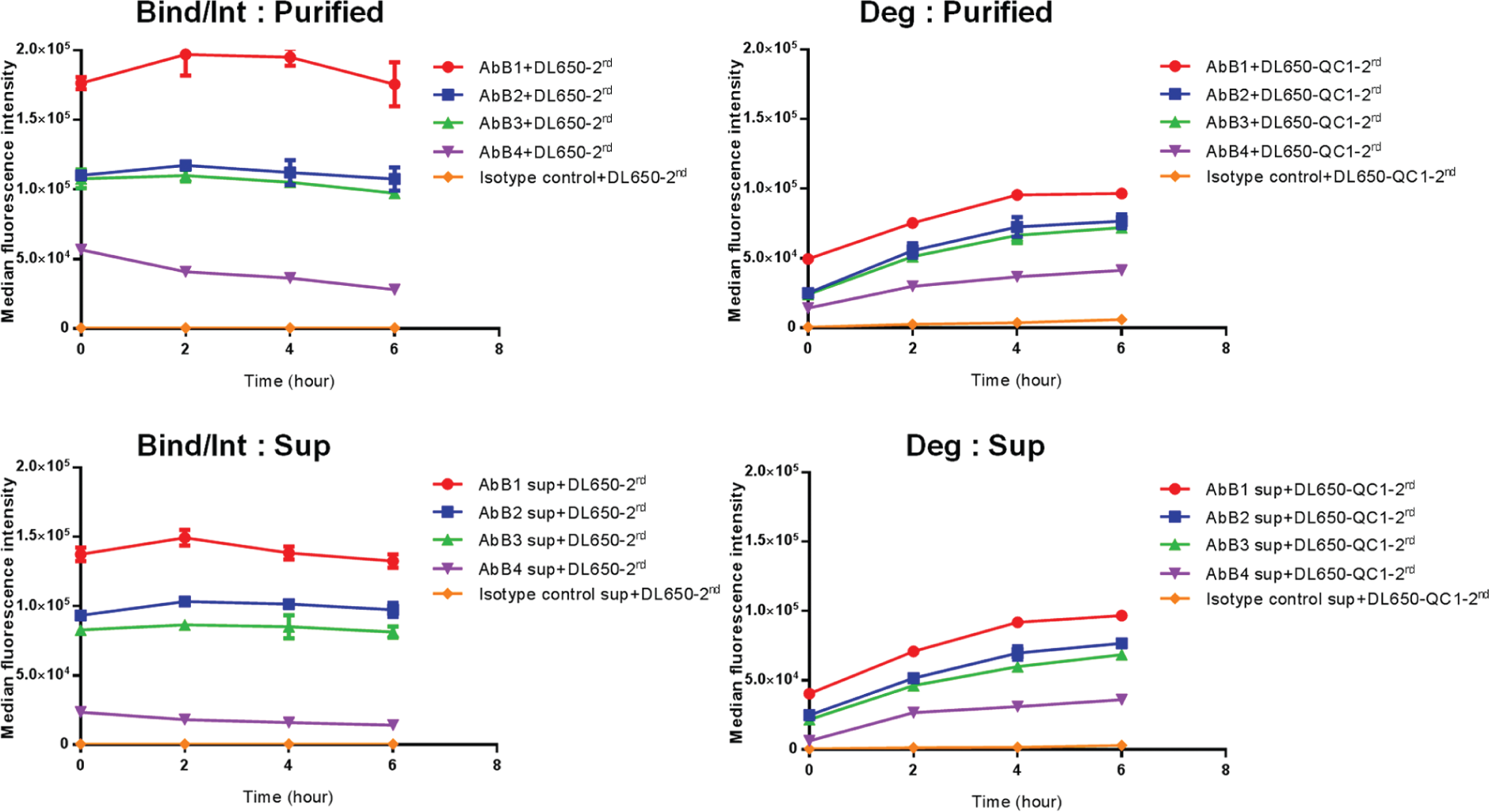
Comparison of purified antibodies with antibodies in cell culture supernatants using indirect labeling with an anti-human secondary Fab. Internalization of the anti-receptor B antibodies (AbB1, AbB2, AbB3, and AbB4) was measured with DL650- and QC1-labeled anti-human secondary Fab using 1 µg/mL of purified antibody (
In conclusion, our data demonstrate that the flow cytometric cell-based assay is a powerful and rapid tool for determining the efficiency of internalization and degradation of antibodies bound to targets on the cell surface. Although we have not yet implemented the assay in the discovery process, we have demonstrated that it can be successfully applied to the measurement of antibody internalization and degradation using samples of antibodies in crude supernatants and that, in principle, the assay should be amenable to high-throughput screening. The protocol could also be modified easily for other applications, such as screening different formats of multi-specific antibodies for the ability to promote internalization and degradation, evaluation of drug conjugation sites or linker design for development of ADCs, or the study of internalizing non-antibody-based proteins.
Footnotes
Acknowledgements
The authors gratefully acknowledge Dr. Juqun Shen (Eli Lilly and Company) for providing antibodies and Dr. Yiwei Zhang (Eli Lilly and Company) for providing Biacore support. We would like to thank Dr. Dale L. Ludwig (Eli Lilly and Company) for support and suggestions.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors are current or previous employees of Eli Lilly and Company.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research and publication of this article are supported by funding from Lilly Research Laboratories.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.