
Abstract
Screening of RNA interference (RNAi) libraries in primary T cells is labor-intensive and technically challenging because these cells are hard to transfect. Chemically modified, self-delivering small interfering RNAs (siRNAs) offer a solution to this problem, because they enter hard-to-transfect cell types without needing a delivery reagent and are available in library format for RNAi screening. In this study, we have screened a library of chemically modified, self-delivering siRNAs targeting the expression of 72 distinct genes in conjunction with an image-based high-content-analysis platform as a proof-of-principle strategy to identify genes involved in lymphocyte function-associated antigen-1 (LFA-1)-mediated migration in primary human T cells. Our library-screening strategy identified the small GTPase RhoA as being crucial for T cell polarization and migration in response to LFA-1 stimulation and other migratory ligands. We also demonstrate that multiple downstream assays can be performed within an individual RNAi screen and have used the remainder of the cells for additional assays, including cell viability and adhesion to ICAM-1 (the physiological ligand for LFA-1) in the absence or presence of the chemokine SDF-1α. This study therefore demonstrates the ease and benefits of conducting siRNA library screens in primary human T cells using self-delivering, chemically modified siRNAs, and it emphasizes the feasibility and potential of this approach for elucidating the signaling pathways that regulate T cell function.
Introduction
T cells play a crucial role in the adaptive immune response to pathogens and microorganisms by migrating into secondary lymphoid organs in search of antigen-presenting cells bearing peptide antigens. 1 An effective immune response is then facilitated by clonal expansion and differentiation of antigen-specific T cells into effector subsets that are licensed to migrate into inflamed target tissues. 1 The migration of T cells through secondary lymphoid organs and inflamed target tissues involves a number of steps, including (a) selectin-mediated rolling of the T cell along endothelial cells that line blood vessels; (b) chemokine-induced “inside-out” activation of T cell integrins, resulting in firm adhesion to integrin ligands on the vessel wall; and (c) ligand-occupied “outside-in” integrin signaling that initiates cytoskeleton-dependent T cell polarization, crawling, and transendothelial migration. 1
The αLβ2 integrin known as lymphocyte function-associated antigen-1 (LFA-1) plays a crucial role in T cell adhesion, migration, and activation. 2 LFA-1 regulates T cell migration into secondary lymphoid organs and nonlymphoid inflammatory sites via interactions with its counterligand, ICAM-1, expressed on endothelial cells. Biochemical, biophysical, and structural studies have shown that LFA-1 is maintained on resting T cells in a bent conformation with low affinity for ICAM-1. 3 However, LFA-1 undergoes a rapid conformational change into at least two extended open conformations in response to chemokine receptor stimulation by chemokines displayed on the surface of endothelial cells. This process is known as inside-out activation and permits binding to ICAM-1, resulting in firm T cell adhesion to the blood vessel wall. 2 A number of intracellular signaling proteins have been shown to be important for inside-out activation of LFA-1 in T cells, particularly the adaptor proteins talin-1 and kindlin-3. 2 Integrins, including LFA-1, also signal bidirectionally such that the ligand-occupied form (i.e., ICAM-1-bound LFA-1) and/or integrin clustering activates intracellular signaling cascades that couple to the cytoskeleton to drive T cell polarization, cell motility, and transendothelial migration. 2 This process is known as outside-in activation, but the intracellular signals that influence this aspect of LFA-1 function are less well defined. Many gaps, therefore, remain in our knowledge of the proteins that regulate LFA-1-mediated T cell adhesion and migration.
The discovery of RNA interference (RNAi) and small interfering RNAs (siRNAs) and its application for loss-of-function studies has revolutionized basic and biomedical science, because the products of individual genes can now be silenced in a specific manner. 4 Furthermore, the development of RNAi libraries and their use for siRNA screening have enabled the rapid and unbiased discovery of novel genes involved in a given pathway in numerous cell types. 4 To date, however, there have been relatively few RNAi library screens performed in primary human T cells. It is likely that this is due to the technical and logistical challenges in scaling up siRNA delivery for large-scale library-screening studies in these cells. Primary T cells are widely known as being hard to transfect because conventional lipid-based transfection methods that effectively deliver siRNAs into adherent cells generally do not work well for these cells. Although electroporation/nucleofection and viral vectors are most commonly used for delivery of siRNAs into primary T cells, 5 their use for screening RNAi libraries is limited because specialist equipment, reagents, and expertise are needed.
Chemically modified, self-delivering siRNAs provide an alternative RNAi strategy because they enable siRNAs to enter cells without the need of a transfection reagent. This type of siRNA has been successfully used in RNAi experiments in a variety of primary cells, including macrophages, neurons, T cells, and in vivo mouse models. The siRNAs are simply incubated with cells for 72–96 h under low-serum or serum-free conditions, after which time the cells are harvested and used for functional analysis and confirmation of gene silencing. This novel siRNA delivery system (Accell siRNA) therefore holds great potential for screening RNAi libraries in primary human T cells. In this study, we demonstrate that self-delivering siRNAs are efficiently delivered into primary T cells, result in significant silencing of gene expression, and can be used for screening of RNAi libraries. Our library-screening strategy identified the small GTPase RhoA as being crucial for polarization and migration of primary human T cells in response to LFA-1 stimulation and other migratory ligands. We also demonstrate that multiple downstream assays can be performed from an individual RNAi screen. This work demonstrates that RNAi library screening can be performed easily in primary human T cells using self-delivering siRNAs for elucidating the signaling pathways that regulate T cell function.
Materials and Methods
Reagents
The self-delivering siRNA library (Accell) was obtained from Dharmacon (part of GE Healthcare, Lafayette, CO) as a single 96-well plate in dried-down format (1 nmol siRNA/well). Other reagents from Dharmacon were Accell siRNA delivery media, Accell SMARTpool siRNAs targeting RhoA, individual Accell siRNAs 1–4 from the RhoA SMARTpool, Accell nontargeting control siRNAs, and Accell Green nontargeting control siRNAs. The following reagents were also used: CellTiter Blue (Promega, Madison, WI); Lymphoprep density gradient solution (Axis Shield, Dundee, Scotland); NucleoSpin RNAII kit (Machery-Nagel, Düren, Germany); RETROscript kit (Ambion, Austin, Texas); Hoechst 33258, cell-permeable Hoechst 33342, and Alexa Fluor 568 goat anti-mouse IgG (Molecular Probes Invitrogen, Paisley, Scotland); staurosporine (Merck Chemicals, Nottingham, UK); recombinant human SDF-1α and interleukin (IL)-2 (Peprotech, London); anti-LFA-1 antibody used for T cell migration assays (clone SPV-L7, specific for the αL chain) (Monosan, Uden, The Netherlands); anti-LFA-1 antibody used for immunofluorescence staining (clone 27, BD Transduction Laboratories, Oxford, UK); recombinant human ICAM-1-Fc and VCAM-1-Fc (R&D Systems, Abingdon, UK); anti-RhoA antibody (Cytoskeleton, Denver, CO); HRP-labeled goat anti-mouse and goat anti-rabbit secondary antibodies (Cell Signaling Technology, Danvers, MA); and goat anti-mouse IgG, phalloidin-TRITC, α-tubulin-FITC, anti-actin, Y-27632, poly-L-lysine solution, goat anti-human Fc fragment-specific IgG, and fibronectin (Sigma, St. Louis, MO).
Reconstitution of the siRNA Library and Preparation of Daughter Plates
The self-delivering siRNA library was reconstituted with siRNA resuspension buffer (Dharmacon) under sterile conditions according to the manufacturer’s recommendations with minor modifications. In brief, 100 µL/well of 1× siRNA resuspension buffer was added to the wells of the plate to give a siRNA concentration of 10 µM. Ten daughter plates were then prepared immediately by dispensing 10 µL of the siRNAs into 96-well plates using a handheld electronic multichannel pipette. The plates were sealed with 96-well plate adhesive covers and stored at −20 °C.
T Cell Culture
Primary human T cells were isolated from buffy coat blood packs obtained from the Irish Blood Transfusion Service (St. James’s Hospital, Dublin, Ireland) and expanded with phytohemagglutinin and IL-2 as described. 6 Flow cytometry was used to confirm that the cell population was >97% CD3+ T cells using a phycoerythrin (PE)-conjugated anti-CD3 antibody from BD Biosciences (clone HIT3a; San Jose, CA; data not shown).
Transfection of T Cells with the siRNA Library or with Individual siRNAs
T cells were harvested after 5 days of culturing with IL-2. The cells were resuspended in IL-2-supplemented siRNA delivery medium at a final concentration of 2×106 cells/ml. During this time, a siRNA library daughter plate was defrosted, centrifuged briefly, and stored on ice until further use. The cells in siRNA delivery medium were added to the plate (100 µL/well) to give a final cell number of 2×105 cells and 0.9 µM siRNA. The plate was then incubated for 72 h at 37 °C. Evaporation and so-called edge effects (when differences are observed in the behavior of cells placed in the outer wells of multiwell plates compared to cells in the inner wells) were minimized during this period by adding sterile dH20 between the wells of the plate and placing the plate in a shallow bath of dH20 in a humidified incubator. The cells were transferred to a V-bottomed 96-well plate and centrifuged for 5 min at 1500 rpm. Cell pellets were resuspended in 120 µL RPMI-1640 supplemented with 10% fetal calf serum (FCS), penicillin/streptomycin, and IL-2 (complete cell culture medium). Aliquots of 20 µL were then taken from the wells and used for (1) a T migration assay on an anti-LFA-1 antibody; (2) a CellTiter Blue viability assay; (3) a total cell count; and (4) adhesion to ICAM-1 in the absence or presence of the chemokine SDF-1α. The siRNA screen was performed three times, each time using T cells isolated from a single blood donor. When indicated, T cells were also transfected with self-delivering siRNAs (1 µM) in 48-well (250 µL) or 24-well (500 µL) plates using the same conditions as described above, and they were incubated for 72 h at 37 °C.
Quantitative Real-Time PCR
Total RNA was isolated from cells using a NucleoSpin RNAII kit according to the manufacturer’s instructions. First-strand complementary DNA synthesis was performed using a RETROscript kit according to the manufacturer’s instructions. Gene expression assays for GAPDH were performed using an Applied Biosystems 7900HT real-time PCR system (Applied Biosystems, Cheshire, UK). Relative gene expression of treated samples compared to untreated control samples was calculated using the comparative CT method (Applied Biosystems). The data were normalized to the housekeeping gene human β-actin.
Flow Cytometry
Delivery of siRNAs into T cells was determined after 72 h by flow cytometry. T cells were washed in phosphate-buffered saline, run on a CyAn flow cytometer (Beckman Coulter, London, UK), and analyzed with FlowJo software (TreeStar, Ashland, OR).
Confocal Microscopy
T cells were harvested and blocked with 3% bovine serum albumin (BSA) in PBS before being incubated with an anti-LFA-1 antibody (BD Transduction Laboratories) to label the plasma membrane. After two washes in PBS, the cells were incubated with Alexa Fluor 568 goat anti-mouse IgG and counterstained with Hoechst 33258. The cells were washed twice in PBS and fixed with 4% PFA. Fixed cells were allowed to settle on Nunc LabTek glass slides coated with poly-L-lysine. Imaging was performed on a Zeiss LSM 510 confocal microscope (Carl Zeiss, Hertfordshire, UK) under 63× magnification using oil immersion.
Fixed T Cell Migration Assays and High Content Analysis
For migration of T cells on anti-LFA-1, standard flat-bottomed 96-well plates were coated with 5 µg/ml goat anti-mouse IgG followed by an anti-LFA-1 antibody (clone SPV-L7), as described.
7
The plate was washed with PBS, and 80 µL of complete cell culture medium was added. Twenty microliters of the cell suspension from the siRNA library plate were then added to the plate. The plate was incubated for 1 h at 37 °C, after which time the cells developed a polarized morphology and migrated (
T Cell Adhesion to ICAM-1
T cell adhesion to ICAM-1 in the absence or presence of SDF-1α was performed using a static adhesion assay as described, 8 with modifications. In brief, two 96-well plates were coated with 5 µg/ml goat anti-human Fc fragment-specific IgG followed by 1 µg/ml ICAM-1-Fc. The plates were washed with PBS and blocked with 0.5% BSA in RPMI-1640 for 30 min at 37 °C, and the wells were filled with 50 µL of complete cell culture medium. Twenty microliters of cells were removed from the wells of the siRNA library plate and added to both ICAM-1-coated plates. The plates were then filled with an additional 30 µL of complete cell culture medium in the presence or absence of SDF-1α (the final concentration of SDF-1α in ICAM-1-coated wells was 100 ng/ml). The plates were incubated for 30 min at 37 °C. The wells were then gently filled with PBS, sealed with adhesive 96-well plate covers, inverted, and centrifuged for 15 s at 6 g (the acceleration and brake on the centrifuge were set to 4; the maximum setting is 9). Unbound cells were removed by keeping the plate inverted and gently flicking the liquid out of the wells. Adherent cells were fixed with 4% paraformaldehyde containing 2 µg/ml Hoechst 33342. The number of cells in each well was enumerated using the IN Cell Analyzer under 10× magnification as described. 6
CellTiter Blue Viability Assay
Twenty microliters of the cell suspension were removed from each well of the siRNA library plate and added to a 96-well plate containing 80 µL/well complete cell culture medium. CellTiter Blue solution (20 µL) was then added to the wells, and the plate was incubated for 4 h at 37 °C. The cells were transferred to a F96 microwell black polystyrene plate, and the levels of fluorescence were quantified with a fluorometer (Wallac VICTOR, PerkinElmer, Waltham, MA). Background fluorescence values were determined by incubating 100 µL of complete cell culture medium with 20 µL of CellTiter Blue and subtracting these values from all other readings. We ensured that the fluorescence values were in the linear range by preparing a standard curve of nontransfected T cells plated at different concentrations in duplicate (from 2×106 cells/ml down to 0×106 cells/ml in twofold serial dilutions) and assaying for CellTiter-Blue fluorescence. Where indicated, 1 µM staurosporine was preincubated with nontransfected T cells for 4 h prior to the addition of CellTiter Blue as a positive control to reduce cell viability.
Total Cell Count
Twenty microliters of the cell suspension was removed from each well of the siRNA library plate and added to a 96-well plate containing 80 µL/well complete cell culture medium. An equal volume of 8% paraformaldehyde containing 2 µg/ml cell-permeable Hoechst 33342 was then added to the wells and incubated overnight at room temperature to fix the cells and label the nucleus. The number of cells per well was enumerated with the IN Cell Analyzer as described. 6
Analysis of siRNA Screening Data and Generation of Heat Maps
Z-scores were calculated for each data set as described. 9 The Z-score is a common statistical method for analysis of high-throughput screens and expresses a sample value as the distance from the mean in multiples of the standard deviation. We defined effects mediated by specific siRNAs as being statistically significant if they displayed a Z-score value of +/− 2 in three out of three screens. Z-score data were imported into Spotfire (Tibco) and represented pictorially as a heat map using a categorical coloring method. siRNAs with a Z-score of −2 or greater were colored green, whereas siRNAs with a Z-score of +2 or greater were colored red. siRNAs that had no effect were colored black. Alternatively, when indicated, the mean Z-score and standard deviations for each siRNA were graphed using GraphPad Prism 5 software.
Live T Cell Migration Assays on Anti-LFA-1
Individual wells of a 96-well plate were coated with anti-LFA-1 antibodies, as described above. T cells were added to the wells at a final concentration of 1.5×105 cells/ml in complete cell culture medium. Live cell imaging was performed using the IN Cell Analyzer equipped with a heated stage set at 37 °C. Bright-field images were acquired every 30 s under 20× magnification. The images were imported into ImageJ, and merged and saved as a movie file (
Transwell Migration Assays
Transwell migration assays were performed using the ChemoTx 96-well plate system from Neuro Probe (Gaithersburg, MD) supplied with 3 µm membrane pore diameters. Filters were first coated with 5 µg/ml goat anti-human Fc fragment-specific IgG, followed by 1 µg/ml ICAM-1-Fc. After washing the filters with sterile PBS, T cells at a concentration of 2×106 cells/ml in 0.5% BSA/RPMI-1640 were added to the upper side of the filter, whereas 30 ng/ml SDF-1 (diluted in 0.5% BSA/RPMI-1640) was placed in the lower chambers. The plates were incubated for 3 h at 37 °C, after which time cells that migrated through the pores into the lower chambers were recovered. Transwell assays were set up using triplicate wells for each condition. Negative controls were run in parallel for all experiments, including analysis of T cell migration through uncoated filters and without chemokine in the lower chambers. Recovered cells were fixed with an equal volume of 8% PFA containing 2 µg/ml Hoechst 33342 and counted with the IN Cell Analyzer, as described. 6
SDS-PAGE and Western Blotting
Proteins were resolved by SDS-PAGE, transferred to polyvinylidene fluoride membrane by Western blotting, and probed with the indicted primary and horseradish peroxidase (HRP)-labeled secondary antibodies. 6 Densitometry was used to determine the intensity of bands on the films using the Fusion FX chemiluminescence system and Bio ID software (Vilber Lourmat, Australia).
Statistics
Statistical analysis was performed using two-tailed Student t tests (GraphPad Prism 5). Effects were deemed to be significant at p < 0.05.
Results
Description of the siRNA Library Plate and Confirmation of siRNA Delivery and Gene Silencing in Primary Human T Cells
We evaluated a self-delivering siRNA library targeting the expression of 72 distinct genes as a “proof-of-principle” screening strategy in primary human T cells. The library, supplied as a single 96-well plate, consisted of self-delivering SMARTpool reagents (Accell siRNAs) wherein each well contained a mixture of four distinct siRNA designs targeting a single gene. The siRNAs were selected to target a variety of intracellular signaling pathways, and these targets are depicted in Figure 1 . The majority of the siRNAs in the library targeted the expression of kinases (41 targets, including cyclin-dependent kinases, serine/threonine kinases Akt1 and 2, and mitogen-activated protein kinases) and protein/lipid phosphatases or their regulatory subunits (14 targets, including the CDC family of protein phosphatases and the lipid phosphatase PTEN). Other siRNAs targeted the expression of transcription factors (p53, Myc, HIF-1α, and Rb1), the small GTPase RhoA, the transmembrane adaptor protein Nck1, and the dehydrogenase enzyme GAPDH. Also included in the library were self-delivering nontargeting control siRNAs (supplied as a SMARTpool as well as individual siRNAs 1–4) and a FAM-labeled self-delivering siRNA targeting PPIB (Cyclophilin B) ( Fig. 1 ). The FAM-labeled PPIB siRNA served as a useful reagent to assess delivery of the siRNAs into the cells. After reconstituting the library and preparation of daughter plates as outlined in the Materials and Methods section, primary T cells were incubated with the siRNAs for 72 h. The cells were then harvested, and siRNA delivery and silencing of gene expression were evaluated. As shown in Figure 2A , the FAM-labeled PPIB siRNA was delivered into 100% of the primary T cells, as determined by flow cytometry. Because flow cytometry cannot distinguish whether the siRNAs were internalized into the cells or were adsorbed to the cell surface, siRNA delivery was verified using confocal microscopy; incubation of primary human T cells with Accell Green nontargeting control siRNAs clearly demonstrated that these siRNAs were present in the cytoplasm of cells and did not co-localize with the plasma membrane-localized integrin LFA-1 (note that Accell Green nontargeting control siRNAs were not present in the siRNA library) ( Fig. 2B ). Similar to the FAM-labelled PP1B siRNA, flow cytometry also confirmed 100% uptake of these fluorescently labeled siRNAs into primary human T cells (data not shown). In addition, real-time PCR revealed that the expression of GAPDH was reduced by ~70% when primary T cells were incubated with siRNAs targeting this enzyme in comparison to cells incubated with nontargeting control SMARTpool siRNAs or nontransfected cells ( Fig. 2C ). Self-delivering siRNAs are therefore readily taken up into hard-to-transfect primary human T cells and result in significant silencing of gene expression.
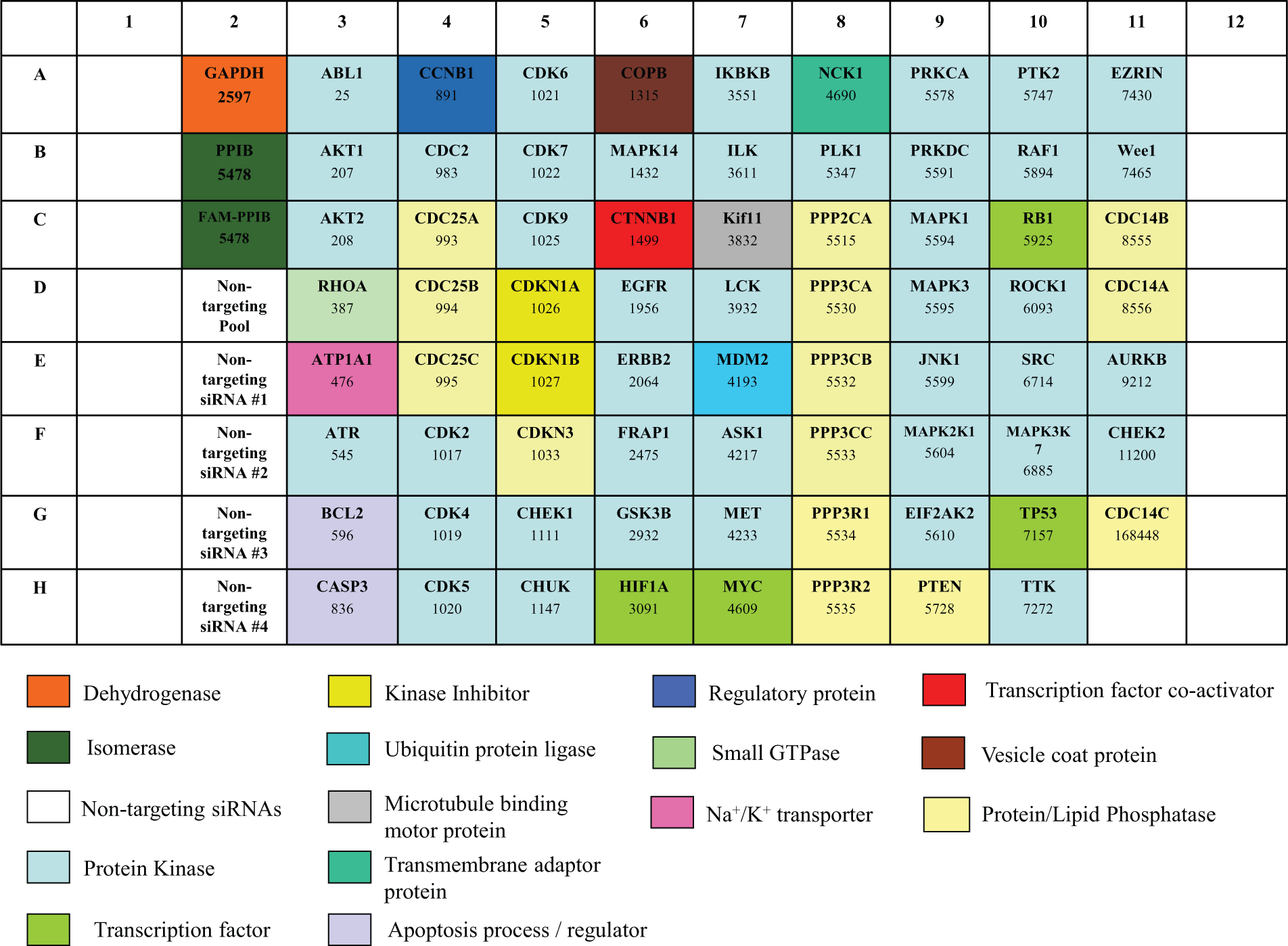
Layout of the self-delivering small interfering RNA (siRNA) library plate. The genes targeted by the siRNAs and the Entrez gene identification number are indicated in each well of the plate. The function of each gene was obtained from the NCBI Gene database (www.ncbi.nlm.nih/gene), GeneCards database (www.genecards.org), or PANTHER database (www.pantherdb.org), and they are segregated by color coding.
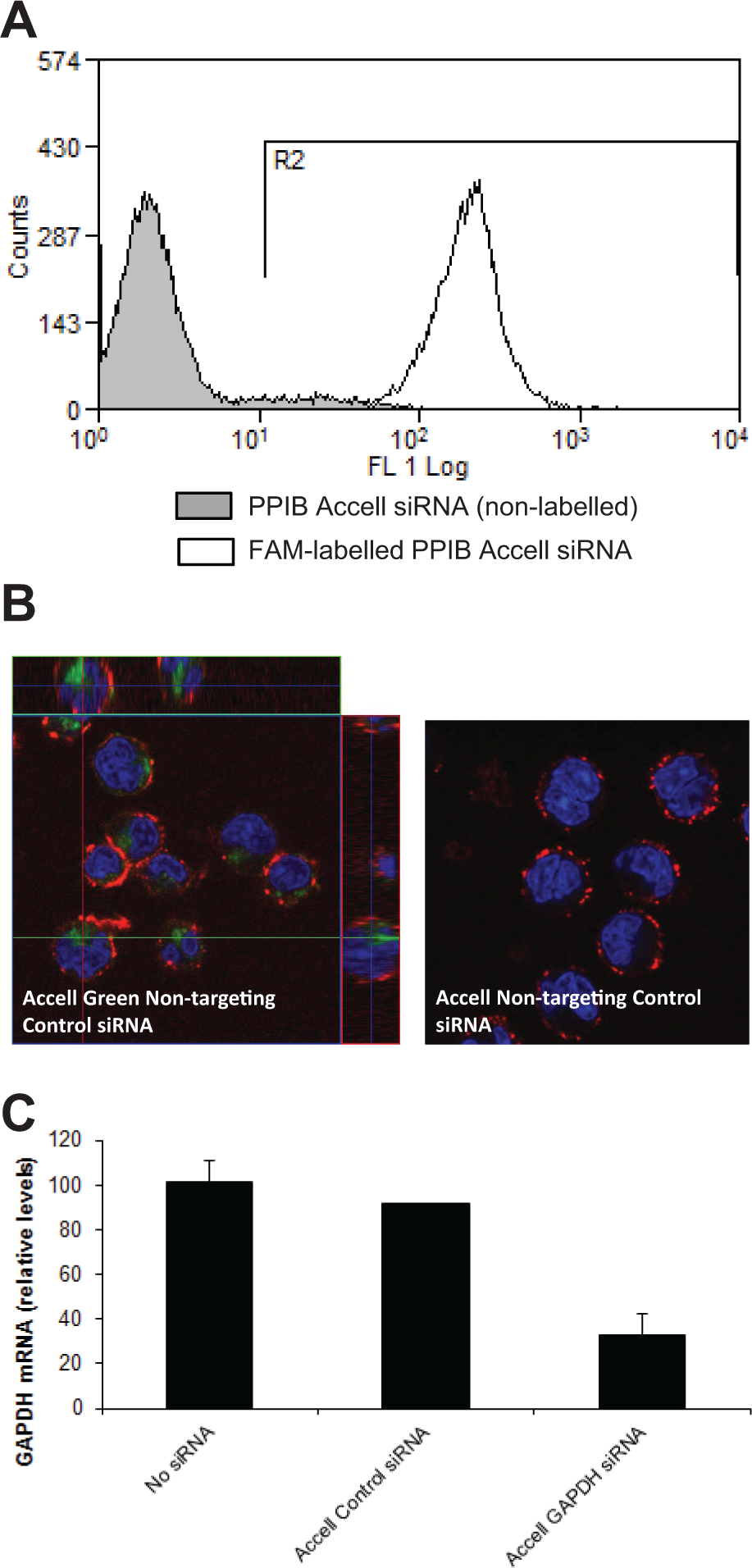
Delivery of self-delivering small interfering RNAs (siRNAs) to primary human T cells and silencing of gene expression. (
Identification of Genes That Regulate LFA-1-Mediated T Cell Migration
The siRNA library was next used to screen for genes that regulate LFA-1-mediated T cell migration. We used our well-characterized in-vitro model of T cell migration wherein an immobilized stimulating anti-LFA-1 antibody is used to crosslink this integrin and trigger T cell motility.
10
Hence, primary T cells were incubated on anti-LFA-1 to allow the cells develop a migratory phenotype following transfection with the siRNA library. As shown in
The cells were subsequently fixed and stained with phalloidin-TRITC, α-tubulin-FITC, and Hoechst 33258 to label F-actin, α-tubulin, and the nucleus, respectively. We then used an imaged-based high content analysis (HCA) platform in conjunction with our multiparametric toolbox assay, 7 using F-actin and α-tubulin staining to analyze the morphology of the T cells, to ascertain whether any siRNAs affected the migratory phenotype. We have previously demonstrated that this novel HCA approach enables measurements of thousands of cells to be acquired rapidly and thus facilitates population-level analysis of cells (~8000 cells/well in every RNAi screen), whereas our multiparametric analysis of T cell morphology that is based on both F-actin and α-tubulin staining results in improved ability to discriminate morphological behavior. 7 The parameters that were used to measure the morphology of the T cells included 1/(Form factor), Nuclear Displacement, Cell Area, and Gyration Radius. 1/(Form factor) is a roundness index, with values ranging from 1 to infinity, where 1 is a perfect circle. The 1/(Form factor) parameter increases as T cells adopt a migratory phenotype and is thus a useful measure of T cell migration. 7 The Nuclear Displacement parameter describes the position of the nucleus relative to the center of the cell body. In nonmigrating/nonpolarized T cells, the nucleus is usually positioned in the center of the cell body, but it translocates toward the lamellipodia (and away from the center of the cell body) in actively migrating T cells. Hence, Nuclear Displacement values also increase as T cells actively polarize, and therefore this is another useful measure of T cell migration. 7 Cell Area and Gyration Radius (a measure of the spread of the cell) were also used for quantification of T cell morphology, because we have previously shown that the values of both of these parameters increase when T cells are stimulated to migrate on anti-LFA-1 and other ligands. 7
The quality of the three RNAi screens was assessed using the Z’-factor, 9 which yielded a value of 0.68, indicating that the screens were robust and of sufficient quality (data not shown). We then identified siRNAs that affected the various T cell morphology parameters using the Z-score system. The Z-score is a common statistical method for analysis of high-throughput screens and expresses a sample value as the distance from the mean in multiples of the standard deviation. 9 siRNAs were defined as being statistically significant if they exceeded a Z-score value of +/− 2 in three out of three screens. The Z-score data for each morphological parameter were displayed as a heat map ( Fig. 3 ). siRNAs with a Z-score of −2 or greater were colored green (i.e., siRNAs that decreased a particular parameter by >2 standard deviations from the mean), whereas siRNAs with a Z-score of +2 or greater (i.e., siRNAs that increased a particular parameter by >2 standard deviations from the mean) were colored red. As shown in Figure 3A , siRNAs targeting the GTPase RhoA increased 1/(Form factor) and Nuclear Displacement parameters when morphological analysis was based on either F-actin or α-tubulin staining, implying that T cell polarization was increased. Indeed, visual inspection of RhoA siRNA-treated T cells confirmed that these cells were elongated relative to other samples, including cells treated with nontargeting control siRNAs, and had elongated uropods ( Fig. 3B ). Although siRNAs targeting RhoA increased 1/(Form factor) and Nuclear Displacement, significant differences in Cell Area were not observed, and an increase in Gyration Radius was observed only when RhoA siRNA-treated cells were analyzed using α-tubulin staining and not F-actin staining ( Fig. 3A ). None of the other siRNAs in the library significantly influenced T cell morphology when analyzed using the 1/(Form factor), Nuclear Displacement, Cell Area, or Gyration Radius parameters.
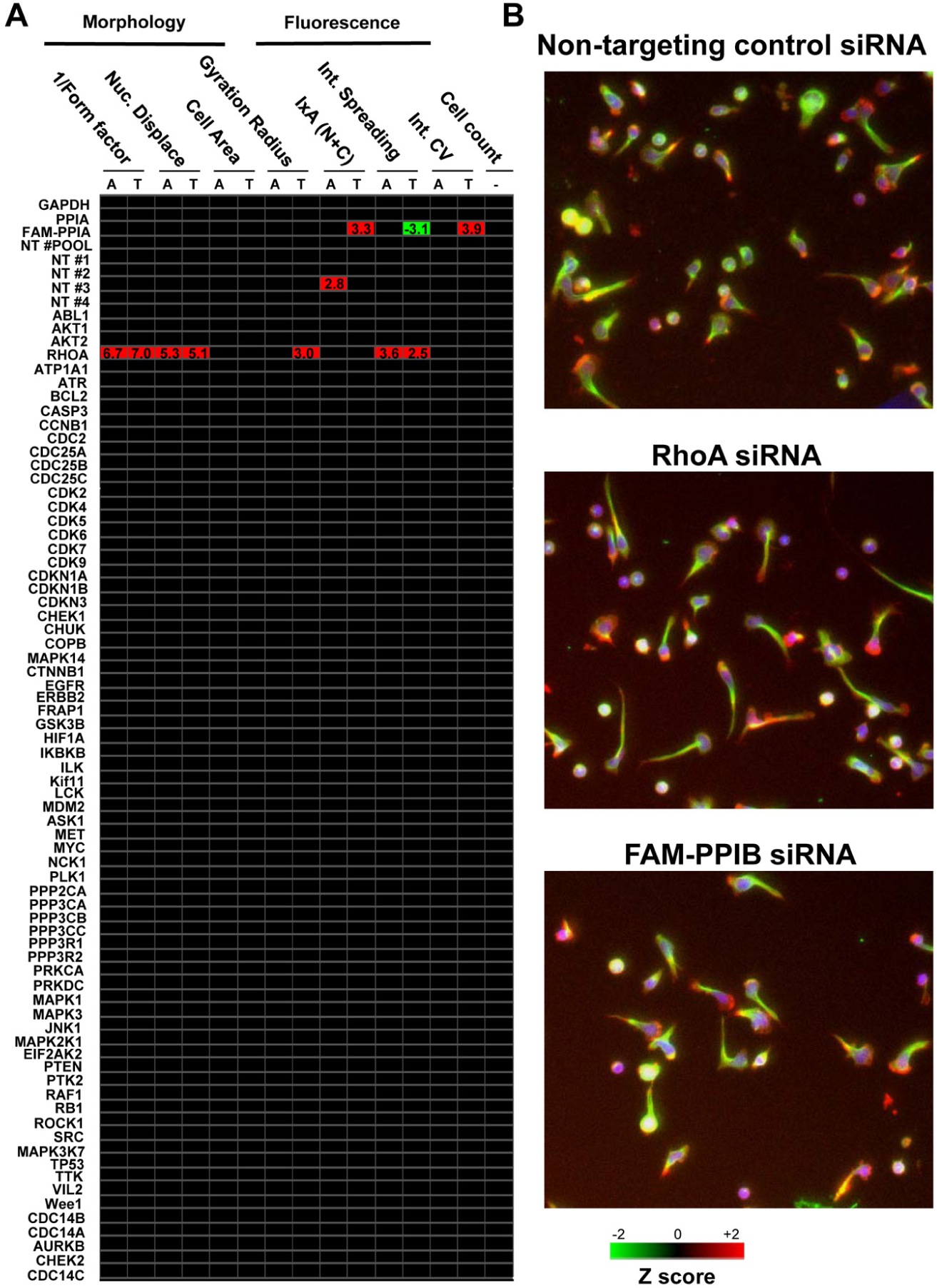
Screening of a self-delivering small interfering RNA (siRNA) library for identification of genes that regulate LFA-1-mediated primary human T cell migration. (
The multiparameter toolbox that we have used for analysis of T cell migration also incorporates a number of fluorescence intensity and fluorescence distribution parameters. 7 These include (1) the IxA (N+C) parameter, which measures the integrated intensity of fluorescence pixels in both the nucleus and cytoplasm; (2) the Intensity Spreading parameter, which estimates the extent of fluorescence intensity near the boundary of the cell; and (3) the Intensity Coefficient of Variance (CV), which measures the variation of the fluorescence intensity in the cytoplasm and thus is a useful readout of the homogeneity of staining within the cell. We investigated whether any of the siRNAs affected these parameters. Interestingly, primary T cells that were incubated with siRNAs targeting RhoA resulted in an increase in the Intensity Spreading of both the F-actin and α-tubulin cytoskeleton ( Fig. 3A ). This implies that silencing of this GTPase increases the localization of the F-actin and α-tubulin cytoskeleton near the cell boundary. As siRNAs targeting RhoA did not affect IxA (N+C) or Intensity CV, we conclude that silencing of this GTPase did not affect the fluorescence intensity of the labeled F-actin/α-tubulin cytoskeleton or the homogeneity of staining of these cytoskeletal components. Interestingly, the FAM-labeled siRNA targeting PPIB significantly affected all three fluorescence intensity/distribution parameters when α-tubulin staining (but not F-actin staining) was used for the analysis ( Fig. 3A ). In particular, the IxA (N+C) and Intensity CV parameters were increased, and the Intensity Spreading parameter was decreased. An increase in the IxA (N+C) parameter implies that the fluorescence intensity of α-tubulin staining significantly increases on addition of the FAM-labeled siRNA. The finding can be explained by the fact that the fluorescence channel used to detect the FITC-labeled α-tubulin stain additionally detects the FAM-labeled siRNA targeting PPIB inside the T cells. The detection of the FAM-labeled siRNAs in the cells by fluorescence microscopy therefore complements the confirmation of siRNA delivery to primary T cells using flow cytometry ( Fig. 2B ). The FAM-labeled siRNA decreased the Intensity Spreading parameter and increased the Intensity CV parameter when α-tubulin staining was used for the analysis ( Fig. 3A ). This is likely due to the fact that the FAM-labeled siRNA localizes to the cytosol (thus decreasing the relative intensity of fluorescence staining at the cell boundary where α-tubulin is localized) and results in a higher CV of fluorescence staining inside the cells. Of note, the FAM-labeled siRNA targeting PPIB did not affect any of the morphology parameters such as 1/(Form factor) or Nuclear Displacement.
Additional Assays Can Be Performed from Self-Delivering siRNA Screens
A notable advantage of performing RNAi screens in suspension cells in comparison to adherent cells is that any surplus of cells remaining after gene silencing can be used for additional assays without requiring extensive manipulation such as trypsinization. Because less than 20% of the cell population in each well was used for the migration assays on anti-LFA-1, the remainder of cells were used for other assays, including cell viability, total cell count, and chemokine-induced inside-out LFA-1 activation (i.e., adhesion to ICAM-1 in the absence or presence of the chemokine SDF-1α). As shown in
Validation and Characterization of RhoA as a Crucial Regulator of Polarization and Migration in Primary Human T Cells
Because this RNAi screening strategy identified RhoA as a regulator of T cell polarization on anti-LFA-1, the role of this GTPase in T cell polarization/migration was validated and characterized in further detail. Using primary human T cells from five independent donors, we demonstrated by Western blotting that self-delivering SMARTpool siRNAs targeting RhoA consistently reduced expression levels by ~85% ( Figs. 4A and 4B ). Because the SMARTpools comprise four distinct siRNA designs targeting a single gene, each of the four RhoA-targeting siRNAs from this SMARTpool was tested for its ability to silence RhoA expression in parallel. Three out of the four siRNAs from this SMARTpool (siRNAs 1, 2, and 3) effectively silenced RhoA expression levels by 70–90% at the protein level and thus were “active” siRNAs within the SMARTpool ( Figs. 4A and 4B ). In contrast, siRNA 4 targeting RhoA was more variable with regard to silencing efficiency in the same experiments ( Figs. 4A and 4B ). As expected, both the SMARTpool and siRNAs 1, 2, and 3 targeting RhoA increased T cell polarization on anti-LFA-1, as demonstrated by an increase in the 1/(Form factor) and Nuclear Displacement parameters, whereas siRNA 4 was the least effective ( Figs. 4C and 4D ). Representative images of these phenotypes on anti-LFA-1 are shown in Figure 5A , with elongated uropods clearly evident following silencing of RhoA expression. Reassuringly, silencing of RhoA expression strongly correlated with cellular phenotype when Nuclear Displacement ( Fig. 5B ) or 1/(Form factor) (data not shown) was used as the morphology parameter.
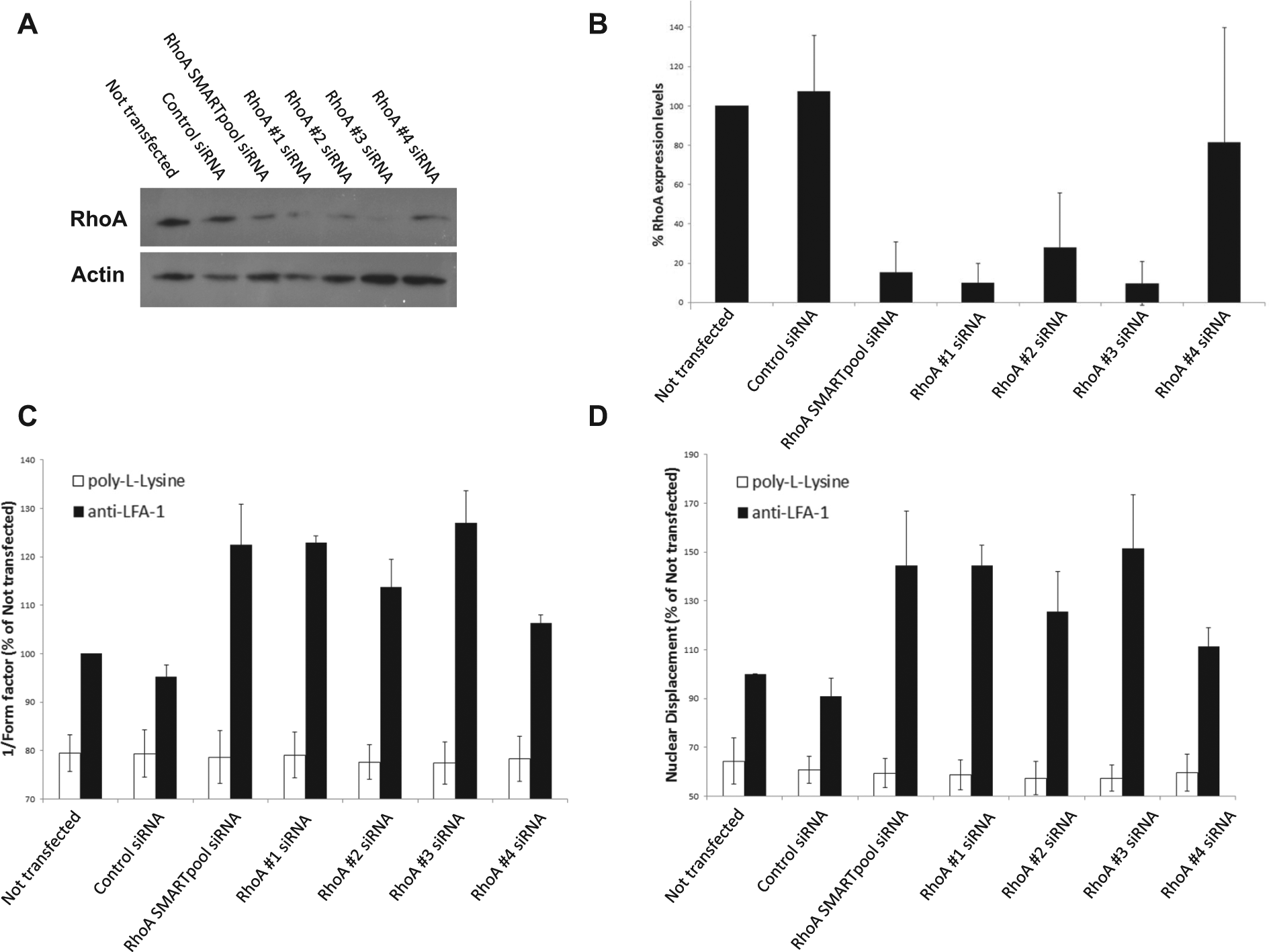
Silencing of RhoA expression using self-delivering small interfering RNAs (siRNAs) promotes an elongated morphology on anti-LFA-1. (
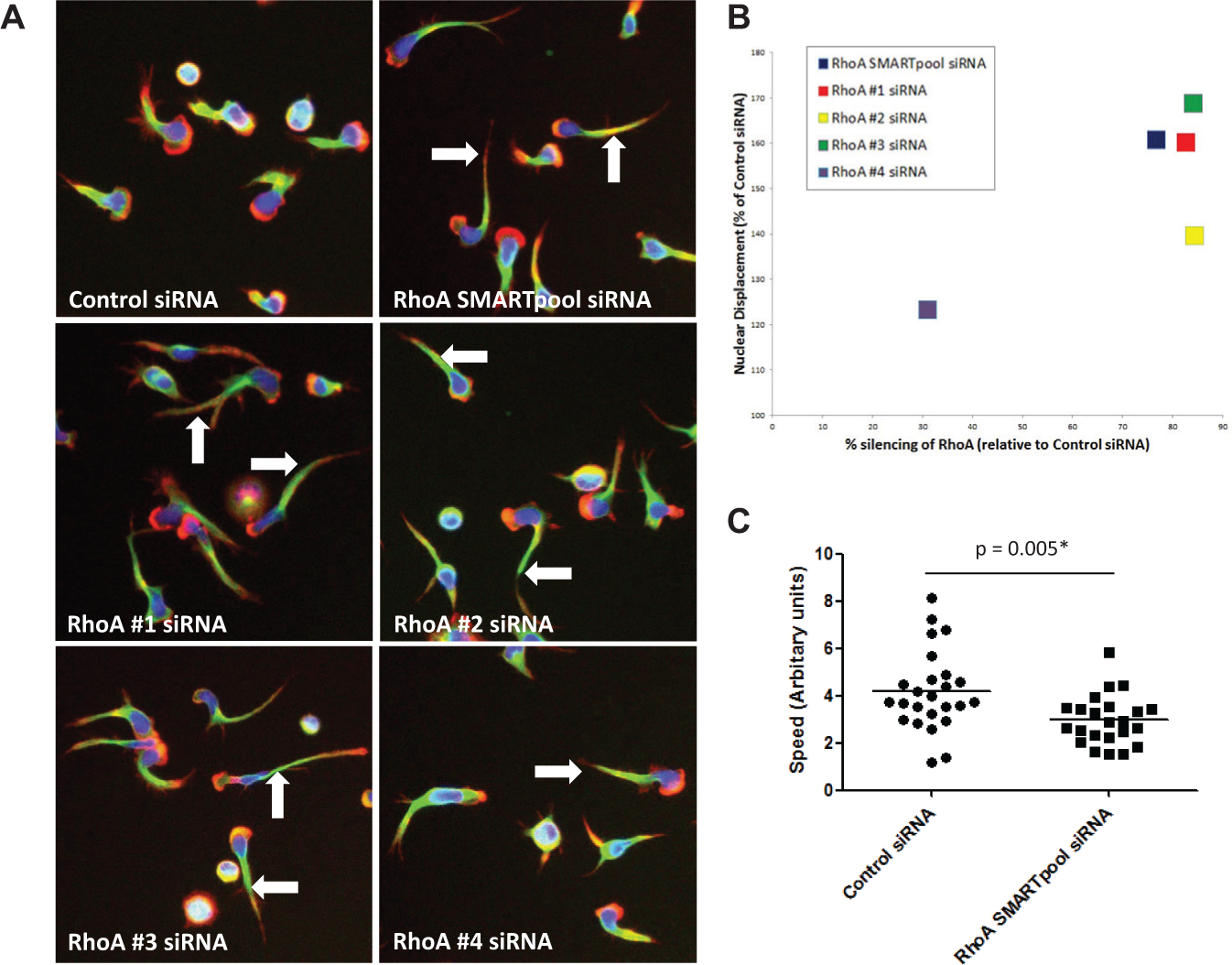
Silencing of RhoA expression using self-delivering small interfering RNAs (siRNAs) promotes an elongated morphology on anti-LFA-1 and perturbs T cell migration speed. (
The impact of RhoA silencing on T cell migration was next investigated. Whereas T cells incubated with nontargeting control siRNAs polarized and migrated over the anti-LFA-1 substrate (
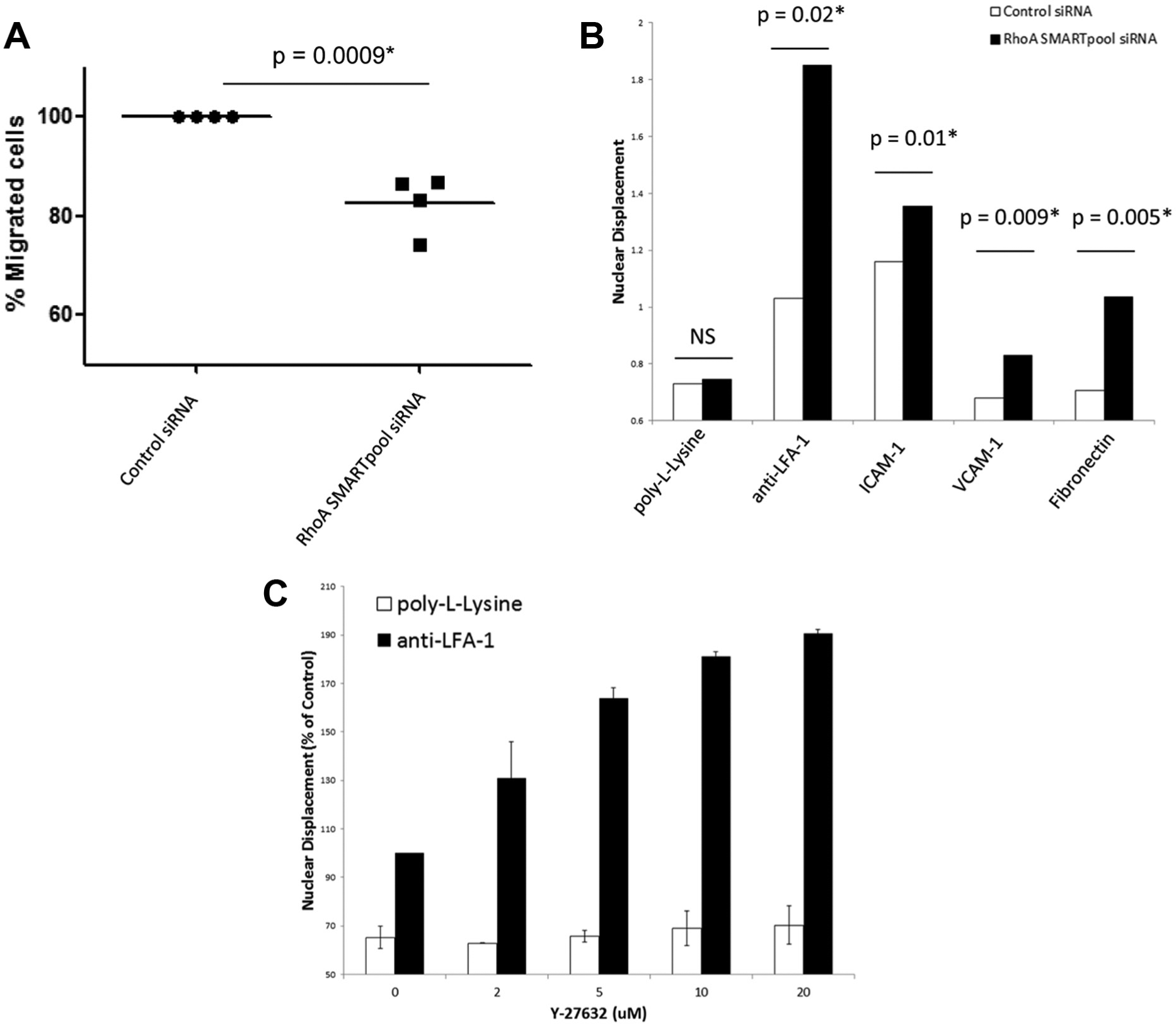
RhoA regulates T cell motility and polarization on β1/β2 integrin ligands. (
Off-target effects of siRNAs have been reported in a number of studies, including RNAi screens. 11 Although three out of the four distinct siRNAs targeting RhoA produced an identical phenotype of elongated uropods in primary human T cells when tested separately and therefore were highly unlikely to be off-target effects, an additional experiment was performed to rule out any potential off-target effects. A key downstream effector of RhoA in mammalian cells is the serine/threonine kinases ROCK 1 and 2; 12 inhibiting ROCK 1/2 should therefore produce a similar phenotype as silencing of RhoA. The ROCK 1/2 pathway was inhibited with the pharmacological inhibitor Y-27632, and T cell polarization on anti-LFA-1 was analyzed. Treating primary human T cells with Y-27632 in a dose-dependent manner resulted in an elongated morphology on anti-LFA-1 that was identical to RhoA-depleted T cells ( Fig. 6C ). Hence, the phenotype observed following silencing of RhoA expression is not an off-target effect in primary human T cells.
Discussion
In this study, we have screened a self-delivering siRNA library to identify genes regulating LFA-1-mediated T cell migration, cell viability, and chemokine-induced inside-out activation of LFA-1 in primary human T cells. To our knowledge, these are the first RNAi library screens to be performed in primary human T cells using self-delivering, synthetic siRNAs. Although the vast majority of RNAi screens generally use a single assay/readout for elucidating gene function, we demonstrate here that multiple downstream assays can be performed from a single screen after gene silencing in primary T cells. This approach offers obvious advantages of maximizing use of all the cells and the potential for addressing numerous biological questions with RNAi libraries.
Self-delivering siRNAs (Accell siRNAs) are proprietary, chemically modified siRNAs that are taken up into cells without the need of a transfection/delivery reagent. The siRNAs are simply mixed with the target cells and are ideal for silencing of gene expression in primary T cells, which are widely known as being hard to transfect. 5 In addition, the availability of these siRNAs in library plate format makes them particularly suitable for screening studies. A number of RNAi screens have been performed in T cells to identify novel genes regulating NF-κB activation,13,14 migration15,16 differentiation, 17 cytokine production, 18 apoptosis/necroptosis,19,20 alternative splicing of CD45, 21 surface expression of CD46, 22 HIV infection/replication,23,24 and survival/proliferation of leukemic T cells.25,26 With the exception of one study, which used lipid transfection to deliver siRNAs into Jurkat leukemic T cells for RNAi library screening, 19 all of these used viral vectors or electroporation to deliver the siRNAs into the T cells. Screening of RNAi libraries in T cells using viral vectors or electroporation is very challenging and labor-intensive, however, because specialist expertise (e.g., in the handling of viral vectors), equipment, and reagents are required. Furthermore, most RNAi screens performed in T cells to date have used transformed cell lines, which are not representative of normal primary T cells. We show here that the self-delivering siRNAs are efficiently delivered into primary human T cells, result in silencing of gene expression, and can be used as a novel screening tool for loss-of-function studies in primary T cells. Self-delivering siRNA libraries therefore offer an alternative strategy to viral vectors or electroporation when undertaking RNAi screening in hard-to-transfect cell types.
We identified the GTPase RhoA from the siRNA library screens as a regulator of T cell polarization and migration when primary human T cells were stimulated through LFA-1 (a β2 integrin) or β1 integrins; siRNAs targeting this GTPase inhibited T cell migration primarily because of a failure of these cells to detach their uropods from the substratum, leading to an elongated phenotype. Also, knockdown of RhoA expression in T cell lines using siRNAs or treating primary T cells/T cell lines with the C3 toxin from Clostridium botulinum (which catalyzes ADP ribosylation and inactivation of Rho family members) yields broadly similar phenotypes,15,27–30 with defects reported in contraction of the acto-myosin cytoskeleton in the uropods of these cells. It is important to highlight, however, that transformed T cell lines harbor numerous mutations and therefore can behave differently than primary human T cells, whereas the C3 toxin does not discriminate between the Rho A, B, and C isoforms. Our data unequivocally demonstrate for the first time that RhoA regulates polarization and migration in primary human T cells and that self-delivering siRNA library screens can be used to identify regulators of T cell migration in more physiologically relevant cell types.
Our screening assays also demonstrated that RhoA does not regulate chemokine-induced inside-out activation of LFA-1, and hence this GTPase appears to influence postintegrin activation events regulating T cell migration. However, two groups used RhoA-targeting cell-permeable peptides to demonstrate a role for this GTPase in inside-out activation of LFA-1/adhesion to ICAM-1 in T cells under shear flow.31,32 Although the reasons for these apparent differences between our study and those described in Refs. 31,32 are unknown at present, it has been reported that certain GTPases and their regulators can influence chemokine-induced T cell adhesion to integrin ligands under shear flow but have no obvious role when assayed under static conditions. 33 Therefore, further work will be needed to clarify the role of RhoA in integrin activation.
It is noteworthy that while siRNAs targeting ROCK1 (a downstream effector of RhoA) were also present in this library, this did not yield a similar phenotype as RhoA siRNAs or the ROCK 1/2 inhibitor Y-27632. We found a measurable but small effect of siRNAs targeting ROCK1 on T cell polarization (data not shown), but this was below our stringent Z-score threshold level set at +/− 2, suggesting possible redundancy with ROCK2 and/or partial knockdown of this target.
Self-delivering siRNA libraries have great potential for novel gene discovery in primary human T cells. One area that would benefit from this technology is the identification of host genes required for HIV infection/replication in primary human T cells, which are the physiological target of this virus. To date, RNAi screens for host genes regulating HIV infection have been performed in nonphysiological cell types transfected with receptors to enable viral infection (reviewed in Ref. 34 ) and in transformed T cell lines.23,24 Surprisingly, very little overlap in host genes required for HIV infection/replication were observed in all of these screens, with the differences observed attributed to the cell types, experimental procedures, and assay readouts used in the respective studies. 34 RNAi library screening in primary human T cells using self-delivering siRNAs may help to resolve these discrepancies and elucidate host factors required for HIV infection/replication in a physiologically relevant cell type. Another area that would benefit from siRNA library screening would be the identification of genes that regulate the survival and proliferation of primary leukemic T cells in cancer patients. For example, screening of a tyrosine kinase siRNA library for molecular vulnerabilities in leukemia cells isolated from cancer patients is an exciting area that has been shown to guide novel treatment approaches. 35
Although we have used this self-delivering siRNA library in conjunction with an automated high content image analysis platform to identify genes regulating human T cell migration, other plate-based assays and readouts can be coupled easily with siRNA libraries for screening purposes. These include enzyme-linked immunosorbent assays (ELISAs) to quantify cytokines or 96-well plate transwell assays to monitor T cell migration. Because we have shown here that multiple downstream assays can be performed from a single siRNA screen, a feasible and potentially exciting approach would be to screen subgenomic siRNA libraries (e.g., kinase libraries) or even whole-genome siRNA libraries to identify genes regulating cytokine secretion in response to T cell activation and T cell migration in response to inflammatory stimuli, including chemokines and integrin ligands. Because cytokine secretion and migration are central for T cell adaptive immune responses, such comprehensive biological readouts from individual RNAi screens will have broad implications in our understanding of the pathways that regulate T cell function and may uncover novel therapeutic targets for treating immunological disorders, cancer, and infection. 5
Footnotes
Acknowledgements
We thank the Irish Blood Transfusion Service for generously providing buffy coat blood packs. We thank Louise Baskin for providing siRNA reagents and critical reading of the manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Devin Leake is an ex-employee of Dharmacon (part of GE Healthcare), which supplied the self-delivering RNAi library. Annaleen Vermeulen is an employee of Dharmacon (part of GE Healthcare), which supplied the self-delivering RNAi library.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.