
Abstract
High-content screening (HCS) using RNA interference (RNAi) in combination with automated microscopy is a powerful investigative tool to explore complex biological processes. However, despite the plethora of data generated from these screens, little progress has been made in analyzing HC data using multivariate methods that exploit the full richness of multidimensional data. We developed a novel multivariate method for HCS, multivariate robust analysis method (M-RAM), integrating image feature selection with ranking of perturbations for hit identification, and applied this method to an HC RNAi screen to discover novel components of the DNA damage response in an osteosarcoma cell line. M-RAM automatically selects the most informative phenotypic readouts and time points to facilitate the more efficient design of follow-up experiments and enhance biological understanding. Our method outperforms univariate hit identification and identifies relevant genes that these approaches would have missed. We found that statistical cell-to-cell variation in phenotypic responses is an important predictor of hits in RNAi-directed image-based screens. Genes that we identified as modulators of DNA damage signaling in U2OS cells include B-Raf, a cancer driver gene in multiple tumor types, whose role in DNA damage signaling we confirm experimentally, and multiple subunits of protein kinase A.
Keywords
Introduction
Image-based high-content (HC) RNA interference (RNAi) screening is an effective experimental approach to elucidate the functions of genes on the systems level by investigating the phenotypes of a large number of living cells in culture. HC perturbation assays generate large amounts of high-resolution image data that are converted into multidimensional numeric data by image-processing software. The resulting features are numeric representations of a variety of phenotypic readouts of cells, such as cellular morphology measurements or the intensity of stains of cellular DNA or specific proteins, and they permit the identification of genes that are involved in complex biological pathways. Although univariate computational methods (i.e., methods that make use of only a single feature) can suffice to identify a limited number of the most salient hits in high-content screening (HCS), it was conclusively demonstrated that multivariate hit identification methods (i.e., methods that exploit multiple numeric features) outperform univariate methods. 1
A recent review lists more than a dozen studies in which multivariate techniques were applied to HC RNAi screens. 2 In the vast majority of these and other relevant studies, multivariate analysis was either a complicated, multistep procedure that involved an extended sequence of separate computational steps for hit identification and dimensionality reduction,3 –7 lacking nongreedy/ad hoc feature selection,8,9 or both.10–12
As a potential result, the majority of researchers pursuing HCS still rely on univariate methods to analyze multidimensional screening data. Analyzing multidimensional data with univariate methods is a self-imposed bottleneck that makes HC assays factually low content. A meta-analysis of 118 published articles to investigate how many features were actually used for hit identification in different HC screens found that even as recently as 2012, only 25% of HC screens were analyzed using multivariate methods, rendering the information content of these experiments much lower than their potential. 13 We believe that the reason for the underwhelming popularity of multivariate methods in HCS is their extensive complexity. A powerful but easy-to-use multivariate method would encourage more researchers to get more actual content out of their HC screens.
Moreover, to gain real insight into the biological mechanisms of identified hits from these HC screens, secondary screens and follow-up experiments are required. The majority of dimensionality reduction methods previously used in HCS, such as factor analysis 14 or principal component analysis, 5 construct novel “meta-features” by linear combination of the original features. Although these techniques successfully reduce the screening data’s dimensionality, they do not necessarily reduce the actual number of features (and therefore the number of phenotypic readouts) required for hit identification. Hence, even after dimensionality reduction, a large number of features need to be rescreened in secondary screens. Selecting the minimum number of the most informative original features at the most informative time points would greatly reduce the effort in biological follow-up experiments because only a subset of the original phenotypic readouts at a limited number of time points would have to be recorded without any significant loss of important biological information.
To address these challenges, we developed the multivariate robust analysis method (M-RAM), a novel computational technique for the multivariate analysis of biological perturbation screens, and applied this method to an HC screen recently performed in our laboratory to discover novel regulators of the DNA damage response (DDR). 15 M-RAM consists of two components: dRIGER, which condenses the effects of all of the short hairpin RNAs (shRNAs) for any particular gene into a single value for each of the features analyzed in the images, and logistic regression paired with the least absolute shrinkage and selection operator (Lasso), an effective, integrated regularization method for feature selection. 16 M-RAM predicts hits and simultaneously selects a limited number of the most informative original features and time points in one single step. Therefore, this method is fast, elegant, and easy to use. We anticipate that M-RAM will find wide acceptance in the HCS community because of its simplicity and interpretability.
Materials and Methods
HCS
To investigate new aspects of the DDR in molecular detail, an RNAi-based HC screen was performed, as described previously.
15
In brief, U2OS cells in 44 384-well plates (
Directional RIGER
A new, extended derivation of RNAi Gene Set Enrichment (RIGER),
17
directional RIGER (dRIGER), was used to transform normalized (see
Mathematically, positional hit scores (PH) and miss scores (PM) were calculated at each position i in a rank-ordered list of length L based on the ranks of the screened shRNAs targeting the gene of interest,
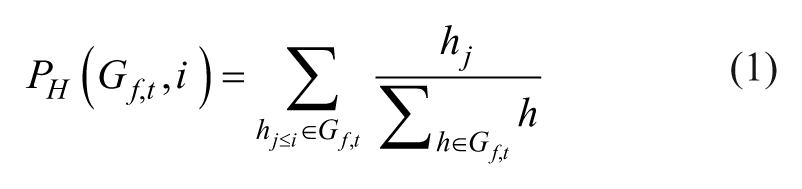
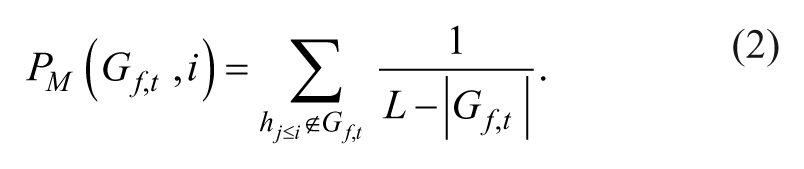
Similarly, inverse positional ES were computed to test for rank enrichment at the bottom/end of the rank-ordered list using an inverse shRNA rank set
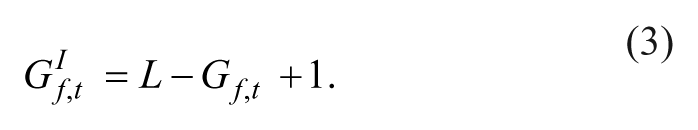
Finally, dES were computed as
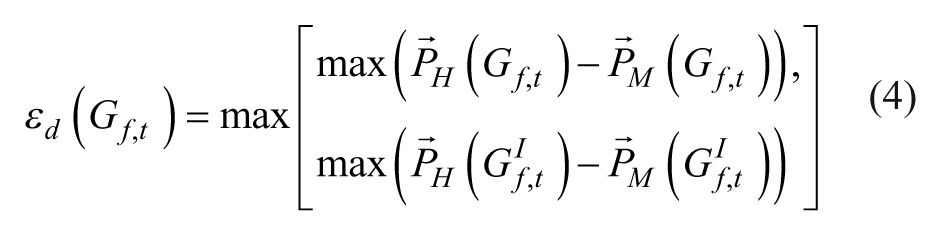
and multiplied with −1 if the inverse directional ES was greater than the directional ES.
A Java implementation of the dRIGER algorithm is available at http://yaffelab.mit.edu/driger/.
Logistic Regression and Lasso
A logistic regression model with Lasso regularization 16 (Lasso model) was used for integrated feature selection and hit identification. Feature weights were computed as
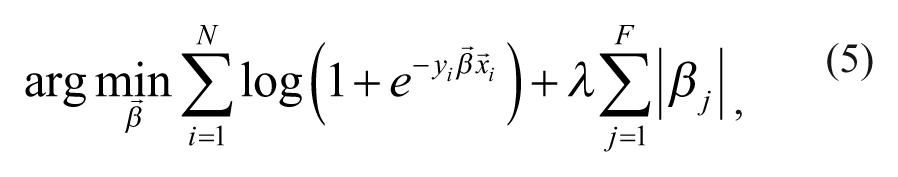
where
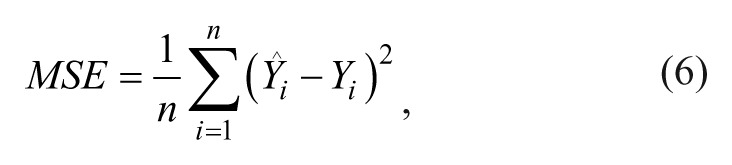
where n is the number of observations in the test data,
Network Analysis
SteinerNet,
19
an implementation of the Prize-Collecting Steiner Tree (PCST) algorithm, was used to produce a focused view of a protein-protein interaction network of interest. Interactions and genes were annotated with edge costs and node prizes, respectively, and fed into SteinerNet (see
Results and Discussion
To identify novel molecular components of the DDR after IR, we performed an image-based HC RNAi screen, looking for unknown DDR modulators in seven functional categories (kinases, phosphatases, chromatin modifiers, RNA binding proteins, DDR modulators, oncogenic regulators, and miRNA machinery). For this multidimensional HC assay, we screened five distinct phenotypic readouts (DNA content, γH2AX, pHH3, CC3, and tubulin) at four time points (before IR and 1, 6, and 24 h after IR) to systematically quantify both temporal and spatial changes in the DDR, thus enabling a sophisticated understanding of the signal transduction network that governs the cell’s response to DNA damage.
dRIGER Transforms shRNA-Level into Gene-Level Data
To capture the consistency of the differential knock-down effects of multiple shRNAs targeting the same specific gene, we developed dRIGER, an extension of the Gene Set Enrichment Analysis–based RIGER.17,18 We developed this method because RIGER was originally designed for continuous signal-to-noise ratios or (log) fold-changes. Inherently, RIGER does not capture the enrichment of ranks of shRNAs targeting the same specific gene toward the bottom of a rank-ordered list of all screened shRNAs. Our new method, dRIGER, computes dNES to quantify the enrichment of ranks of shRNAs targeting the same specific gene toward both the top and the bottom of a rank-ordered list of all screened shRNAs, therefore capturing the consistency of both increased and decreased phenotypic knock-down responses.
We applied dRIGER to all genes on all screened plates to compute dNES for each feature at each time point. To demonstrate how dRIGER captures both statistical location and statistical spread of differential knock-down phenotypes of shRNAs targeting specific genes, we computed dNES for the integrated γH2AX intensity feature 1 h after IR for a small number of selected genes. We chose Brd4, H2AFX, and the negative control luciferase because the phenotypic responses to H2AFX and Brd4 knock-down are well characterized.15,20 As expected, knock-down of H2AFX substantially decreased recorded γH2AX intensity 1 h after IR, and Brd4 knock-down substantially increased it ( Fig. 1A ). Although the majority of shRNAs targeting Brd4 and H2AFX induced a consistent phenotypic effect, outliers existed in both cases. Negative control knock-downs induced a wide range of phenotypic effects, from increased to decreased γH2AX intensities ( Fig. 1A ). dRIGER effectively captured these variable phenotypic effects and assigned high dNES to the H2AFX and the Brd4 knock-down but a low dNES to the negative control knock-down ( Fig. 1B ). dRIGER successfully quantified statistical location and statistical spread—or the lack thereof—for known DDR modulators and negative controls. At the same time, dRIGER transformed shRNA-level data (67,584 rows) into gene-level data (10,892 rows), which led to a more than sixfold reduction of our data’s dimensionality. All subsequent analyses were performed on gene-level data.
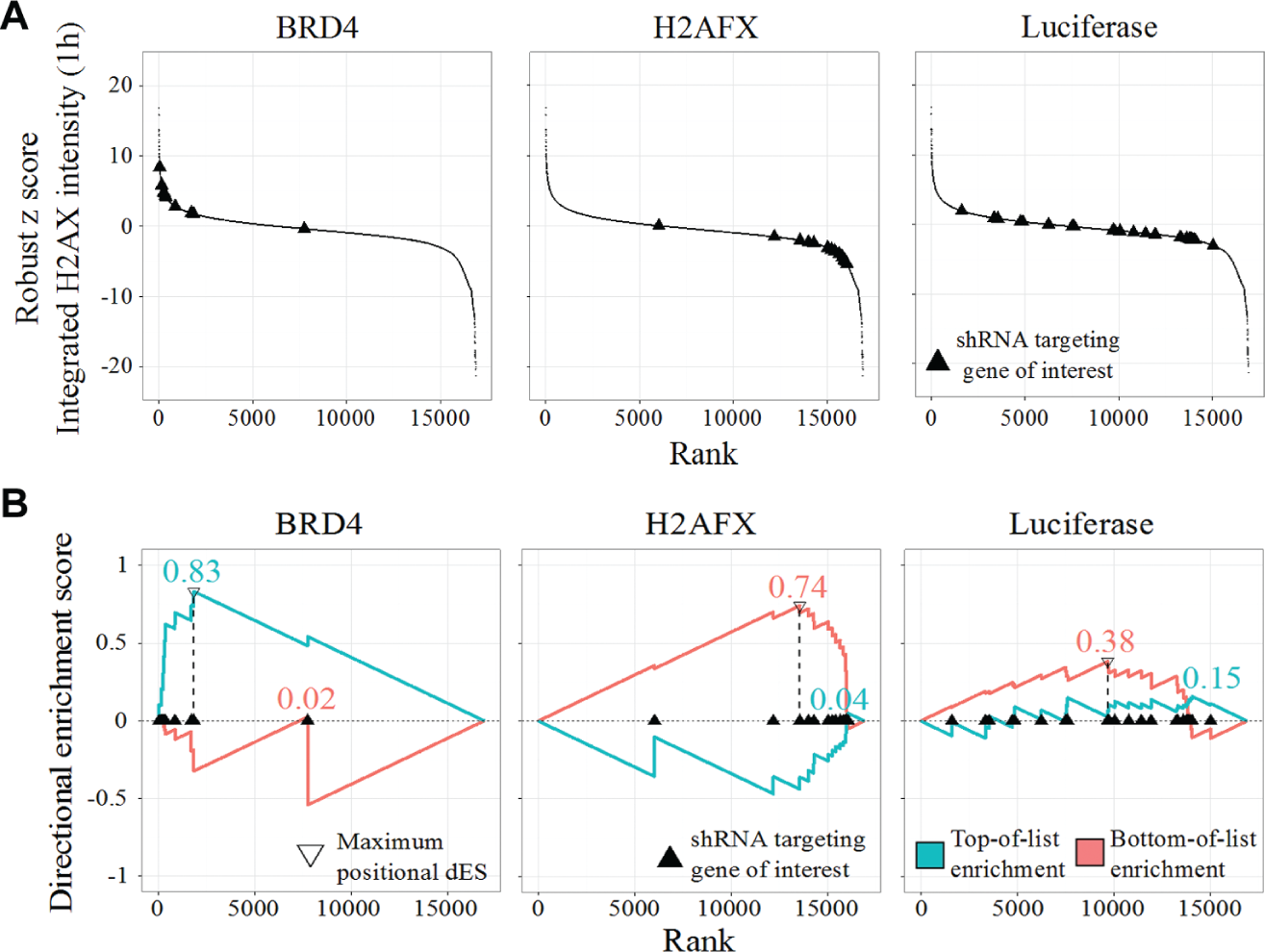
Directional RNA interference gene enrichment ranking (dRIGER) captures the consistency of differential effects of multiple short hairpin RNAs (shRNAs) and transforms shRNA-level data into gene-level data. (
Feature Selection with the Lasso
As M-RAM’s explicit purpose is to generate more reliable hypotheses for follow-up experiments, we wanted to select the phenotypic readouts and time points for which these follow-up experiments would prove most successful from the set of all screened readouts and time points ( Fig. 2A ). To select the features that were most predictive for DDR modulators and discard features mainly capturing noise, we used a logistic regression model with Lasso regularization (Lasso model).
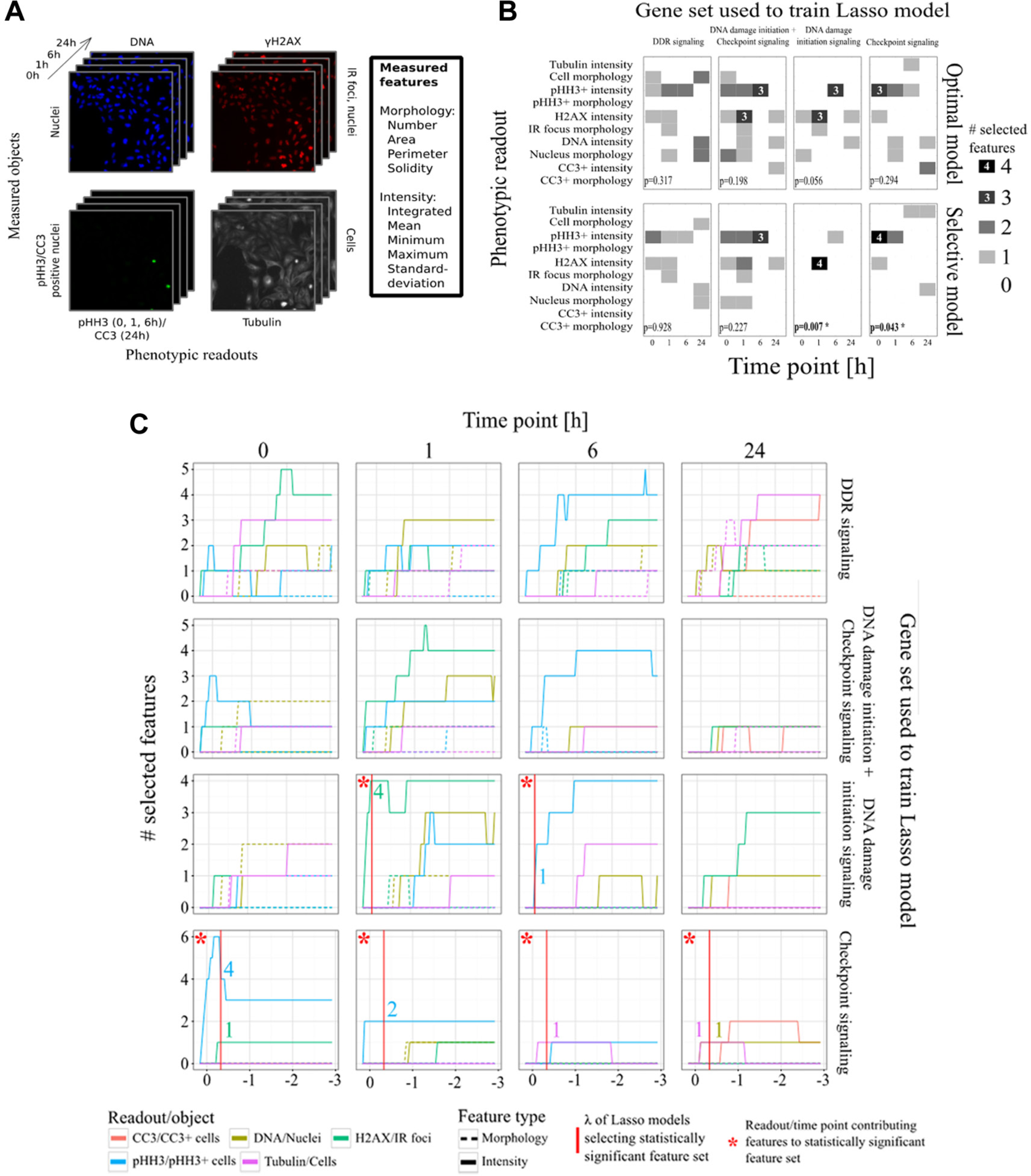
Logistic regression with least absolute shrinkage and selection operator (Lasso) regularization selects the most informative phenotypic readouts and time points that best capture the differences between knocked-down genes and negative controls. (
First, we wanted to investigate whether a feature set existed that was able to capture a putative “über phenotype” shared among all the knock-downs of a large set of functionally diverse DDR modulators. We trained our Lasso model on a training set consisting of 17 genes known to play a prominent role in various aspects of the DDR (
We hypothesized that the different and diverse functions of the various DDR modulators used as positive observations in the Lasso model’s training set were the reason for the lack of statistical significance of the selected feature set and postulated that our predictive models might be better at successfully capturing knock-down phenotypes of more functionally coherent gene sets. Knock-down of genes that are functionally coherent (i.e., that participate in a similar process within the larger set of molecular events that constitute the DDR) is likely to induce similar phenotypic responses that can be captured by automated microscopy and subsequently numerically captured in the extracted features. We therefore trained Lasso models for DNA damage initiation signaling, checkpoint signaling, and, as a stringent control, the union of these two, to test our hypothesis. As before, GFP, RFP, and lacZ served as negative observations in the training sets. All Lasso models converged (
Features selected by the selective Lasso models trained on DNA damage initiation and checkpoint signaling genes. a
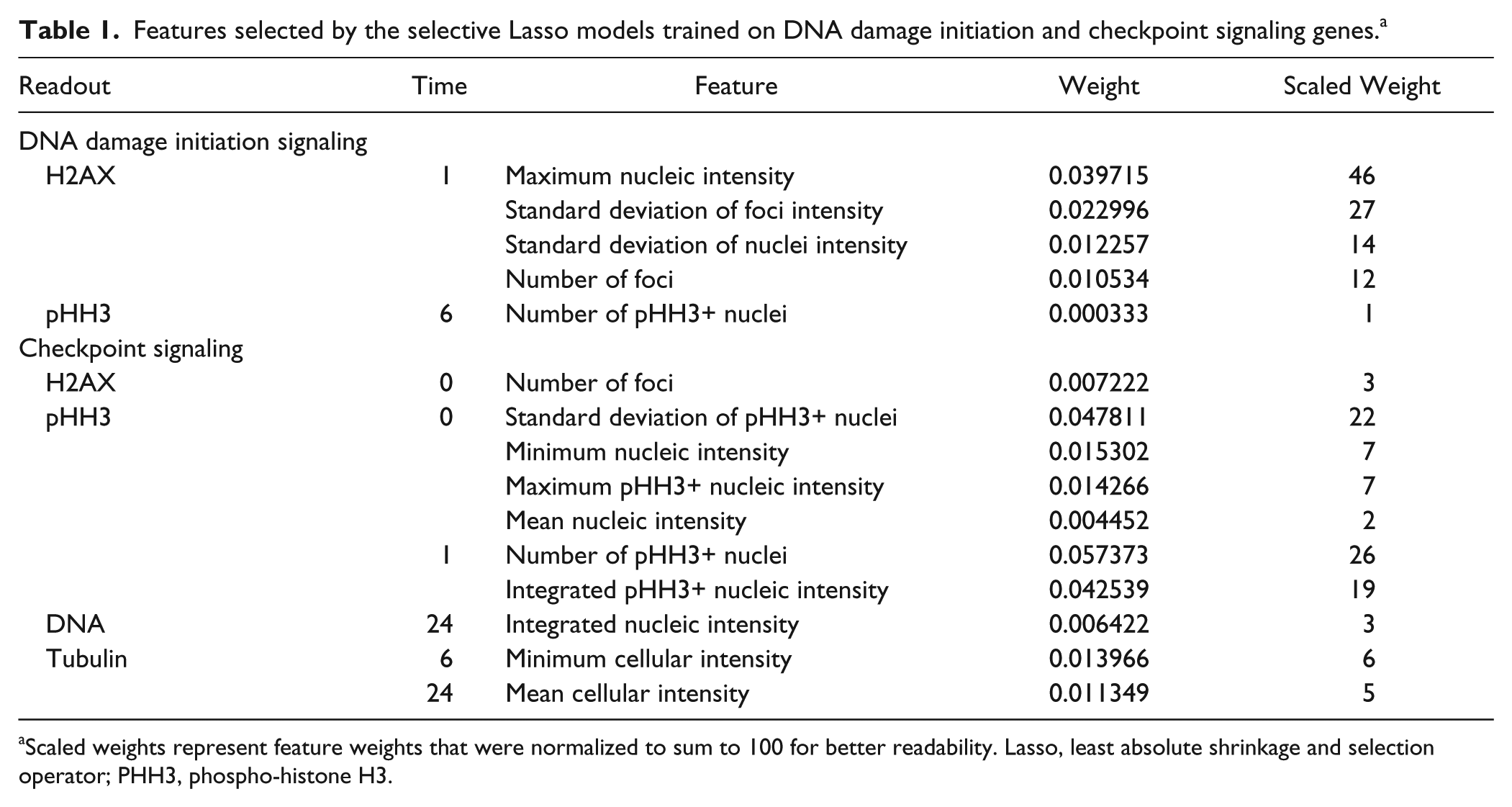
Scaled weights represent feature weights that were normalized to sum to 100 for better readability. Lasso, least absolute shrinkage and selection operator; PHH3, phospho-histone H3.
Surprisingly, only two of the five selected features were canonical features likely to be picked manually. These two features, the number of γH2AX foci 1 h after IR and the number of pHH3-positive nuclei 6 h after IR, received the lowest feature weights in the Lasso model. The three remaining features, all γH2AX features 1 h after IR (maximum nucleic intensity, standard deviation of IR foci intensity, and standard deviation of nucleic intensity), received significantly higher weights ( Table 1 ). These features either directly captured information about the statistical spread of γH2AX intensities (standard deviations) within segmented nuclei or foci or were highly sensitive to outliers and increased statistical spread (maximums). This analysis reveals that the statistical spread of intensities within each object better captured knock-down phenotypes of DNA damage initiation signaling genes than estimators for statistical location such as average γH2AX intensity.
One potential cause for the importance of statistical spread estimators over statistical location estimators is the wide variety of RNAi-induced changes on the single-cell level. The microenvironment of cells that are subject to RNAi can be a potential source of the stochasticity of differential phenotypic responses. 12 Additional contributors to this intranuclear or intrafoci variation in γH2AX intensity include varying levels of shRNA integration and expression or stochastic effects of equally expressed shRNAs on protein expression, particularly if these effects result in local alterations in chromatin structure or DNA damage repair efficiency. Indeed, image analysis on the single-cell level visually confirmed a high variability of phenotypes of single cells that were targeted by the same shRNA. 21 Imperfect knock-down and puromycin selection can also lead to multiple subpopulations of cells that exhibit more variable and convoluted phenotypic effects at the single-cell level. We therefore propose that features that capture statistical spread might be able to better quantify the resulting variability of knock-down effects and thus better identify hits in RNAi screens.
It should be noted that the entirety of phenotypic information in an HC screen can be captured on the single-cell level only by analyzing distributions of populations of individual cells in screened wells. However, as each well frequently contains large numbers of individual cells, single-cell analysis would potentially increase the computational effort required to analyze the data by multiple orders of magnitude. We consider statistical spread in combination with statistical location as a good compromise to estimate characteristics of cell population distributions in HC screens such as ours, in which the richness and amount of data render single-cell analysis impractical.
The selective Lasso model for DNA damage initiation signaling did not select γH2AX intensity features at the 6 h time point, meaning that γH2AX features did not reliably differentiate DNA damage initiation signaling genes from negative control genes at this time point. In our previous study, 15 we simply hand selected three image features (integrated γH2AX intensity, number of IR foci per nucleus, and mean IR foci area) at 1 h and 6 h after IR as a metric to rank chromatin modifier genes using quartile thresholding. This basic thresholding method selected Brd4 as top hit, as did M-RAM. However, some of the chromatin modifier genes ranked in the top and bottom quartile using these metrics were not in the top or bottom quartile of M-RAM–ranked genes. Because half of the thresholds in our previous analysis were applied to imaging features from the 6 h time point, a time point shown by M-RAM to be entirely unpredictive, the lack of perfect agreement in gene ordering between these two lists can be attributed to the ad hoc nature of our selection process for image features and time points used in our previous analysis.
To learn if a simple method such as quartile thresholding would exhibit increased performance after automatic feature selection, we dropped the 6 h time point as suggested by our model. Quartile thresholding of the three features at the 1 h time point alone led to a relative increase in sensitivity by 11.2% and a relative decrease in specificity by 0.53% as compared with thresholding at both time points. Therefore, even simple hit identification methods such as quartile thresholding may benefit from a priori feature selection.
The selective Lasso model for checkpoint signaling also produced a statistically significant readout profile with a confidence level of 95% ( Fig. 2B ). Sixty percent of the profile’s features were derived from the pHH3 readout, and two-thirds of these specifically captured pHH3 before IR, although the highest-scoring features were selected at the 1 h time point ( Table 1 ). The high prevalence of pHH3 features before IR likely reflects the importance of CHEK1 and CHEK2 in cell cycle control even in the absence of exogenous DNA damage. This finding suggests that intrinsic DNA damage in an unperturbed cell cycle in these cells is already sufficient to control cell cycle progression rates through CHEK1 and CHEK2.
Furthermore, we investigated whether images of cells treated with shRNAs targeting genes in the training set for DNA damage initiation signaling (H2AFX, ATM) and cells treated with shRNAs targeting genes in the training set for checkpoint signaling (CHEK1, CHEK2) would have similar phenotypes within their respective functionally coherent groups. Indeed, knock-down of γH2AX and ATM led to a decrease in IR-induced γH2AX foci 1 h after IR, whereas knock-down of CHEK1 and CHEK2 resulted in an increased number of mitotic cells (
The readout profiles of the control model trained on the union of DNA damage initiation and checkpoint signaling genes were not statistically significant ( Fig. 2B ). Therefore, we conclude that statistical significance of the selected feature sets depends on functional coherence of the positive observations in the training sets. This finding is important because it shows that broad computational approaches to identify complex phenotypes cannot be blindly performed using a diverse set of genes, which are important in various different parts of a biological process. Instead, only genes that function together to control a limited portion of a complex phenomenon are likely to be useful in training predictive models that capture their more well-defined phenotypes. To capture a complex biological process in its entirety, it will likely be necessary to use smaller subsets of the whole, each representing a functionally coherent subcomponent.
M-RAM Identifies DDR Modulators Missed by Univariate Methods
We used the selective Lasso model for DNA damage initiation signaling with the selected feature set (
Table 1
) to identify novel DDR modulators. Intuitively, this Lasso model ranked all screened genes based on how much their knock-down phenotype resembled the knock-down phenotypes of genes in the DNA damage initiation signaling training set (
Best-ranked hits that resemble the knock-down phenotype of DNA damage initiation signaling genes (top) or the opposite phenotype (bottom). a

MRAM, multivariate robust analysis method; 2BHM, second-best hairpin method; cAMP, cyclic adenosine monophosphate.
To establish a baseline for comparisons with M-RAM, we applied a popular method for identifying hits in HC RNAi screens, the second-best hairpin method (2BHM; see
No sound justification exists to select the second-best shRNA, and not the best, third best, or any other. Selecting one arbitrary, single shRNA makes the implicit assumption that all other shRNAs with stronger or weaker effects do not contribute useful information. A single shRNA, by definition, can be a measure of only statistical location but not statistical spread. High spread implies inconsistent knock-down effects that should decrease the confidence in an identified hit. This highly important aspect of hit identification is completely lost using 2BHM but captured by shRNA aggregation methods such as dRIGER.
To compare M-RAM’s and 2BHM’s classification performance, we performed leave-one-out cross-validation on the training set of DNA damage initiation signaling genes. The selective Lasso model outperformed 2BHM (area under the receiver-operating characteristic curve of 0.83 and 0.77, respectively; Fig. 3A ). M-RAM consistently ranked independent caffeine controls closer to the top of the hit list (where one would expect knock-downs that decrease γH2AX) than 2BHM ( Fig. 3B ). In addition, it ranked Brd4 and selected protein phosphatase 2 (PP2A) subunits 22 closer to the bottom of the list (where one would expect knock-downs that increase γH2AX) than 2BHM ( Fig. 3C ).
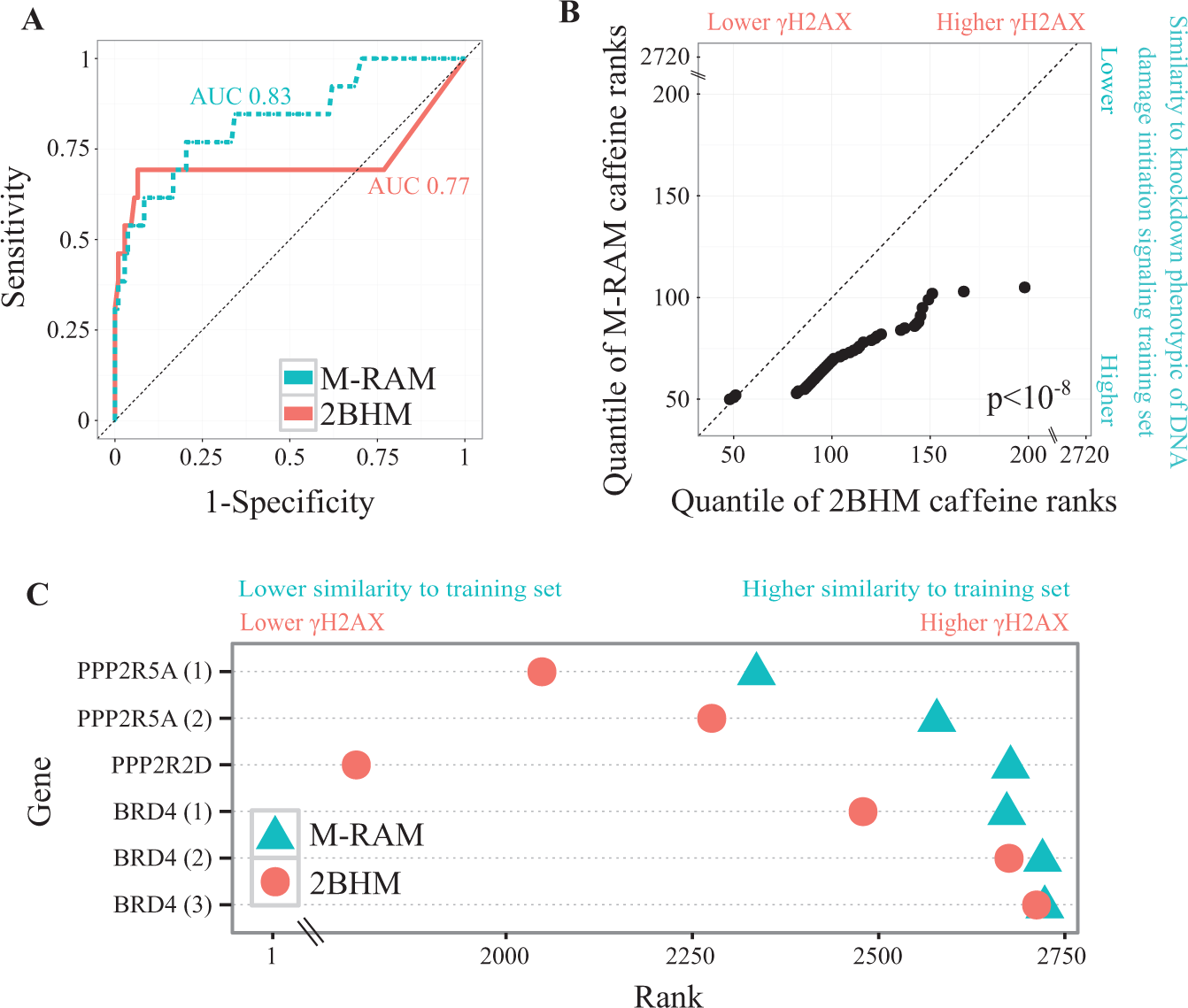
Multivariate robust analysis method (M-RAM) outperforms second-best hairpin method (2BHM). (
Lastly, we used dRIGER to rank screened genes based on their integrated γH2AX intensity 1 h after IR to investigate whether dRIGER provides potential performance gains over 2BHM, even in the absence of systematic feature selection. The top and bottom of the list of screened genes ranked by their dNES were heavily enriched in genes known to be involved in the DDR and oncogenic processes (
Network Analysis Puts Identified Hits into Context
To generate even more reliable hypotheses about how the hits previously identified by M-RAM potentially interact among themselves and with known DDR modulators, we investigated how these hits could be tied into known protein-protein interaction networks that were enriched with kinase-substrate predictions. We anticipated that the most tightly connected network structures would suggest potential mechanisms of DDR signaling. For this purpose, we employed the PCST, a network flow algorithm successfully applied in the biological domain 23 by using M-RAM to assign prize values to genes in the shRNA screen.
First, we constructed a base network from four sources: a prior knowledge network (PKN), the screened genes, the filtered STRING interactome, and Scansite
24
predictions. We defined a small, tightly connected network, the PKN, that represented well-established DNA damage initiation signaling genes20,25,26 (
Because the filtered base network was still far too complex to allow its intuitive interpretation and visualization, we employed the PCST to extract the most confident subnetwork. We rewarded high confidence in a gene with high node prizes and high confidence in an interaction with low edge cost. The PCST extracted a network consisting of the 6 genes from the PKN, 35 screened genes, and 6 genes from the filtered STRING interactome, which we visualized with a hive plot
28
(
B-Raf Is Involved in the DDR
M-RAM identified the knock-down phenotype of B-Raf in U2OS cells as similar to that of Brd4 (
Table 2
, bottom 10 hits), a chromatin-modifying protein whose knock-down we recently showed results in expanded chromatin architecture and enhanced DNA damage signaling with elevated γH2AX foci intensity.
15
Importantly, B-Raf’s knock-down phenotype was ranked significantly worse by 2BHM because no single shRNA induced an exceptional signal, although the vast majority of its shRNAs had a highly consistent, positive effect (
To independently verify the knock-down effects of B-Raf on γH2AX foci intensity, we used additional shRNA sequences against B-Raf that differed from those used in the HC screen (see
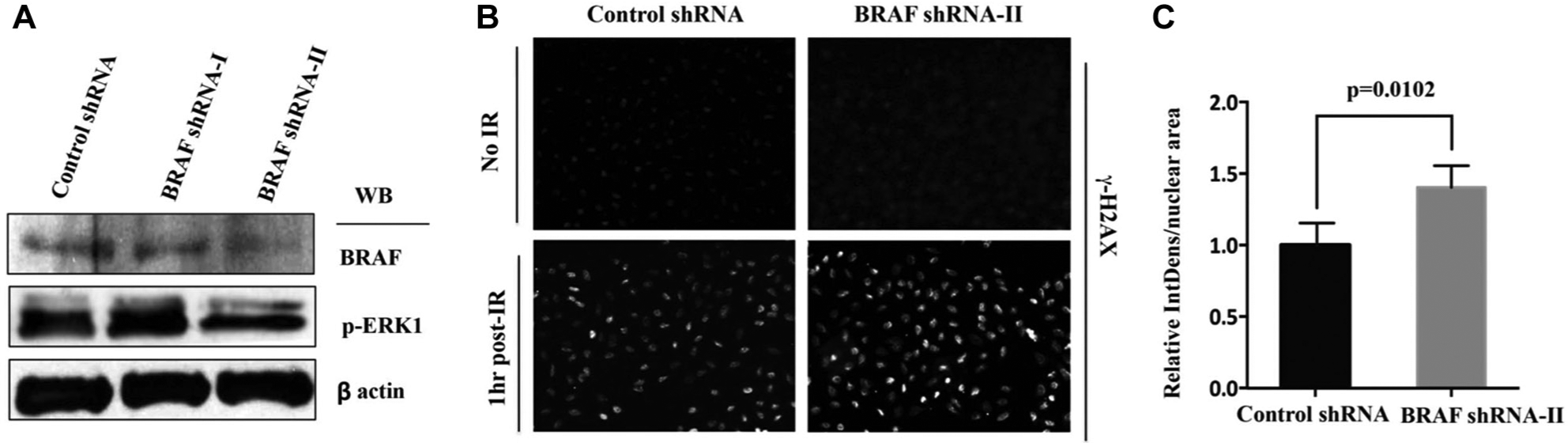
Validation of B-Raf as a modifier of the DNA damage response in U2OS cells. (
M-RAM also identified DNA damage signaling alterations following knock-down of various components of protein kinase A (PKA) complexes. These components were missed by 2BHM for the same reasons described above (
In conclusion, we conducted an image-based HC RNAi screen to identify novel regulators of the DDR. We then proceeded to develop M-RAM, a novel computational method to tap the full potential of this and similar HC screens. Employing dRIGER, an enhanced version of RIGER, we significantly reduced the dimensionality of the screening data. We transformed shRNA-level data into gene-level data, capturing consistency and variability of differential shRNA effects, and achieved a nearly sevenfold reduction in dimensionality. Lasso models selected the most predictive features at the applicable time points. In the case of DNA damage initiation signaling, the feature selection step resulted in a more than 50-fold dimensionality reduction. Functional coherence of training sets—that is, the specific selection of genes for a training set that function together within a single process within a much larger multiprocess phenomenon such as the DDR—was required to select statistically significant feature sets. The resulting selective logistic regression model generated a rank-ordered list from which hits could be selected for further analysis and verification. Canonical DDR regulators were highly clustered toward the top and bottom of this hit list. Comparison of the sensitivity and specificity of M-RAM with the 2BHM demonstrated that our method provides superior performance. In addition, our method ranked independent controls better than 2BHM. Lastly, we applied a PCST to a network consisting of our weighted hits, Scansite predictions, a PKN and the filtered STRING interactome to narrow down the hit list and generate hypotheses about how the hits might modulate the DDR. M-RAM identified both B-Raf and specific subunits of PKA as hits that were missed by 2BHM. Follow-up experiments further verified that B-Raf knock-down in U2OS cells indeed markedly increased γH2AX 1 h after IR, a finding that has potentially important clinical applications for the addition of radiation therapy in the treatment of B-Raf mutant tumors that are being concurrently treated with B-Raf inhibitors. 32
We believe that M-RAM has two important advantages over other published multivariate approaches for the analysis of HC screens. First, M-RAM elegantly combines hit identification and feature selection in one single computational step. Other multivariate approaches treat feature selection and hit identification as a multistep procedure in the HCS data analysis pipeline, complicating implementation and interpretation of results. Furthermore, M-RAM requires only the selection of the Lasso tuning parameter λ. The appropriate λ can be easily determined using cross-validation and by computing the statistical significance of the resulting readout profiles. Although we did not implement a rigid binary computational classification for hit selection, as we believe this is best done in consultation with biologists familiar with the process being studied, we do provide an explicit method for doing so using the resulting rank-ordered list to place the genes in a relevant signaling network based on preexisting knowledge with a PCST algorithm.
Second, our method provides integrated feature selection, not dimensionality reduction such as principal component analysis or factor analysis. The inherent objective of computational methods for the analysis of HC screens is to generate hypotheses for follow-up experiments from primary HC data. It is essential to reduce the number of screened phenotypic readouts and the number of time points without losing essential biological information to save experimentalists the effort of rescreening all of them. Our method efficiently selects the most predictive phenotypic readouts at the most predictive time points, therefore vastly simplifying confirmatory experiments. Hence, we believe that M-RAM will find more widespread adoption than other published multivariate approaches.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by National Institutes of Health (NIH) grants U54-CA112967, R21-NS063917, and R01-ES015339 to M.B.Y., and a pilot grant from the Center for Environmental Health Sciences NIH grant P30-ES002109. J.R. was supported by the International Fulbright Science and Technology Award, the Howard Hughes Medical Institute International Student Research Fellowship, the Hugh Hampton Young Memorial Fund Fellowship, and a David H. Koch Graduate Fellowship. Y.D. was supported by a postdoctoral fellowship from the Mazumdar-Shaw International Oncology Fellows Program at Koch Institute for Integrative Cancer Research at MIT. K.K. and T.E. were supported by Marshall Plan Scholarships.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.