
Abstract
Demethylation of histones by lysine demethylases (KDMs) plays a critical role in controlling gene transcription. Aberrant demethylation may play a causal role in diseases such as cancer. Despite the biological significance of these enzymes, there are limited assay technologies for study of KDMs and few quality chemical probes available to interrogate their biology. In this report, we demonstrate the utility of self-assembled monolayer desorption/ionization (SAMDI) mass spectrometry for the investigation of quantitative KDM enzyme kinetics and for high-throughput screening for KDM inhibitors. SAMDI can be performed in 384-well format and rapidly allows reaction components to be purified prior to injection into a mass spectrometer, without a throughput-limiting liquid chromatography step. We developed sensitive and robust assays for KDM1A (LSD1, AOF2) and KDM4C (JMJD2C, GASC1) and screened 13,824 compounds against each enzyme. Hits were rapidly triaged using a redox assay to identify compounds that interfered with the catalytic oxidation chemistry used by the KDMs for the demethylation reaction. We find that overall this high-throughput mass spectrometry platform coupled with the elimination of redox active compounds leads to a hit rate that is manageable for follow-up work.
Keywords
Introduction
The reversible methylation of specific histone lysine residues is increasingly being recognized as a key epigenetic, posttranslational modification that controls gene transcription. Lysine methylation is “written” by a family of 52 lysine methyltransferase (KMTs) that use the methyl-donating substrate S-adenosylmethionine to add one, two, or three methyl groups to the ε-nitrogen of the lysine side chain. 1 Conversely, lysine methylation is “erased” by a family of 26 lysine demethylases (KDMs) that remove these methyl groups using two distinct oxidative mechanisms. 2 The methylation status of specific histone lysine residues recruits a variety of enzymes, transcription factors, and auxiliary proteins containing one or more of the approximately 200 known methyl-lysine (KMe) “reader” modules that recognize modified or unmodified lysine. 2
The misregulation of lysine methylation is increasingly being implicated in an array of diseases and most notably in cancer. 3 The development of small-molecule inhibitors for KMTs and KDMs is critical for enabling the interrogation of their roles in oncogenesis. Uncertainty surrounding the druggability of KMTs has been addressed by the development of several examples of nanomolar and subnanomolar inhibitors for G9a, SMYD2, DOT1L, and EZH2. 3 In the case of DOT1L and EZH2, inhibitors selectively killed multiple cell lines bearing genetic alterations that create dependencies on these enzymes, and drugs against both of these target KMTs have progressed to human clinical trials. 4 Recently, the KDM1A inhibitor GSK2879552 has advanced into the clinic for the treatment of advanced myeloid leukemia and small cell lung cancer (https://clinicaltrials.gov/).
The KDMs can be separated into two subfamilies, the lysine-specific demethylase (LSD) family and the Jumonji domain–containing family (JmjC), both of which oxidize methyl groups, releasing them as formaldehyde. Only two enzymes comprise the LSD family, KDM1A (also known as LSD1, AOF2) and KDM1B (also known as LSD2). 2 Both are flavin adenine dinucleotide (FAD)–dependent enzymes that use FAD+ to oxidize mono- and dimethylated lysine to an imine intermediate that then undergoes hydrolysis. 5 KDM1A has been implicated in a number of cancers, including acute myeloid leukemia (AML) and small cell lung carcinoma. 6 JmjC is a larger family of 24 enzymes 2 that uses iron and 2-oxoglutarate to oxidize the methyl group. 7 Among the JmjC family, the demethylase KDM4C (also known as JMJD2C or GASC1) has received attention for several genetic alterations, including 9p23-24 amplifications8–11 and chromosomal translocations creating IgH-KDM4C fusions, 12 that are postulated to drive the progression of lymphoma, medullablastoma, and esophageal, breast, and lung cancers.
Despite the putative dependence of certain cancers on aberrant KDM activity, there are few quality chemical probes having the potency and selectivity necessary for testing disease hypotheses. KMD1A is inhibited by compounds bearing cyclopropylamine and propargylamine pharmacophores that form covalent adducts with the FAD+ cofactor, inactivating the enzyme. In fact, the Food and Drug Administration–approved antidepressant tranylcypromine, a known inhibitor of the FAD-dependent enzymes monoamine oxidase A (MAO-A) and monoamine oxidase B (MAO-B), has been shown to inhibit KDM1A biochemically and in cells. 13 JmjC family enzymes have been shown to be inhibited by compounds that are competitive with 2-oxoglutarate and chelate the active site iron. These compounds have low micromolar potency and only show modest selectivity over other iron-dependent demethylases and other 2-oxoglutarate and iron-dependent enzymes such as the hypoxia-inducible factor (HIF) prolyl hydroxylase family.14,15 Recent work has shown that it may be possible to achieve a reasonable potency, selectivity, and cell activity with 2-oxoglutarate–competitive iron chelators; however, additional examples are needed before this inhibition strategy can be considered applicable across the entire JmjC target class. Examination of the known inhibitors against the LSD and JmjC families reveals very limited chemical diversity and modalities of inhibition. Reversible substrate-competitive inhibitors are clearly lacking despite this being a viable approach in the inhibition of the KMTs G9a and SMYD2, 3 which share the same substrate/product ligands (albeit in reverse order) as many KDMs. The lysine-binding channels of KDM1A (pdb code 2HKO) 16 and KDM4A (pdb code 2YBS) 17 from published X-ray crystal structures reveal pockets that could potentially be druggable.
Diversity screening is a powerful method for the identification of novel inhibitory scaffolds and could be a method for the discovery of KDM inhibitors with additional modes of inhibition. However, nearly 10 years after the discovery of the KDM enzyme families, there are few examples of successful screening campaigns for this enzyme class in the literature. This may be attributed to either chemical intractability of KDMs with respect to potent and selective active site–directed ligands, compound interference with respect to the assays used to detect methylation, or the high sensitivity of KDMs to compounds interfering with the reduction/oxidation (redox) enzymatic mechanism. The latter two possibilities would populate hit lists with bad-acting compounds and render hit validation a challenging exercise.
We sought to address these concerns by selecting the FAD-dependent demethylase KDM1A and the iron- and 2-OG–dependent demethylase KDM4C and screening them against a collection of 13,824 compounds using a high-throughput self-assembled monolayer desorption/ionization (SAMDI) mass spectrometry assay. The use of mass spectrometry ensured few false positives or negatives would result from interference of signal detection (see below), and a simple assay for the detection of redox activity 18 was employed in the dose-response phase of follow-up to rapidly triage the high-throughput screening (HTS) hits, yielding a small selection of compounds worthy of follow-up studies.
Materials and Methods
Protein Purification
Bulk quantities of KDM enzymes were produced by BPS Biosciences (San Diego, CA). Human KDM1A (GenBank accession no. NM_015013) amino acids 158–end (852) were expressed in Escherichia coli and purified using an N-terminal GST tag and size exclusion chromatography. The GST tag was cleaved and the KDM1A was determined to be 93% pure by capillary electrophoresis (Agilent Bioanalyzer, Santa Clara, CA) and stored in 40 mM Tris-HCl (pH 8), 110 mM NaCl, 2.2 mM KCl, 1 mM dithiothreitol (DTT), 1 mM EDTA, 0.05% Tween-20, and 20% glycerol. Human KDM4C (GenBank accession no. BC143571) amino acids 2–372 were expressed in baculovirus-infected Sf9 cells and purified using an N-terminal GST tag and size exclusion chromatography. KDM4C was determined to be 94% pure by capillary electrophoresis (Agilent Bioanalyzer) and was stored in 40 mM Tris-HCl (pH 8.5), 110 mM NaCl, 2.2 mM KCl, and 20% glycerol. Purification details for the protein used for crystallography are further outlined in the supplementary materials section.
Reagents
The following peptides were synthesized by 21st Century Biochemicals (Marlborough, MA) and high-performance liquid chromatography (HPLC) purified to greater than 95% purity. H3K4 substrate peptides for KDM1A were based on the human histone H3 sequence, residues 1–21, with C-terminal biotin appended to a terminal lysine. All methylation states (me0, me1, me2, and me3) of H3K4 were produced. H3K9 substrate peptides for KDM4C were based on the human histone H3 sequence, residues 1–22, with C-terminal biotin appended to a terminal lysine. All methylation states (me0, me1, me2, and me3) of H3K9 were produced. Tranylcypromine, N-oxalylglycine, 4-carboxy-2,2′-bipyridine, ammonium iron(II) sulfate hexahydrate (AIS), sodium ascorbate, nicotinamide adenine dinucleotide (NAD+), FAD+, tris(2-carboxyethyl)phosphine hydrochloride (TCEP), DTT, bovine serum gelatin (BSG), and formic acid were purchased from Sigma-Aldrich (St. Louis, MO). Resazurin was purchased from Life Technologies (Carlsbad, CA). All other buffer reagents and chemicals were purchased from VWR (Radnor, PA). Polyproylene V-bottom 384-well microplates and 384-well black polystyrene microplates were purchased from Greiner Bio-One (Monroe, NC).
KDM1A Demethylase Assays
Assays were performed in a 50-µL volume in 384-well V-bottom polypropylene microplates (Greiner Bio-One) at 25 °C. Optimized 1× assay buffer was 50 mM HEPES (pH 8.0), 20 mM NaCl, 100 nM FAD+, 1 mM DTT, 0.002% Tween-20, and 0.005% BSG. For compound screening, 35 µL KDM1A158–852 (final concentration, f.c. = 0.1 nM) was added using a Multidrop Combi (Thermo Scientific, Waltham, MA) and preincubated with 1 µL compound (f.c. = 10 µM) for 30 min, and subsequently 15 µL peptide (f.c. = 700 nM) was added by the Multidrop Combi to begin the reaction. Reactions proceeded for 90 min and were stopped by the addition of 5 µL formic acid (f.c. = 0.5%) using a Multidrop Combi.
KDM4C Demethylase Assays
Assays were performed in a 50-µL volume in 384-well V-bottom polypropylene microplates (Greiner Bio-One) at 25 °C. Optimized 1× assay buffer was 50 mM HEPES (pH 8.0), 100 µM sodium ascorbate, 20 mM NaCl, 10 µM ammonium iron sulfate, 1 mM TCEP, 0.002% Tween-20, and 0.005% BSG. For compound screening, 35 µL KDM4C2–372 (final concentration, f.c. = 5 nM) was added using a Multidrop Combi and preincubated with 1 µL compound (f.c. = 10 µM) for 30 min, and subsequently a 15-µL cocktail of peptide (f.c. = 500 nM) and 2-OG (f.c. = 15 µM) was added by Multidrop Combi to begin the reaction. Reactions proceeded for 150 min and were stopped by the addition of 5 µL formic acid (f.c. = 0.5%) using a Multidrop Combi.
General SAMDI Procedure
A 2-µL sample of the stopped demethylase reactions was transferred to a SAMDI array coated with 384 biotin-neutravidin spots using a 384-channel pipet station with tip washing in water between plate transfers. The SAMDI arrays were incubated for 1 h in a humidified chamber, followed by washing of the surface with deionized ultra-filtered water (DIUF) and drying under nitrogen. A matrix of 30 mg/mL 2′,4′,6′-Trihydroxyacetophenone monohydrate (THAP) in acetone was applied by dispensing 50 nL on each spot in the array. SAMDI–mass spectrometry (MS) was performed using reflector-positive mode on an AB Sciex 5800 MALDI TOF-TOF (Matrix-assisted laser desorption/ionization time-of-flight-time-of-flight) mass spectrometer (AB Sciex, Framingham, MA). The percent conversion of substrate to product was calculated using the ratio of substrate area under curve (AUC) over the sum of substrate and product AUC peptide peaks.
Redox Assay
A cocktail of 50 mM HEPES (pH 7.5), 50 mM NaCl, 1 mM DTT, and 0.5 mM resazurin was freshly prepared, taking caution to minimize exposure of the solution to light, and then 50 µL was added to a black 384-well polystyrene microplates containing 1-µL spots of compounds using a Multidrop Combi. The reaction was incubated in the dark for 60 min at 25 °C, and then resorufin fluorescence was measured in a Spectramax M5 (Molecular Devices, Sunnyvale, CA) using an excitation of 544 nm and an emission of 590 nm. A redox active control (compound 2 from Lor et al. 18 ) was added to each plate and the percent redox activity based on the resorufin fluorescence of this compound and a DMSO control. A compound was considered redox active versus KDM1A or KDM4C if it had at least 20% redox activity at the IC50 measured in the KDM enzyme assay.
Data Analysis
Enzyme kinetics and parameters such as Km and kcat were determined using Prism software (GraphPad Software, San Diego, CA) to analyze Michaelis-Menten fits of steady-state enzyme velocities. Screening data were processed using Core Informatics (Branford, CT) and IC50 values, and Hill slopes were generated using four-parameter fits. The quality and robustness of the assay were determined by analysis of the Z′ factor 19 and performance of the 50% inhibition and IC50 control compounds.
KDM4C Crystallography
See supplemental information for X-ray crystallography methods.
Results
KDM1A Assay Development
KDM1A is widely reported in the literature to demethylate H3K4me2 and H3K4me1 but not H3K4me3.
20
This activity was recapitulated in vitro by incubating biotinylated H3K4me3 and H3K4me2 peptides with KDM1A and capturing the resultant products on a SAMDI array coated with biotin-neutravidin. Incubation of KDM1A with an H3K4me3 peptide did not produce any demethylated product. However, in the reaction that was initiated with H3K4me2, the appearance of both H3K4me1 and H3K4me0 peptides was observed (
Fig. 1A
). For the ease of assay development, we selected the H3K4me1 peptide for further optimization so that only a single demethylation step was being observed. Next, the effect of pH, ionic strength, and reducing agent was tested to identify optimal buffer conditions for KDM1A (
Fig. 1B
). In agreement with previously published observations,
5
optimal enzymatic activity was observed at pH 8, and HEPES was superior to Tris as a buffer system. KDM1A activity was tolerant of up to 25 mM NaCl, and at greater concentrations of NaCl, the activity diminished; therefore, 20 mM NaCl was chosen for the final buffer formulation. The effect of KCl was also studied and was found to have a similar effect on activity as NaCl (
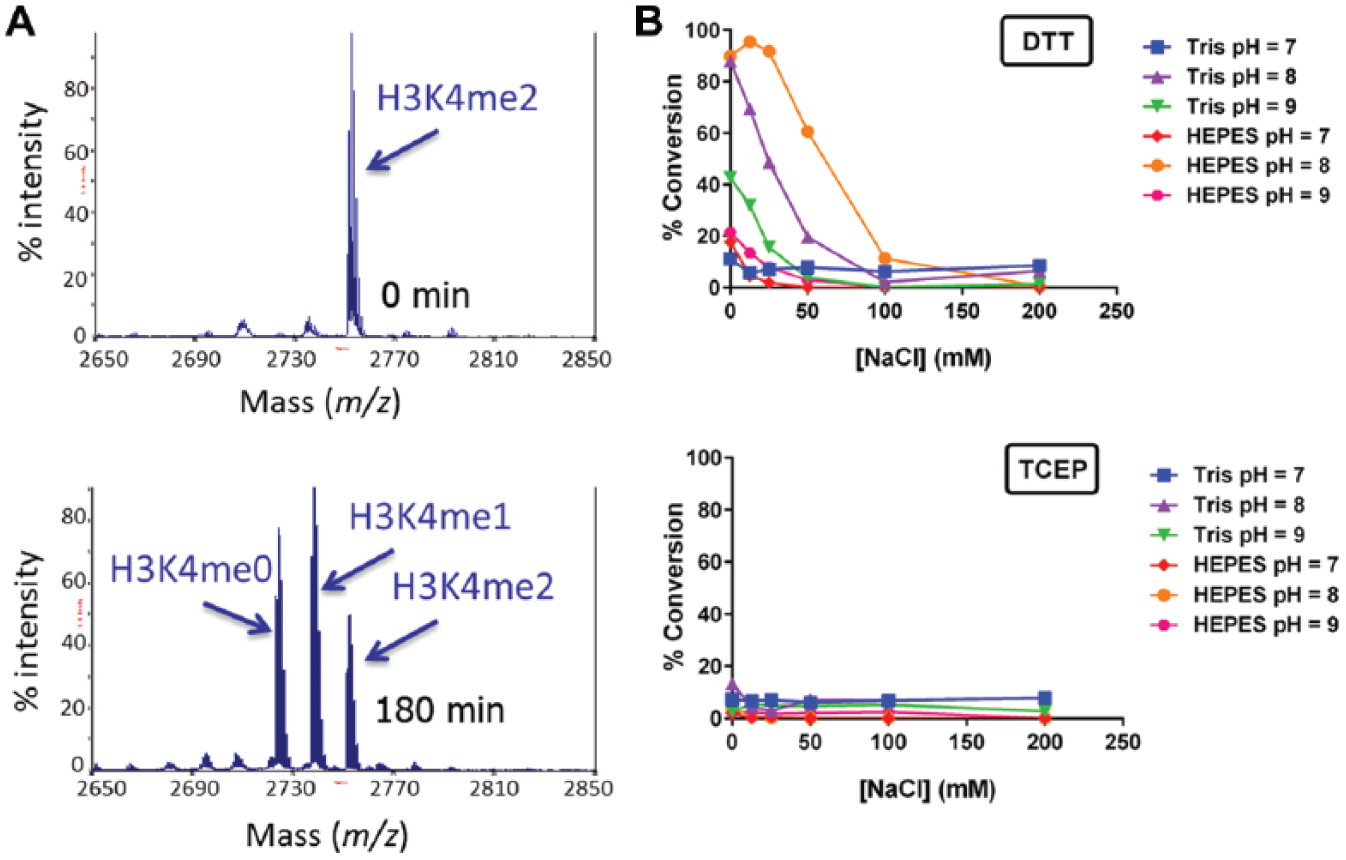
Initial activity testing of KDM1A and buffer optimization. (
With an optimal buffer selected, steady-state enzyme kinetics were subsequently investigated. A titration of H3K4me1 peptide was performed, sampling the reaction over several time points, and velocities were calculated using the linear portions of the progress curves. Michaelis-Menten fitting of the velocity data yielded the following kinetic parameters for the H3K4me1 peptide: Km = 705 ± 92 nM and kcat = 11.4 ± 0.5 min−1 ( Fig. 2A ). These data are within range of previously reported kinetic parameters (Km = 3.0 ± 0.3 µM and kcat = 3.2 ± 0.1 min−1) measured using a peroxidase-coupled assay on a nonbiotinylated peptide encompassing the same residues. 5 To develop an assay that would be sensitive to inhibitors of various modalities (e.g., peptide competitive or uncompetitive), we opted to fix the H3K4me1 peptide concentration at Km for further assay development.
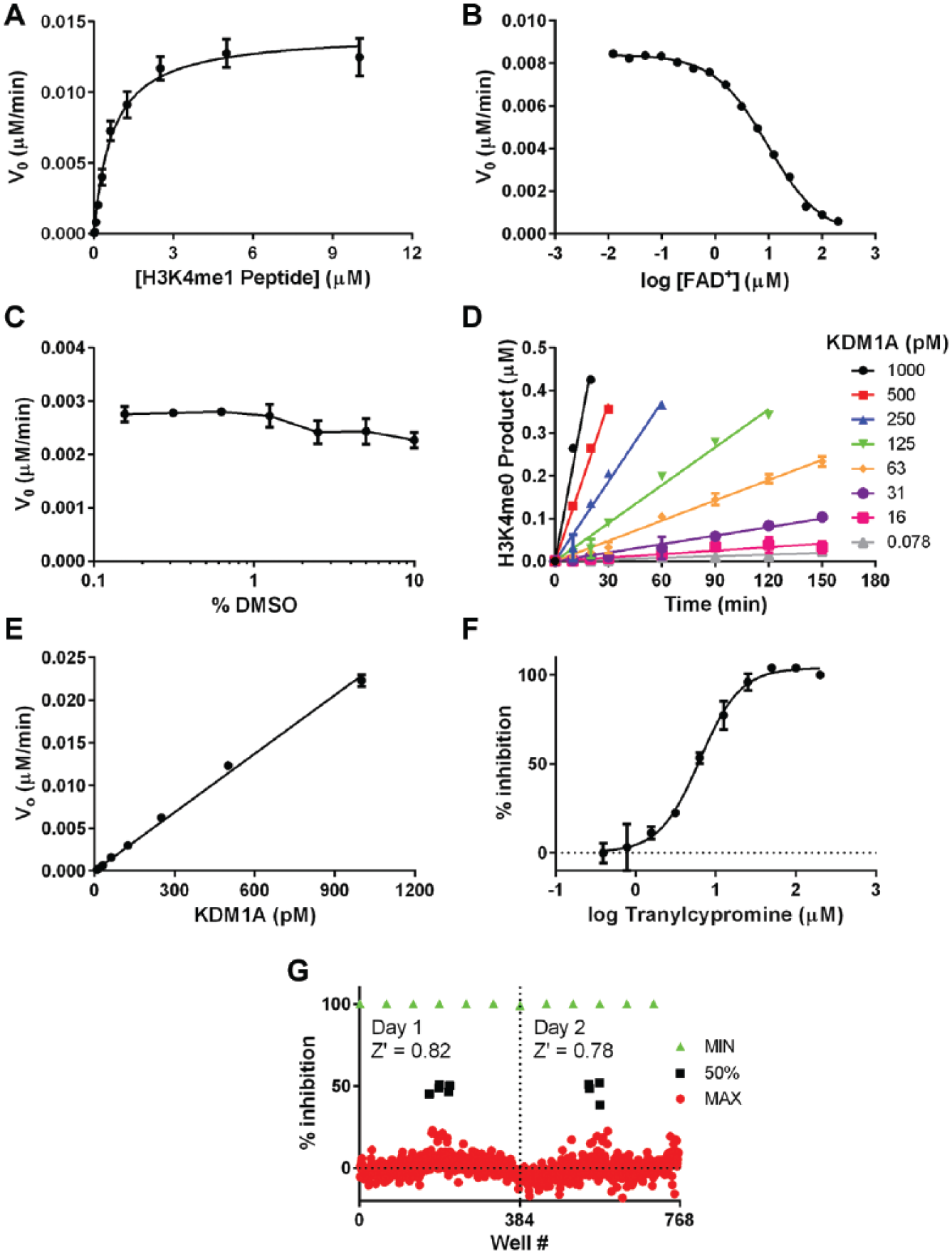
Developing a robust and sensitive assay for KDM1A. (
No additional FAD+ was required for KDM1A activity, indicating that similarly to previously reported studies,5,20 FAD+ also copurifies with our enzyme preparation. 21 To investigate this effect further, we titrated FAD+ and observed that addition of FAD+ did not substantially enhance the reaction rate but rather began to inhibit the enzyme appreciably at a concentration of >1 µM ( Fig. 2B ). In consideration of this, we included 100 nM FAD+ in the final assay. Next, we determined that KDM1A could tolerate DMSO up to at least 10% v/v with little impact on reaction velocity ( Fig. 2C ).
Finally, we investigated the linearity of the reaction as a function of time and enzyme concentration using the finalized assay conditions (optimized buffer described above with 700 nM H3K4me1 peptide substrate, 100 nM FAD+ cofactor, and 2% DMSO). Linearity of product formation versus time was lost after approximately 50% of the substrate was demethylated. The reaction velocity also increased linearly with enzyme concentration up to at least 1 nM enzyme ( Fig. 2D , E ). On the basis of these results, we selected an enzyme concentration of 100 pM and a reaction time of 90 min for compound screening to target roughly 25% conversion to product, keeping the assay well within the linear range of both the product versus time and velocity versus enzyme curves.
To test the performance of the optimized assay, the inhibition of KDM1A by the tool compound tranylcypromine was measured under the finalized assay conditions. A concentration-response curve of this compound yielded an IC50 of 7 ± 1 µM ( Fig. 2F ). While this value was reproducible under our standardized assay conditions, it should not be considered reflective of the compound’s true potency. Due to the nature of the irreversible inhibition of tranylcypromine, a kinact/Ki study should be performed to understand the true affinity of this compound for KDM1A. 22 Nevertheless, tranylcypromine can act as a reliable 50% inhibition and IC50 quality control diagnostic inhibitor for inclusion on each assay plate under standardized assay conditions, particularly when adhering to strict rules around preincubation time with enzyme and length of the assay. Last, uniformity analysis of plates containing mock compound wells and high/mid/low controls performed on consecutive days showed that the assay was robust, having an average Z′ of 0.80 ( Fig. 2G ).
KDM4C Assay Development
KDM4C is reported to demethylate H3K9me3 and H3K9me2 but not H3K9me1.
23
This activity was recapitulated in vitro by incubating biotinylated H3K9me3 and H3K9me2 peptides with KDM4C and capturing the resultant products on a SAMDI array coated with biotin-neutravidin. In the reaction with H3K9me3, we observed the formation of H3K9me2 and H3K9me1, and no H3K9me0 product was detected (
Fig. 3A
). For ease of assay development, we selected the H3K9me2 peptide for further optimization so that only one demethylation step was being observed. Next, the effect of pH, ionic strength, and reducing agent was tested to identify optimal buffer conditions for KDM4C (
Fig. 3B
). As with KDM1A, optimal enzymatic activity was observed at pH 8 and only in HEPES. It was surprising to see that no enzymatic activity was detected in Tris, considering its popular use in assays for many types of enzymes. The explanation for this unexpected result was discovered during crystallization trials. Crystals grown in the presence of Tris without addition of the 2-oxoglutarate mimic N-oxalylglcine (OGA), contained a molecule of Tris coordinated to the active site metal (
Fig. 3C
). Therefore, we surmise that Tris inhibits the enzyme by competing with the 2-oxoglutarate substrate for binding; we measured the IC50 of this inhibition and found it to be approximately 11 mM (
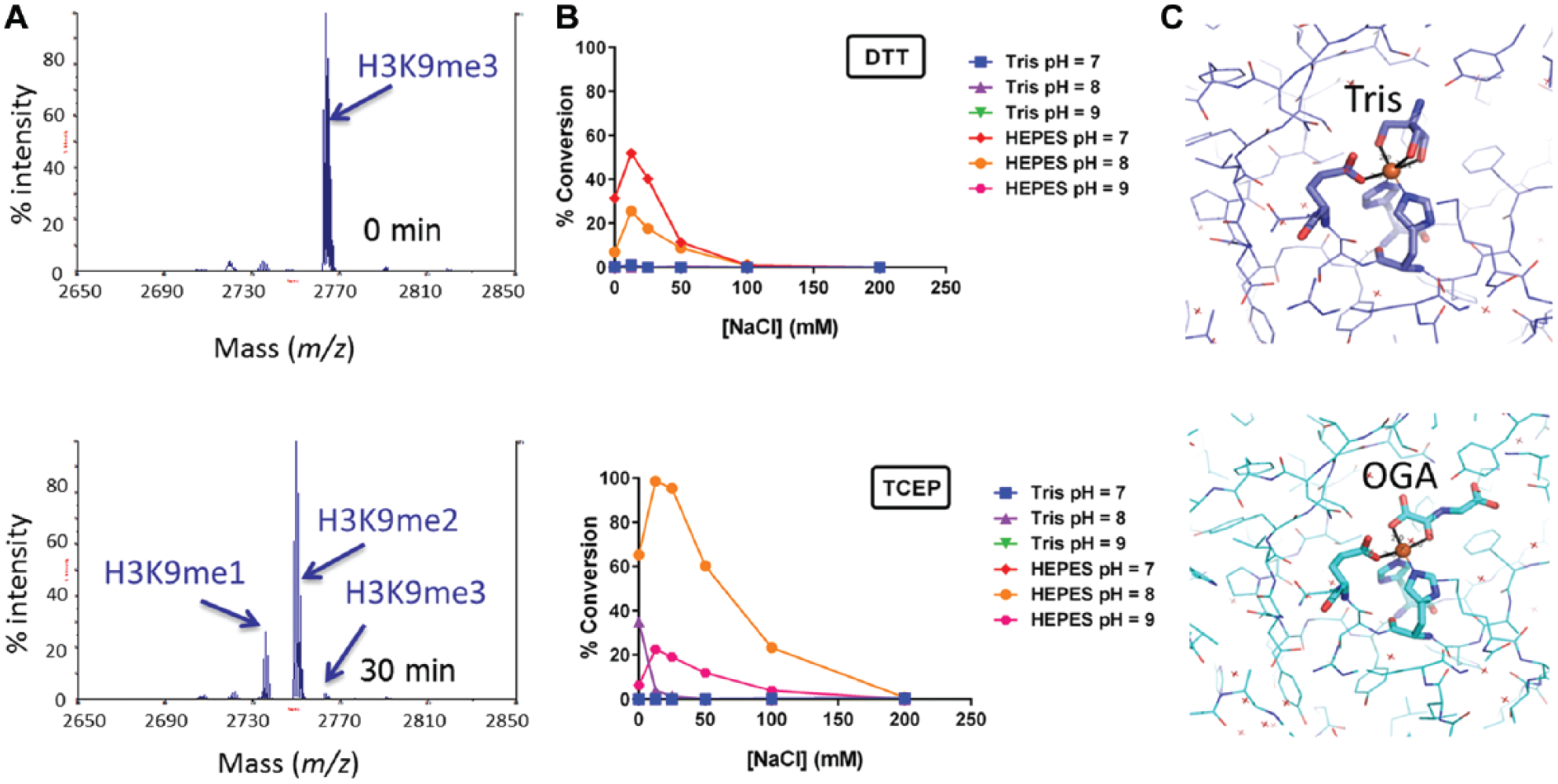
Initial activity testing of KDM4C and buffer optimization. (
With an optimal buffer selected, a double titration of H3K9me2 peptide and 2-oxoglutarate was performed to establish balanced assay conditions
24
so that the assay would be sensitive to inhibitors of various modalities (e.g., peptide or 2-oxoglutarate competitive/uncompetitive). Global fitting yielded a peptide Km of nearly 500 nM and 2-oxoglutarate Km of close to 15 µM (
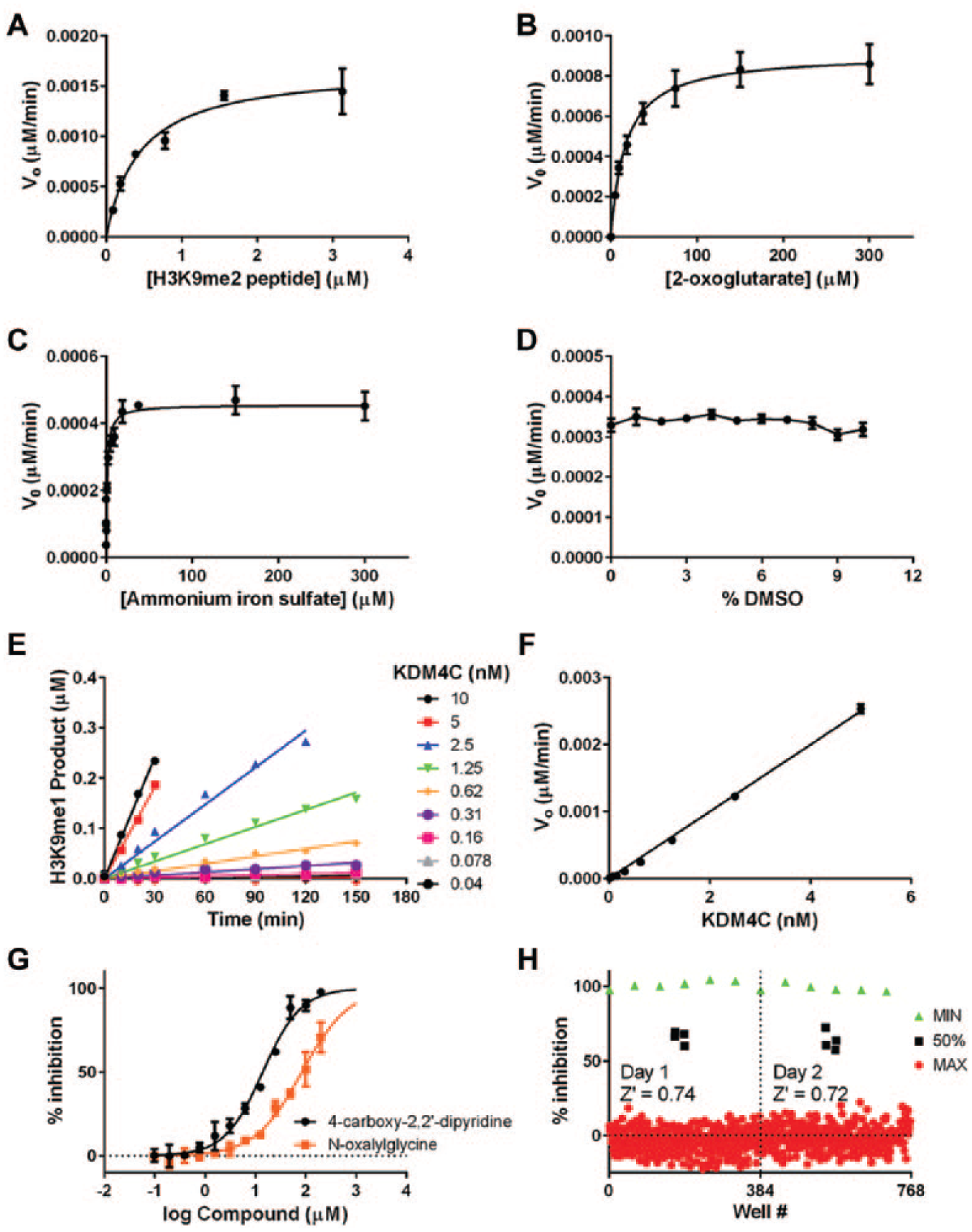
Developing a robust and sensitive assay for KDM4C. (
Finally, we investigated the linearity of the reaction as a function of time and enzyme concentration using the optimized assay conditions (optimized buffer described above, 700 nM H3K9me2 substrate, 15 µM 2-oxoglutarate, and 10 µM iron). Linearity of the product formation versus time is lost after approximately 50% of the substrate is demethylated ( Fig. 4E ), and reaction velocity increases linearly with enzyme concentration up to at least 5 nM enzyme ( Fig. 4F ). On the basis of this result, we selected an enzyme concentration of 2.5 nM and a reaction time of 90 min for compound screening to target roughly 30% conversion to product, keeping the assay well within the linear range of both the product versus time and enzyme versus velocity curves and maximizing the signal-to-background ratio.
The inhibition of KDM4C by 4-carboxy-2,2′-dipyridine (CDP) and N-oxaylglycine (NOG) was measured using the optimized assay, and concentration-response curves yielded respective IC50 values of 14 ± 2 µM and 84 ± 11 µM (
Fig. 4G
). These values were reproducible and within accepted values in the literature. CDP is known to chelate iron using bidentate coordination between the pyridine nitrogen and the metal, and to ensure this interaction was specific to the iron in the active site and not iron that was free in solution, we determined the IC50 at several iron concentrations spanning 10 to 200 µM. There was no significant impact on CDP IC50 as the iron concentration was raised (
Screening KDM4C and KDM1A against a Library of Diverse Compounds
With optimized KDM1A and KDM4C screening assays in hand, we next screened a subset of a diversity library to assess overall assay performance and ability to batch process plates. A collection of 13,824 diverse, small molecules were arrayed in duplicate within 384-well microplates and screened at a final concentration of 10 µM in batches of approximately 25 plates per day. Plates with Z′ values below 0.5 or where the specified 50% inhibition control value was outside of the accepted 30% to 70% inhibition range were rejected and rescreened. Overall, the KDM1A and KDM4C assays were quite robust, having respective average Z′ values of 0.86 ± 0.06 and 0.79 ± 0.05 (
Fig. 5A
,
B
). The overall distribution of percent inhibition values for the compounds against each is shown in
Figure 5
, and a run-based cutoff of 3 standard deviations yielded a hit rate of 1.8% for KDM1A and 1.6% for KDM4C (
Fig, 5C
,
D
). A chemistry assessment was performed on the hits, and a subset was chosen to be followed up with concentration-response studies.
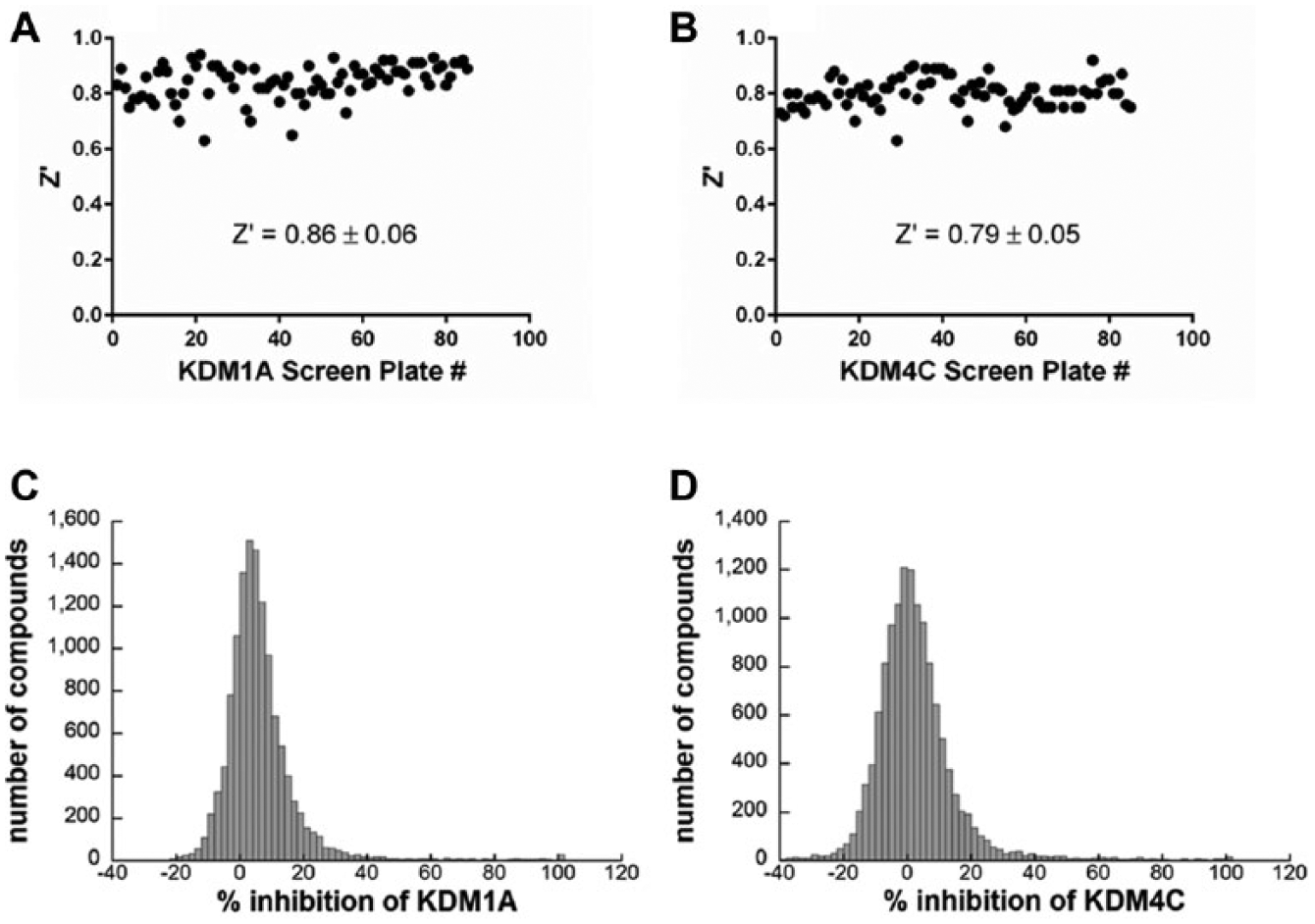
Performance of the KDM1A and KDM4C assays during a screen of 13,824 compounds. The average Z′ value of the screen of 85 plates for (
Employing an Assay for the Detection of Nonspecific Reduction/Oxidation to Triage the Screening Hits
A general concern when screening enzymes that rely on reduction/oxidation chemistry in their catalytic cycle is the hypersensitivity to inhibition by compounds that possess redox potential. Since both the LSD family and JmjC demethylases must oxidize the methyl group to initiate its release, we adapted a simple assay for the detection of redox activity 18 immediately downstream of the concentration-response phase of hit follow-up to rapidly eliminate nuisance compounds with intractable modes of inhibition. The assay monitors the conversion of resorufin to resazurin by compounds in the presence of DTT and is run in 384-well plates. Since some compounds produce atypical redox dose-response curves that are not fit by standard four-parameter fits, all redox curves were manually examined and compounds were eliminated for being redox active if they had 20% redox activity at the IC50 for enzyme inhibition. For both KDM1A and KDM4C, the majority of the most potent hits (IC50 < 1 µM) were redox active. However, as the potency of the hits decreased into the single- and double-digit micromolar range, a lower proportion of the hits was determined to be redox active ( Fig. 6 ). Overall, the number of KDM1A hits designated for follow-up was reduced from 232 to 138 compounds, and for KDM4C, it was reduced from 149 to 79 compounds, eliminating 50% to 60% of compounds from the hit validation process using a single assay to identify an intractable mode of inhibition.

Initial phase of hit triage. An assay for the detection of redox activity was performed on (
Discussion
There are several HTS-compatible assay technologies that have been reported for the study of KDMs, including enzyme-coupled detection of formaldehyde 25 and peroxide, 26 which are formed as a result of oxidation, and detection of peptide demethylation using protease-coupled microfluidic mobility shift technology, 27 time-resolved fluorescence resonance energy transfer (TR-FRET),28,29 and RapidFire chromatography coupled with mass spectrometry.30,31 The first two of the above-mentioned assay technologies rely on enzyme-linked detection of reaction products and fluorescent readouts, which can invite a high number of false positives or negatives if the compounds interfere with the enzyme coupling system or the detection of fluorescence from the assay well. TR-FRET is less prone to fluorescent artifacts, but the reliance of this technology on antibodies against methyl-lysine is a weakness given what is known about the specificity of antibodies against histone peptides. 32 Mass spectrometry is relatively artifact free, is highly quantitative, and has the advantage of allowing one to simultaneously detect all levels of methylation (me0, me1, me2, and me3) of lysine. Recent advances in the throughput of this technique make it an attractive technique for the KDM families. As opposed to other assay formats such as TR-FRET/AlphaScreen and formaldehyde detection, no counterscreens are needed to account for compounds interfering with the mass spectrometry assay technology, and chemistry assessment of the hits is not complicated with a high number of false positives.
In this study, we have developed sensitive, high-throughput KDM1A and KDM4C assays using self-assembled monolayer desorption/ionization mass spectrometry33,34 to detect the demethylation of peptide substrates for the H3K4 and H3K9 marks. SAMDI is an efficient sample preparation technique that purifies 384 samples in seconds prior to mass spectrometry. This technique bypasses the need for cumbersome chromatography steps such as those used in RapidFire MS, thereby making it faster than solid-phase extraction columns that often clog or malfunction and interrupt screening runs. The throughput of SAMDI is about 3-fold higher than that of RapidFire, taking about 20 min to analyze one 384-well plate. In SAMDI, peptide substrates are immobilized through a neutravidin-biotin interaction to a self-assembled monolayer after the enzymatic reaction occurs, then washed and subsequently released from the monolayer using laser desorption in a MALDI TOF-TOF mass spectrometer. The ratio of substrate and product is quantified to generate percent conversion data, which are less prone to assay noise and artifacts affecting the peptide.33,34 While 50-µL reactions were performed here to match a compound plating paradigm used for our methyltransferase assays, SAMDI reactions can be miniaturized further since only 2 µL of sample is needed for MS analysis.
34
SAMDI mass spectrometry provided quantitative enzyme kinetic data, leading to a greater understanding of two members of the KDM family, and enabled the establishment of screening conditions at substrate Km values. The assays were highly sensitive, with as little as picomolar to single-digit nanomolar amounts of enzyme needed for the generation of robust screening data. In addition, we have detected activity of other KDM enzymes such as KDM2B, KDM4A, KDM5A, KDM5B, and KDM6A on a variety of methylated lysine marks on several biotinylated histone peptides (
The optimized KDM1A and KDM4C assays were used to screen a collection of 13,824 compounds, yielding an initial hit rate of less than 2%, which is well below that of previously reported screening campaigns using alternative formats such as formaldehyde detection, in which the hit rate is as high as 10%. 35 To address false positives stemming from the ability of compounds to nonspecifically affect the oxidative mechanism of these enzymes, we ran a simple assay for the detection of redox activity 18 in parallel with the concentration-response curves for enzyme activity. Comparison of compound IC50 versus redox activity eliminated approximately half of the hit populations for each enzyme, reducing the effective hit rates to less than 1%.
In summary, KDM hit finding can appear to be a challenging task, and the pathway to validated inhibitors can often be overwhelmed by compounds with intractable modes of inhibition due to weaknesses of current assay technologies and susceptibility of the enzymatic mechanism to redox chemistry. By selecting mass spectrometry, which is a relatively artifact-free assay technology, and immediately counterscreening for redox activity in the hit confirmation phase, hit validation can be streamlined and made to be more efficient. Resources can be focused on a smaller and more meaningful set of compounds with a higher probability of yielding tractable inhibitors. In this example, we show that this is a viable strategy against a member of the LSD family, KDM1A, and the JmjC family, KDM4C, and we are left with a manageable set of compounds for each enzyme to be further validated by lower throughput methods such as biophysical measurements, mechanism of action studies, selectivity screening, elucidation of structure-activity relationships, and X-ray crystallography.
Footnotes
Acknowledgements
We thank Andrew Santospago and Christina Majer for assistance with compound management and integration of mass spectrometry data into the Epizyme database.
Declaration of Conflicting Interests
T. Wigle, K. Swinger, J. Campbell, E. Admirand, P. Boriack-Sjodin, K. Kuntz, R. Chesworth, M. Porter Scott, M. Moyer and R. Copeland are current or former employees and shareholders of Epizyme. M. Scholle and J. Sherrill are current employees and shareholders of SAMDI Tech.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.