
Abstract
Human lysine demethylase (KDM) enzymes (KDM1–7) constitute an emerging class of therapeutic targets, with activities that support growth and development of metastatic disease. By interacting with and co-activating the androgen receptor, the KDM4 subfamily (KDM4A–E) promotes aggressive phenotypes of prostate cancer (PCa). Knockdown of KDM4 expression or inhibition of KDM4 enzyme activity reduces the proliferation of PCa cell lines and highlights inhibition of lysine demethylation as a possible therapeutic method for PCa treatment. To address this possibility, we screened the ChemBioNet small molecule library for inhibitors of the human KDM4E isoform and identified several compounds with IC50 values in the low micromolar range. Two hits, validated as active by an orthogonal enzyme-linked immunosorbent assay, displayed moderate selectivity toward the KDM4 subfamily and exhibited antiproliferative effects in cellular models of PCa. These compounds were further characterized by their ability to maintain the transcriptionally silent histone H3 tri-methyl K9 epigenetic mark at subcytotoxic concentrations. Taken together, these efforts identify and validate a hydroxyquinoline scaffold and a novel benzimidazole pyrazolone scaffold as tractable for entry into hit-to-lead chemical optimization campaigns.
Introduction
Histone-modifying enzymes facilitate the regulation of gene expression by mediating DNA accessibility. Methylation of specific lysine residues within histone tails by methyl transferase activity is known to down-regulate gene expression via formation of transcriptionally silent heterochromatin. Conversely, demethylation by lysine demethylase (KDM) enzymes activates gene transcription by initiating a process of chromatin decondensation, which yields transcriptionally active euchromatin. To date, approximately 20 human KDM enzymes have been identified, each possessing the ability to demethylate mono-, di-, or tri-methylated lysine residues within unstructured regions of histone tails. Together, these enzymes help regulate several disparate and coordinated cellular processes that function normally to maintain homeostasis and abnormally during the development of disease.
The Jumonji domain (Jmjd)–containing KDM4 subfamily comprises five functional members, KDM4A–E. Each isoform contains N-terminal catalytic Jumonji N- and C-domains that can demethylate di- and tri-methylated lysine 9 (KDM4A–E) and lysine 36 (KDM4A–C) of histone H3 (denoted H3K9/K36me2/3). Demethylation requires as co-substrates Fe2+ and α-ketoglutarate (α-KG). Substrate turnover generates succinate, CO2, formaldehyde, and demethylated H3K9/36 as products.
Since their discovery, KDM4 enzymes have been investigated for their ability to regulate pathways that culminate in metastatic disease. KDM4A–C isoforms are implicated1–5 in aggressive phenotypes associated with metastatic prostate cancer (PCa). By associating with and co-activating2,3 the androgen receptor (AR), these isoforms initiate a process of chromatin decondensation that renders AR-regulated gene promoters accessible for transactivation. Ultimately, this process enables PCa cells to grow and divide by expressing factors that give tumor cells selective growth advantage. 3 Initial experiments have demonstrated that knockdown of KDM4C gene expression reduced growth 3 of various PCa cell lines and suggested a novel therapeutic approach for treatment of this often lethal disease.
The extended Jmjd-containing KDM (Jmjd-KDM) superfamily is now recognized to play much wider roles in cancer biology and is implicated in supporting growth phenotypes of several disparate cancer lineages (reviewed in reference 6 ). Given these broad and overlapping associations in cancer biology, KDM enzymes are increasingly found in chemical biology campaigns aimed at designing molecular probes that modulate their activity.
Several small molecules are described as inhibitors of Jmjd-KDMs (reviewed recently by McAllister et al. 7 ). Most are analogs or structural mimics of α-KG, and they inhibit the enzyme by interfering with co-substrate turnover. These inhibitors share structural determinants of binding with α-KG, including Fe2+ chelation and formation of distal active-site hydrogen bonds. 8 Of these, the most characterized include N-oxalylglycine, as well as compounds bearing 8-hydroxyquinoline, 2,4-pyridine dicarboxylic acid, and bipyridyl motifs.1,9–13 In addition, a unique compound (JIB-04) was discovered 14 that noncompetitively inhibits Jmjd-KDMs with respect to α-KG.
These initial reports have fueled considerable interest in designing selective inhibitors of the Jmjd-KDM superfamily. Here, we report our efforts to screen for inhibitors of the human KDM4 family of KDM enzymes. By means of a formaldehyde dehydrogenase (FDH) coupled-enzyme assay, we screened the ChemBioNet (CBN) library 15 for inhibitors of recombinant human KDM4E. As expected, some scaffolds were identified with motifs known to inhibit KDM4 enzymes. In addition, we identified several compound classes with unique chemistries that are as-of-yet undescribed. Inhibitory properties of the active compounds were confirmed by a novel enzyme-linked immunosorbent assay (ELISA) of KDM4E activity. Selectivity was examined by testing for inhibition against two representative enzymes from the extended superfamily of Jmjd-KDM enzymes. All members of the KDM4 subfamily were inhibited with similar potencies, highlighting the challenge of designing isoform-specific inhibitors should such a need arise. However, some selectivity was observed across the two distally related KDM enzymes of the Jmjd-KDM superfamily. Furthermore, select compounds elicited cytostatic responses in KDM4-expressing PCa cell lines, including one compound that enriched levels of the H3K9me3 epigenetic mark relative to untreated cells. Collectively, these compounds represent scaffolds with tractable features that are unexplored in published hit-to-lead campaigns.
Materials and Methods
Chemicals and Reagents
Selected compounds of the CBN library (CBN IDs: 101848, 102735, 207192, 211191, 300553, 303229, 400447, and 402050) were purchased from Molport SIA (Riga, Latvia). The KDM4E inhibitor ML324 was purchased from ActiveMotif (Carlsbad, CA). Enzymology reagents (α-KG), sodium ascorbate, Fe(NH4)2(SO4)2, NAD+, and TMB (ELISA substrate) were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies were purchased from Thermo Fisher Scientific (Waltham, MA) (rabbit pAb H3K9me3, Invitrogen #49-1008), Abcam (Cambridge, UK) (mouse mAb H3K9me3, #ab6001 and rabbit pAb histone H3, #ab1791), BioVision (Milpitas, CA) (rabbit pAb histone H4, #3624-100), or Cell Signaling Technology (Danvers, MA) (HRP-mouse anti-rabbit IgG, #7074, or HRP-rabbit anti-mouse IgG, #7076).
Plasmids, cDNA Clones, and Enzymes
cDNA clones encoding the catalytic domains of human KDM enzymes were purchased from Source Bioscience (Nottingham, UK). The catalytic domain of KDM4A (KDM4Acat) comprising residues 1-359 was cloned into the pQTEV expression vector containing an N-terminal hexahistidine tag. KDM4Bcat (1–347) was cloned into the pET-28a expression vector and expressed as an N-terminal hexahistidine variant, as previously described. 4 KDM4Dcat (1–378) was cloned into the pNH-TrxT expression vector and expressed as a fusion protein bearing an N-terminal hexahistidine plus thioredoxin tag. KDM4Ccat (1–347), KDM4Ecat (1–337), and PHF8cat (plant homeodomain finger protein 8, residues 115–483) were gifts from the Structural Genomics Consortium Oxford (Oxford, UK) and were expressed in Escherichia coli as N-terminal hexahistidine variants either from the vector pNic28-Bsa4 (KDM4C/Ecat) or from pNH-TrxT (PHF8cat). The catalytic domain of KDM2A (1–517) was cloned into pQTEV and expressed as a hexahistidine variant. FDH from Pseudomonas putida was purchased from Sigma-Aldrich.
Enzyme Purification
All KDM enzymes were purified by affinity and size exclusion chromatography as previously described (KDM2A, 16 KDM4A–E,4,9,17,18 and PHF8 19 ). With the exception of KDM4B, all constructs were processed by removal of N-terminal affinity tags prior to experimentation.
KDM Coupled-Enzyme Assay
KDM activities were measured by a fluorescence-based coupled enzyme assay that measures reduction of NAD+ to NADH by the coupling enzyme FDH.20,21 The assay used as KDM4A–E substrate a synthetic octapeptide (Biosyntan GmbH, Berlin, Germany) corresponding to residues 8–15, AR-Kme3-STGGK, of histone H3, where (Kme3) represents tri-methylated lysine 9. A second peptide used as KDM2A/PHF8 substrate corresponded to residues 32–42, ATGGV-Kme2-KPHRY of histone H3, where (Kme2) represents di-methylated lysine 36. KDM4A–E were present at 1.5 µM; all other KDM enzymes were present at 2.0 µM. Details of the assay are described in the High-Throughput Screen section. For validation studies, inhibition data were analyzed using a log[inhibitor] vs normalized response model with variable slope as implemented in GraphPad Prism version 5.01 (GraphPad Software, La Jolla, CA).
High-Throughput Screen
Screening was performed at the Screening Unit of the Leibniz-Institut für Molekulare Pharmakologie (FMP), Berlin. The CBN library was screened to identify inhibitors of KDM4E. The library comprised 32 032 small molecules that were curated based upon diverse chemical features represented in bioactive compounds of the World Drug Index. 15 During the primary screen, compounds were tested singly in a 384-well microtiter plate (MTP) format analogous to previously reported screens against KDM4E.10,21 The final assay was performed by first pipetting 10 µL of a 2-fold concentrated enzyme solution containing purified KDM4E and HEPES buffer into 368 wells of a 384-well MTP. Buffer alone was pipetted into the remaining 16 wells to serve as background controls. Compounds were then transferred (10 µM final concentration, 1% v/v final DMSO concentration) to each well, and the plate was incubated for 10 min at room temperature. Enzymatic reactions were initiated by adding 10 µL of 2-fold substrate solution containing HEPES buffer, H3K9me3 peptide, Fe(NH4)2(SO4)2, α-KG, sodium ascorbate, NAD+, and FDH. Final assay conditions were: KDM4E (1.5 µM), H3K9me3 (50 µM), Fe(NH4)2(SO4)2 (40 µM), α-KG (1 mM), sodium ascorbate (2 mM), NAD+ (1 mM), FDH (0.2 U/mL), and HEPES (20 mM, pH 7.5). Controls for DMSO-treated (1% v/v) enzyme and no-enzyme background (n = 16, each) were included on each plate for calculation of Z’-factors 22 and to measure the robustness of screening. Initial velocities were measured over a period of 10 min from the increase in fluorescence due to FDH-coupled reduction of NAD+ to NADH. Fluorescence measurements (λex = 355 nm, λem = 460 nm) were performed on a Safire 2 plate reader (Tecan Group, Männedorf, Switzerland). Resulting initial velocities were used to calculate Z-scores for each reaction and to identify hits as statistical outliers from the distribution across all plates. 22 Finally, compounds were titrated to calculate IC50 values as described in the Results section. As an initial validation of selected high-throughput screening (HTS) hits, the FDH assay was employed in a 96-well format using a FLUOstar Optima plate reader (BMG Labtech GmbH, Ortenberg, Germany).
KDM4 ELISA (CTH-ELISA)
An orthogonal immunoassay was used as a secondary test to validate KDM4 inhibitors. The assay used as substrate core histones purified from calf thymus (CTH type II-A; Sigma-Aldrich) and quantified directly the status of H3K9 tri-methylation. First, CTH was diluted into coating buffer (100 mM sodium carbonate, pH 8.0) to a final concentration of 0.02 µg/mL. Wells of a 96-well MTP were then coated with 1 µg CTH and incubated overnight at 4 °C with shaking. Wells designated as blanks were coated with buffer alone. The following day, plates were blocked for 2 h at 25 °C with blocking buffer (5% w/v) bovine serum albumin (BSA, AppliChem GmbH, Darmstadt, Germany) in phosphate-buffered saline (PBS) and washed 4 times for 5 min at 25 °C with ELISA wash buffer PBST (PBS plus 0.1% v/v Tween 20). Wells were then conditioned by washing twice for 5 min with KDM reaction buffer (20 mM HEPES, pH 7.5, plus 0.01% v/v Tween 20). Meanwhile, KDM reaction components were prepared as 2-fold concentrated solutions containing a KDM4 isoform plus additives (HEPES, Fe(NH4)2(SO4)2, sodium ascorbate, Tween 20). Concentrated enzyme mixtures were prediluted into water (100 µL 2-fold enzyme solution plus 46 µL H2O for each concentration of inhibitor tested). Inhibitor solutions dissolved in 100% DMSO were then added to enzyme mixtures (4 µL to each dilution of enzyme described above) and incubated on ice for 10 min prior to enzyme activation. Pure DMSO was added to the enzyme mixtures in parallel as a vehicle control. Enzymes were finally activated by a 1:4 dilution of a 4-fold solution containing concentrated α-KG. Final enzyme mixtures (200 µL) comprised: 100 nM KDM4 isoform in 20 mM HEPES, pH 7.5; 10 µM Fe(NH4)2(SO4)2; 1 mM sodium ascorbate; 1 mM α-KG; and 0.01% (v/v) Tween 20. When applicable, DMSO was present at 2% (v/v).
Demethylation reactions were initiated by pipetting, in triplicate, 50 µL of activated KDM4 solutions into appropriate wells of the CTH-coated MTP. Demethylation proceeded for 2 h at 37 °C. Plates were then washed 4 times for 5 min with PBST containing 1% (w/v) BSA (PBST-BSA). Levels of H3K9me3 were detected by addition of primary antibody (rabbit, anti-human pAb to H3K9me3, Invitrogen, #49-1008) diluted 1:500 into blocking buffer and incubation of the plate for 60 min at 25 °C. Plates were then washed 4 times for 5 min with PBST-BSA before adding horseradish peroxidase (HRP)–coupled secondary antibody (mouse, anti-rabbit IgG, CST, #7074). Plates were incubated for 60 min at 25 °C, washed 4 times for 5 min with PBST-BSA, and developed for 20 min at 25 °C by adding ELISA TMB substrate. Finally, reactions were quenched with 0.2 M H2SO4 before reading the absorbance at 450 nm with a FLUOstar Optima plate reader. Raw values were corrected for background absorbance from blank wells, and the percentage of H3K9me3 remaining was expressed relative to wells containing 1 µg CTH. Data were analyzed with GraphPad Prism 5.01 as described above for the FDH-coupled enzyme assay.
Cell Viability Assay
Human PCa cell lines (LnCaP, DU145, and PC-3) originated from ATCC and were maintained and propagated according to accompanied protocols. Cell viability was evaluated by the alamarBlue assay (Thermo Fisher Scientific) according to the manufacturer’s protocol. The assay measures reduction of resazurin to resorufin in healthy cells. For all cell lines tested, 10 000 healthy cells were seeded into wells of an MTP (n = 6, for each test condition) and allowed to attach overnight at 37 °C, 5% CO2. Compounds were added the following day (0.5% final DMSO (v/v)) and incubated for 48 h prior to addition of the alamarBlue reagent. Presence of resorufin was detected by its fluorescence in a Tecan Infinite F200 Pro plate reader (λex = 570 nm, λem = 590 nm). Percentage viability was calculated as the ratio of fluorescence from wells containing cells ± inhibitor relative to the fluorescence from wells containing cells grown in medium alone (healthy cell control) and was normalized relative to cell-free medium (dead cell control).
Nucleosomal ELISA (Nu-ELISA)
Global levels of methylated chromatin from cells grown in the presence of selected inhibitors or DMSO alone were measured by a Nu-ELISA, which quantifies a given epigenetic histone modification relative to a static control. 23 In our implementation, PCa cells (LnCaP, DU145, and PC-3 cell lines; 5 × 105 cells each) were seeded into wells of a 6-well plate containing 2 mL of each respective growth medium. Cells were allowed to attach overnight at 37 °C, 5% CO2. The following day, culture medium was aspirated and replaced with test media: basal (growth medium alone) or growth medium supplemented with KDM4 inhibitors (25 µM ML324, 30 µM CBN 207192, 100 µM CBN 209350, or DMSO alone). The KDM4 inhibitor ML324 was used as a cell-permeable control. 24 Cells were then grown an additional 48 h under test conditions, at which point the medium was aspirated, the cells washed twice with PBS, and the plates flash-frozen in liquid N2 and stored overnight at −80 °C. The following day, the plates were thawed at room temperature, and wells containing freshly lysed cells were resuspended in micrococcal nuclease (MNase) buffer, as previously described. 23 Crude histone extracts were processed 23 by MNase digestion (Sigma-Aldrich) and used to coat the wells of a 96-well MTP. Plates containing the immobilized nucleosomal preparations were then blocked with BSA and probed for levels of the H3K9me3 modification by addition of primary antibody (mouse, anti-human H3K9me3: Abcam #ab6001) diluted 1:100 in blocking buffer (5% (w/v) BSA in PBST). Second, identically-coated plates were probed for static levels of histone H4 (rabbit, anti-human histone H4: BioVision #3624) to serve as a loading control for normalization of H3K9me3 levels detected in the first plate. Separate, identically-coated plates were also probed to evaluate levels of histone H3 (rabbit, anti-human histone H3: Abcam #ab1791) in order to assess whether growth conditions affected global levels of this histone. After a 60 min incubation at 25 °C, plates were washed, probed with HRP-conjugated secondary antibody, and developed as described above for the KDM activity ELISA. The resulting colorimetric profiles from both plates were corrected by subtracting absorbance values from blank wells. Finally, signals arising from H3K9me3 detection were expressed as a ratio relative to signals from identical wells of the paired plate that was probed with anti-H4 IgG. The resulting H3K9me3/H4 ratios were evaluated for statistical significance relative to DMSO-treated samples using a paired Student’s t-test as implemented in GraphPad Prism 5.01.
Results
Identification of Small Molecule Hits
To initiate the HTS, we utilized a fluorescence-based and FDH-mediated coupled enzyme assay to measure inhibition of recombinant human KDM4E. The CBN small molecule library was then screened to search for novel KDM4E inhibitors. Global analysis
25
of the primary screen revealed it to be robust, with a mean Z’ factor of 0.7 ± 0.2 across all plates tested (
Fig. 1A
). The complete data set therefore possessed the statistical rigor needed to proceed with hit identification. For this, we employed a strategy that combined statistical scoring methods with a filtering scheme to exclude false positives (
Fig. 1B
). Of all the compounds tested, 22 generated non-numeric initial velocities that likely arose from auto-fluorescent compounds that saturated the fluorescence detector during data acquisition. After omitting these compounds, 32 010 remained for further analysis. From these data, Z-scores were calculated
22
and plotted as a frequency distribution across the range of plates tested. A compound was designated as an initial hit if its Z-score value was less than −3.5. Some compounds manifested as artifacts with apparent activities less than that of negative controls (samples without enzyme). To account for such potential false positives, samples with relative activity more than 20% below negative controls were omitted. Initially, 828 hits met the criteria for selection when present at a single concentration of 10 µM. Of these, the 704 most active compounds were selected to validate KDM4E inhibition in duplicate (10 µM compound concentration) and to screen for activity against the coupling enzyme FDH. Cross-correlation analysis revealed compounds that favored inhibition of KDM4E over FDH (
Fig. 1C
); 550 compounds were found to inhibit FDH by more than 50% relative to controls and were omitted from further consideration. In total, 154 compounds were identified as KDM4E-specific inhibitors. These 154 compounds were selected to calculate dose-dependent inhibition profiles against KDM4E and against FDH. Compounds were titrated in duplicate from 200 nM to 50 µM, and the resulting inhibition profiles were fit to a four-parameter sigmoidal function to identify values of IC50 with IRLS robust regression methods using the software package Pipeline Pilot (Biovia, San Diego, CA). Compounds were then ranked by IC50 and examined for potential artifacts that could lead to their classification as false positive. Several compounds exhibited responses at a high concentration indicative of assay interference due to their auto-fluorescence. We suspected that fluorescence from these compounds might contribute to the presumptive NADH fluorescence measured during data collection. To confirm this, we compared the fluorescence of each compound at 100 µM in PBS (1% (v/v) DMSO) to an equimolar solution of NADH in PBS and to 100 nM solutions of fluorescein or rhodamine B in PBS. Of the 154 hits, nearly 75% were considered to be false positives based on comparisons between fluorescence profiles (
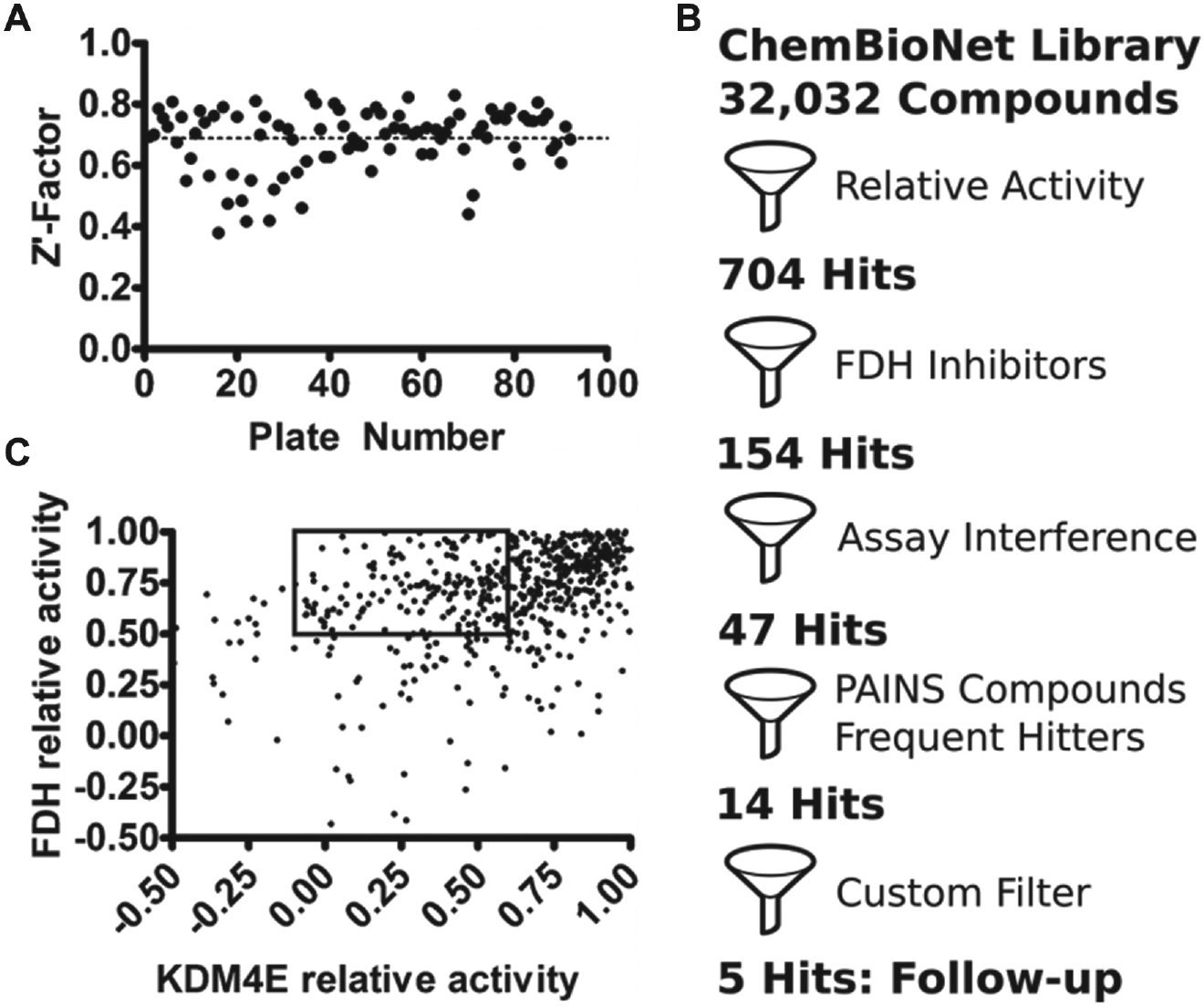
High-throughput screening (HTS) workflow.
Characterization of Selected Hit Scaffolds
Compounds designated as KDM4E inhibitors were further characterized by similarity and by known properties existing in curated databases. By clustering these hits into classes, we evaluated the chemical space selected during screening and rationalized whether the compounds merit further investigation into a hit-to-lead campaign. Because optimization campaigns require significant resources, we defined the essential properties of an ideal hit. At this stage of the screening process, we sought hits that could inhibit KDM4 enzymes in a competitive manner. We avoided compounds that could irreversibly inhibit enzymes as they might not possess the selectivity required to target KDM4 in living cells. Furthermore, we focused on compounds that were not overrepresented as hits from in-house databases nor from curated online databases. Such frequent hitters may arise due to assay interference (PAINS compounds) and could complicate downstream analyses. Finally, we focused on compounds with scaffolds that are suitable for derivatization in order to gain insight into the structure-activity relationship at an early stage.
Classification of the chemical space selected during the HTS resulted primarily in the clusters outlined in
Eight compounds were identified as singletons and could not be assigned to any group. These hits were therefore grouped together into an orphan class (
Validation of Selected Hits
After the initial assessment of HTS hit properties, we identified eight classes of compounds considered tractable for follow-up analysis (
Listed in
Table 1
are the remaining two HTS hits assessed for validation. Prior to this, we assessed each compound for purity. Analytical liquid chromatography–mass spectrometry experiments revealed compound CBN 207192 to be approximately 75% pure, while CBN 209350 was found to be nearly 100% pure (
Inhibition Profiles of Validated KDM4 Inhibitors.
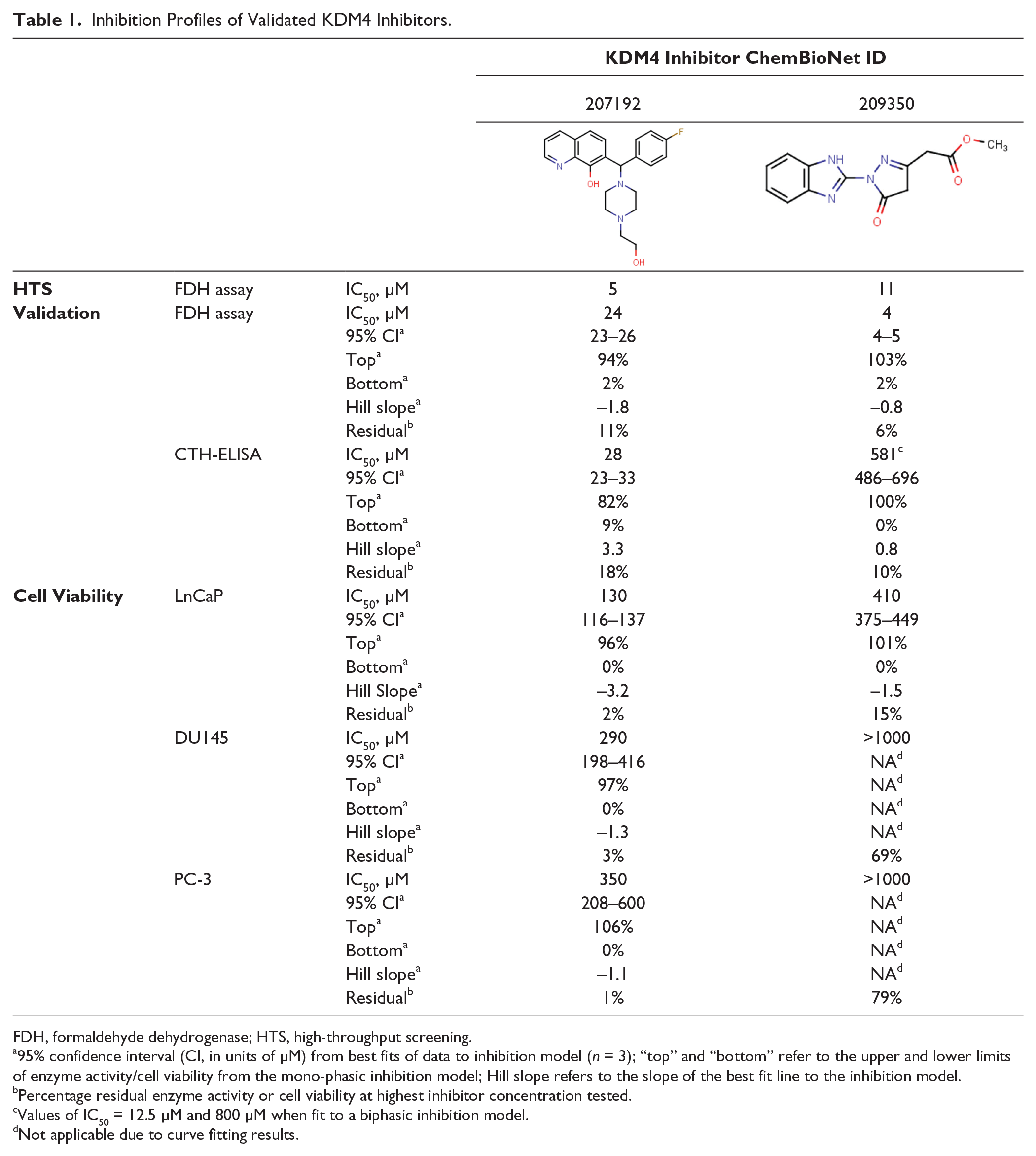
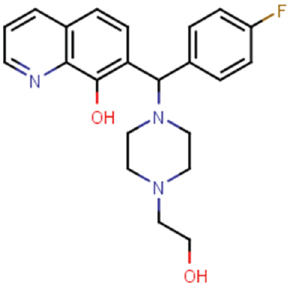
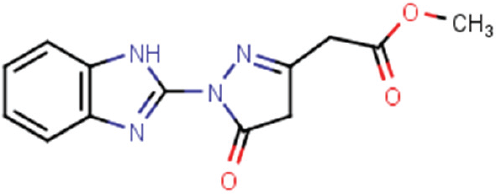
FDH, formaldehyde dehydrogenase; HTS, high-throughput screening.
95% confidence interval (CI, in units of µM) from best fits of data to inhibition model (n = 3); “top” and “bottom” refer to the upper and lower limits of enzyme activity/cell viability from the mono-phasic inhibition model; Hill slope refers to the slope of the best fit line to the inhibition model.
Percentage residual enzyme activity or cell viability at highest inhibitor concentration tested.
Values of IC50 = 12.5 µM and 800 µM when fit to a biphasic inhibition model.
Not applicable due to curve fitting results.
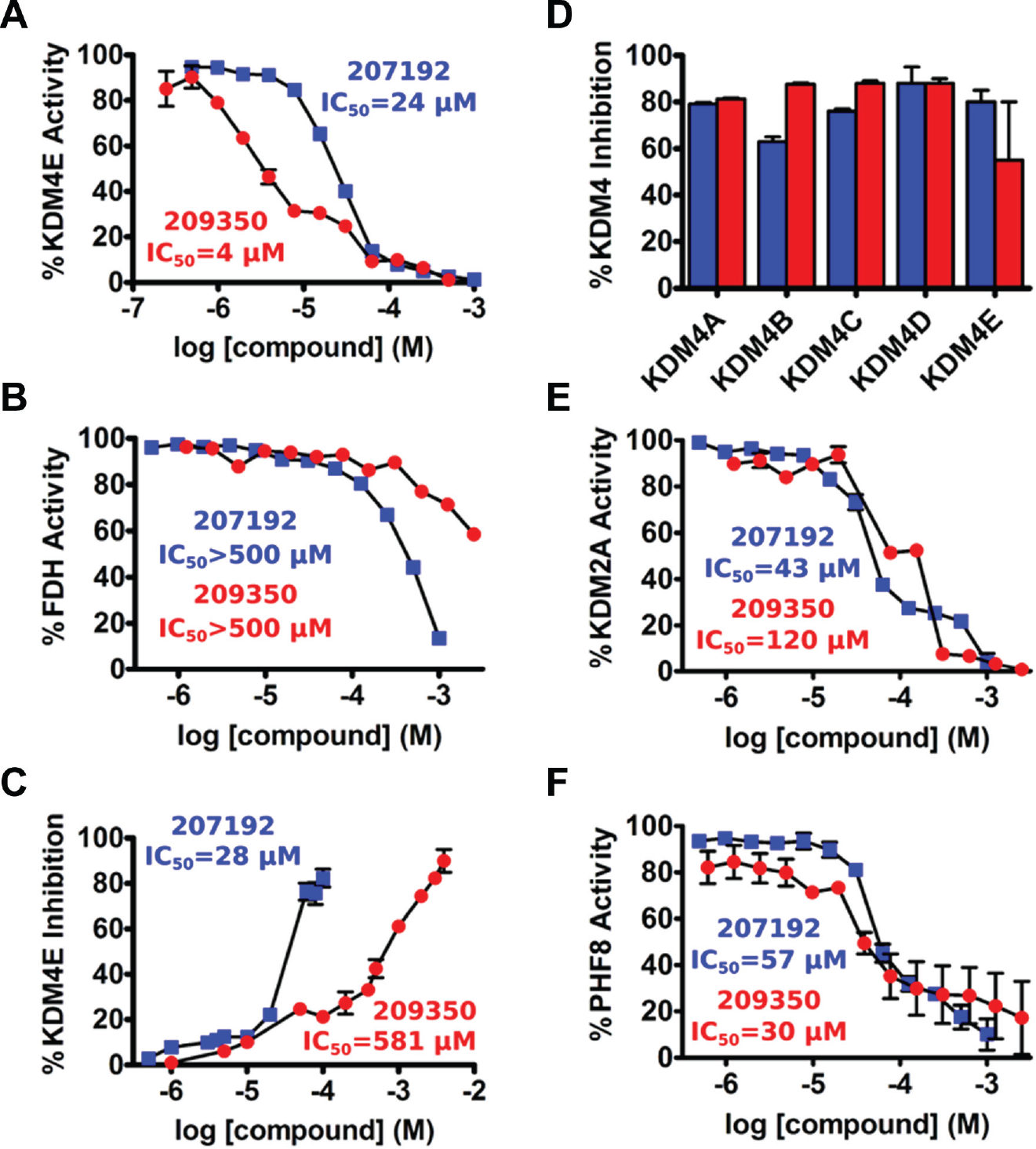
Validation of selected KDM4E inhibitors. Selected high-throughput screening primary hits were either purchased or synthesized and retested for inhibition by orthogonal assays of KDM4E activity.
We further employed an ELISA-based method as a second and independent measure of inhibition that directly measures the methylation status of a core histone substrate. Similar to the confirmatory fluorescence-based assay, both HTS hits were generally found to be active by ELISA (
Fig. 2C
;
Table 1
). Excellent agreement between IC50 values from the fluorescence assay and ELISA was obtained for compound CBN 207192, whereas 209350 exhibited an approximately 100-fold weaker IC50 value by the ELISA. However, 209350 repeatedly yielded unique biphasic inhibition curves in both the fluorescence assay and the ELISA (
KDM Selectivity
Employing the FDH-coupled fluorescence assay and the CTH-ELISA, validated compounds were tested for selectivity within members of the KDM4 family and across two distal members of the Jmjd-KDM superfamily. Values of IC50 indicate that these compounds were not selective within the KDM4 isoform subfamily (
Cell-Based Activities of Validated Hit Scaffolds
We next tested whether validated compounds were active against human PCa model cell lines, including those that overexpress KDM4A/C (LnCaP, DU145, and PC-3 cell lines).2,26,27 All cell lines exhibited cytostatic responses at high inhibitor concentrations ( Fig. 3A–C ; Table 1 ). Compound CBN 207192 was consistently the most potent compound tested, whereas 209350 generally displayed cytostatic effects at higher concentrations (>500 µM). Of the PCa cell lines tested, PC-3 cells exhibited the weakest inhibitory response. The complete CBN library has also been profiled using standard assay settings for cell viability testing to assess the cytotoxicity of each compound against two standard noncancer cell lines: HEK293 and HepG2 (unpublished data). From these data, compound CBN 207192 exhibited some toxicity against the noncancer HEK293 cell line but not against the HepG2 cell line (data not shown). Conversely, CBN 209350 was not found to be toxic to either cell line. This might reflect a generally greater cytostatic potential for CBN 207192. However, since we aim to inhibit KDM4 enzymes, we consider that differences in cytostatic properties could be due to differences in KDM4-dependent growth phenotypes, as well as the amount of KDM4 isoform expressed relative to each cell line.
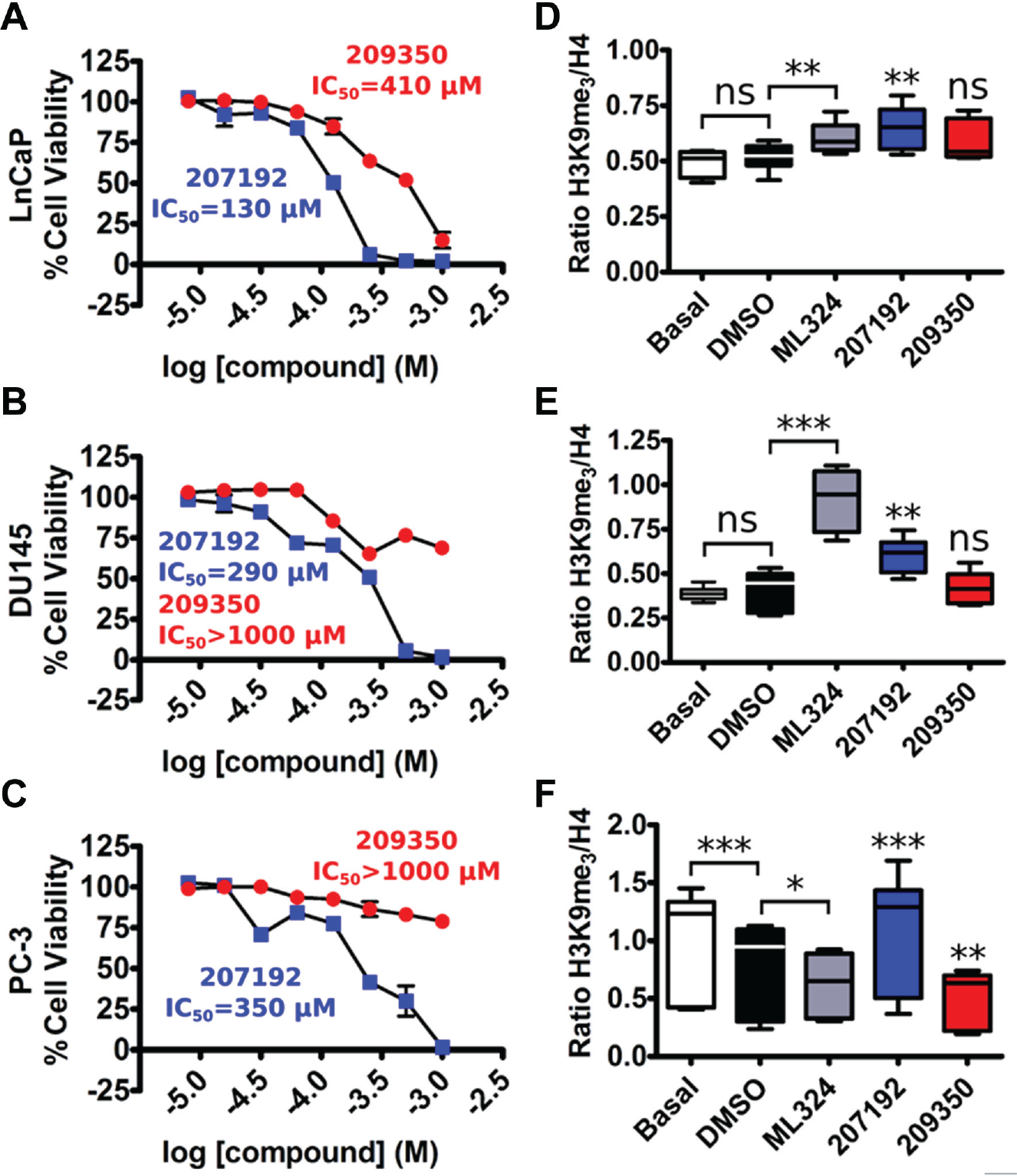
Activities of KDM4 inhibitors in cellular models of prostate cancer.
Methylation Status of Chromatin from Inhibitor-Treated Cells
Finally, we tested whether cytostatic effects observed in the cell-based assays correlated with an increase in global levels of methylated chromatin. The recently described Nu-ELISA technique enables direct quantification of dynamic epigenetic modifications relative to a static normalizing chromatin motif. 23 For this study, we compared dynamic levels of the H3K9me3 mark relative to levels of a static sequence derived from the N-terminus of histone H4. Crude preparations of chromatin from cells grown in basal medium, medium plus DMSO (vehicle control), or medium plus KDM4 inhibitors were processed into nucleosomal units by addition of the enzyme MNase. KDM4 inhibitor concentrations were chosen based on results from cell proliferation assays. We selected concentrations (30 µM CBN 207192 and 100 µM CBN 209350) that did not compromise cell viability but that approached or exceeded the values of IC50 measured by the in vitro FDH assay (24 µM and 4 µM, respectively).
As a cell-permeable control, we used the published KDM4 inhibitor ML324 (IC50 against KDM4E = 29 µM, data not shown), which has been suggested to modulate global levels of chromatin methylation by inhibiting KDM4 enzymes. 24 Methylation of H3K9 was quantified by an ELISA with a primary antibody that specifically detects the H3K9me3 modification. These values were normalized to an identical plate that was probed for static levels of histone H4. Normalized levels of H3K9me3 from cells grown in test conditions were compared relative to cells grown in basal medium plus DMSO.
Chromatin methylation levels profiled by this approach are depicted in
Figure 3
. Global levels of the H3K9me3 epigenetic mark remained unchanged in LnCaP and DU145 cell lines grown in the presence of 0.5% (v/v) DMSO relative to cells grown in basal medium alone (
Fig. 3D
,
E
). Treatment with 25 µM ML324 caused a significant increase in H3K9 methylation relative to treatment with DMSO in both LnCaP and DU145 cell lines. Similar results were observed when these cells were grown in the presence of 30 µM CBN 207192, whereas treatment with 100 µM 209350 did not alter chromatin methylation. By contrast, these same treatments yielded markedly different results in the PC-3 cell line (
Fig. 3F
). This cell line exhibited a highly significant decrease in H3K9 methylation when treated with DMSO alone. Interestingly, both ML324 and 209350 appeared to cause a further decrease in methylation in the PC-3 cell lines compared to DMSO. However, similar to LnCaP and DU145 cell lines, treatment of PC-3 cells with 207192 caused a highly significant increase in H3K9 methylation compared to DMSO. This increase was also significant relative to H3K9me3 levels of PC-3 cells grown in basal medium (p = 0.0195). In parallel, we tested whether fluctuations in global levels of histone H3 accounted for the changes observed in epigenetic levels of H3K9me3 (
Discussion
The discovery that KDM enzymes promote cancer cell growth has prompted several campaigns focusing on inhibitor development and the therapeutic potential that lies therein. Here we describe the identification of two scaffolds that inhibit members of the Jmjd-KDM enzyme superfamily. We validated our HTS campaign as successful by identifying CBN 207192, which bears a scaffold known to inhibit KDM4 isoforms. We also provide initial evidence for selectivity of the benzimidazole pyrazolone scaffold for the KDM4 family of isoforms and demonstrate that both HTS hits exhibit anti-cancer effects by reducing the proliferation of PCa cell lines. These results highlight the promise of compounds 207192 and 209350 as interesting candidates for further investigation by structural and biochemical methods and possibly for immediate entry into hit-to-lead campaigns.
Compound CBN 207192 contains the well-characterized 8-hydroxyquinoline (8-HQ) motif known to chelate iron in the KDM4A active site. Crystal structures of this fragment bound to KDM4A (PDB ID 3NJY, 4BIS) revealed how the motif chelates active site iron.10,11 The substituted ring position observed in 207192 opposes that reported from other 8-HQ inhibitors, revealing a relatively unexplored region for further modification. It most closely resembles a piperazine-substituted 8-HQ inhibitor developed by Schofield and colleagues (PDB ID 3RVH). These similarities suggest that 207192 might chelate the metal center within the KDM4 active site if the hydroxyquinoline ring flips its orientation relative to that observed in the crystal structures noted above ( Fig. 4A , B ). Furthermore, if 207192 chelates iron in this orientation, its piperazine ring might still contact the enzyme as observed in PDB 3RVH.
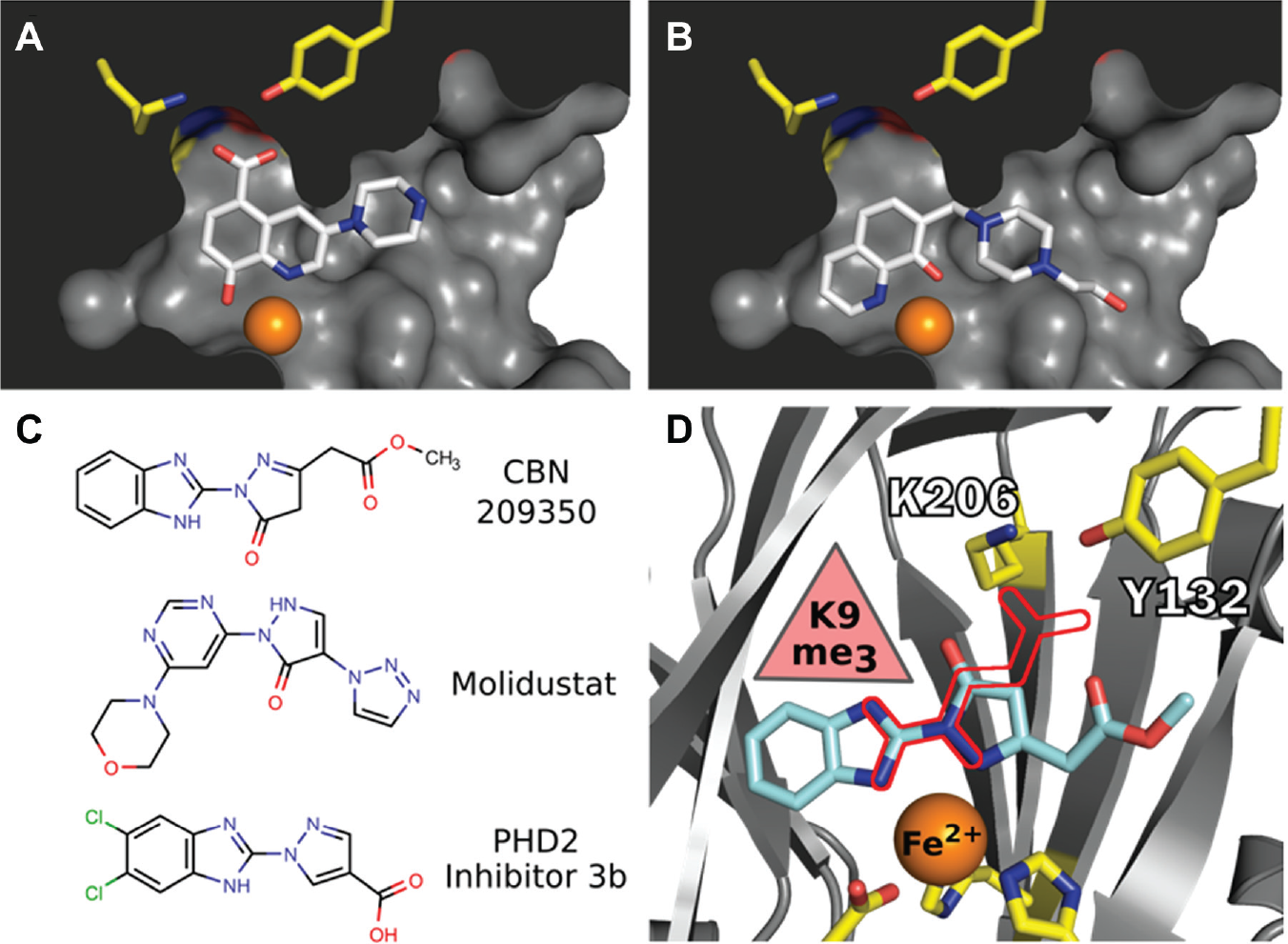
Molecular models of KDM4 inhibitors.
We conclude that the novel KDM4 inhibitor CBN 209350 exhibits features most intriguing for further investigation. First, the compound represents a scaffold with properties that are as-of-yet undescribed in KDM inhibitors. Second, the compound is active in orthogonal enzyme assays and exhibits some specificity toward KDM4 enzymes. Third, it displays cytostatic activity in PCa cell lines at a higher concentration. Fourth, it bears key structural similarities ( Fig. 4C ) to other privileged scaffolds that inhibit Fe2+/α-KG-dependent hypoxia inducible factor prolyl hydroxylase (PHD) enzymes such as the drugs Molidustat 28 and a benzimidazole-2-pyrazole carboxylate described by Rosen and colleagues 29 . Finally, we suggest that its mode of binding ( Fig. 4D ) reflects those observed in crystal structures of a KDM2A-specific inhibitor (triazolopyridine) bound to KDM4A (PDB ID 4URA), of a pyrazolopyridine inhibitor bound to KDM4A (PDB ID 4GD4), or of a diazole inhibitor bound to KDM5B (PDB ID 5FPL). 30 A secondary binding mode observed in a crystal structure containing a benzimidazole fragment bound to the surface of KDM4D (PDB ID 4D6S) may also apply to 209350.
These most tractable scaffolds are active across orthogonal assays of KDM activity. Our use of the CTH-ELISA is advantageous in its simplicity and requires no specialized equipment. Materials used in the assay are familiar to most laboratories, and several measurements of IC50 values are easily performed in a single day. A possible limitation of the assay is the need to work with well-characterized antibodies. We found that results varied with the commercial source of antibody used, indicating an essential need to characterize the antibody prior to use in such experiments. Despite this drawback, we obtained excellent agreement between IC50 values for compound CBN 207192.
Compound CBN 209350 was unique in that the IC50 calculated from the CTH-ELISA was a hundred times larger than that observed from the fluorescence assay. We are still investigating the source of this discrepancy, but it may result from what we identified as a bimodal response in the inhibition profiles. When these data from both assays are fit to a bimodal inhibition model, we observe two values for IC50: a high-affinity value of around 12 µM and a low-affinity value of around 800 µM. It is possible that fluorescence-based assays are more sensitive to measurements of the higher-affinity binding site where artifacts due to ligand fluorescence are less substantial.
Antiproliferative properties of the compounds in the KDM4-expressing PCa cell lines generally agreed with the KDM4E inhibition profiles generated by CTH-ELISA. Compound CBN 207192 was most cytostatic toward PCa cells, followed by 209350. We expected that compounds exerting a cytostatic response in PCa cells mediate their effects at least in part through inhibition of intracellular KDM4 enzymes. The resulting Nu-ELISA profiles from cell lines treated with the cell-permeable KDM4 inhibitor ML324 and our HTS hit CBN 207192 provide some evidence to support this hypothesis. Chromatin methylation profiles from LnCaP and DU145 cells were similar; both exhibited significant increases in the H3K9me3 mark when treated with either compound. Conversely, we were unable to detect significant changes in methylation when these cells were treated with 209350, possibly reflecting hindered passage across the cell membrane. Calculation of physicochemical properties by the SwissADME server (www.swissadme.ch) reveals (
We now aim to evaluate the potential of these selected scaffolds in optimization campaigns. In particular, we will evaluate structure-activity relationships around the benzimidazole pyrazolone scaffold of CBN 209350, to characterize it more thoroughly by enzyme kinetics, to assess its selectivity across a larger panel of Jmjd-KDM enzymes, and to elucidate its mode of binding by determining crystal structures of KDM4-inhibitor complexes. We anticipate that such structural details will help clarify our observation of biphasic inhibition curves. In addition, whether this and derivatives of the same chemotype inhibit cell growth by targeting KDM4 enzymes within the cellular milieu will be further investigated. Use of quantitative real-time PCR, reporter gene assays, and chromatin methylation profiling technologies will be of considerable value with such investigations. Ultimately, we believe that these efforts will generate useful tool compounds for probing epigenetic modifications in cell biology and that they will contribute to new therapeutic approaches to treat malignancies such as PCa.
Footnotes
Acknowledgements
We would like to recognize the talented and valuable contributions of M. Schneider and V. Schwietzke for help in cloning and purifying various KDM enzymes. We would also like to thank G. Illing for influential contributions and support during the conceptual phase of the project.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by an MDC GO-Bio grant awarded to DMC.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.