
Abstract
A variety of covalent modifications of RNA have been identified and demonstrated to affect RNA processing, stability, and translation. Methylation of adenosine at the N6 position (m6A) in messenger RNA (mRNA) is currently the most well-studied RNA modification and is catalyzed by the RNA methyltransferase complex METTL3/METTL14. Once generated, m6A can modulate mRNA splicing, export, localization, degradation, and translation. Although potent and selective inhibitors exist for several members of the Type I S-adenosylmethionine (SAM)-dependent methyltransferase family, no inhibitors have been reported for METTL3/METTL14 to date. To facilitate drug discovery efforts, a sensitive and robust mass spectrometry–based assay for METTL3/METTL14 using self-assembled monolayer desorption/ionization (SAMDI) technology has been developed. The assay uses an 11-nucleotide single-stranded RNA compared to a previously reported 27-nucleotide substrate. IC50 values of mechanism-based inhibitors S-adenosylhomocysteine (SAH) and sinefungin (SFG) are comparable between the SAMDI and radiometric assays that use the same substrate. This work demonstrates that SAMDI technology is amenable to RNA substrates and can be used for high-throughput screening and compound characterization for RNA-modifying enzymes.
Keywords
Introduction
The translation of messenger RNA (mRNA) into proteins is a complex and gated mechanism involving posttranscriptional modifications of specific nucleosides among multiple RNA species. These modifications regulate nuclear export, splicing, translation, stability, and degradation with profound impact on RNA metabolism and function (reviewed in Refs. 1–3). These control mechanisms are referred to as epitranscriptomics,4,5 RNA epigenetics, or simply RNA modification. One of the most prevalent modifications within mRNA is the methylation of the adenine base at the N6 position, resulting in N
6
-methyladenosine (m6A).5,6 The majority of m6A modifications in mRNA are found within a five-residue sequence, with an invariant adenosine and with cytosine and nucleotide variations occurring at the −2, −1, and +2 positions, most commonly 5′-(A/G/U)(G/A)
m6A has emerged as a key mRNA modification that is implicated in hematological cancers and solid tumor pathogenesis. 10 The m6A mark is involved in several aspects of mRNA stability and turnover, and it controls translation of oncogenes that confer growth advantage and migratory behavior.11–13 Knockout of METTL3 and/or METTL14 promotes acute myelogenous leukemia (AML) differentiation and apoptosis, and have been shown to be important for both in vitro and in vivo growth for AML.11–13 In addition, m6A has been shown to be important for differentiation of multiple T-cell lineages, including Tregs 14 and CD4+ cells, 15 which are involved in immunosuppression within tumor microenvironments. 16 Due to the increasing validation of m6A as important in several aspects of cancer biology, there is significant interest in METTL3/METTL14 as a target for drug discovery.
Although both METTL3 and METTL14 belong to the Type I methyltransferase structural superfamily, which uses S-adenosylmethionine (SAM) as a substrate for methyl transfer, structural, biochemical, and biophysical studies demonstrate clearly that METTL3 is the only catalytically active methyltransferase within the dimer.17–19 METTL14, however, is important for m6A production, because the heterodimer is significantly more active than METTL3 alone.17,18,20,21 In addition to stabilizing METTL3, mutagenesis studies and modeling indicate that single-stranded RNA substrates bind to a positively charged groove located at the interface of the two proteins.17–19 Numerous studies have shown that the METTL3/METTL14 dimeric complex is sufficient for enzymatic catalysis in vitro.17–21
To date, no small-molecule inhibitors of any human RNA methyltransferases have been reported except for the universal methyltransferase inhibitors S-adenosylhomocysteine (SAH), a product of the reaction, and sinefungin (SFG), a natural-product SAM analog. 3 Inhibitors of the distantly related RNA methyltransferase from dengue virus have been reported.22,23 In addition, chemical matter for several Type I protein methyltransferase inhibitors has been identified,24–26 and compounds have advanced into clinical trials for three Type I protein methyltransferases (DOT1L, PRMT5, and PRMT1; www.clinicaltrials.gov). Finally, nucleoside analogs for Type I DNA methyltransferases are approved for clinical use. 24 These data in aggregate support the premise that the human RNA methyltransferases are likely to be druggable enzymes. 27 Hence, the combination of biological validation of the METTL3/METTL14 complex as a driver for human disease and the apparent druggability of the enzyme superfamily warrants drug discovery efforts on this target.
Large-scale diversity screening requires a robust, high-quality, and high-throughput assay to be developed. Although an assay using tritiated SAM has been published for METTL3/METTL14, 21 a non-radioactive assay would be desirable for screening efforts. The mass spectrometry–based approach, termed self-assembled monolayer desorption/ionization (SAMDI),28,29 has previously been reported as a label-free method of high-throughput screening for a variety of targets 30 and has been used for characterization of protein methyltransferases.31–34 Here, we report the development of METTL3/METTL14 assays using both radiometric and SAMDI technologies ( Fig. 1 ). This is the first report of a SAMDI assay for an RNA methyltransferase and the first application of SAMDI technology to an RNA substrate.
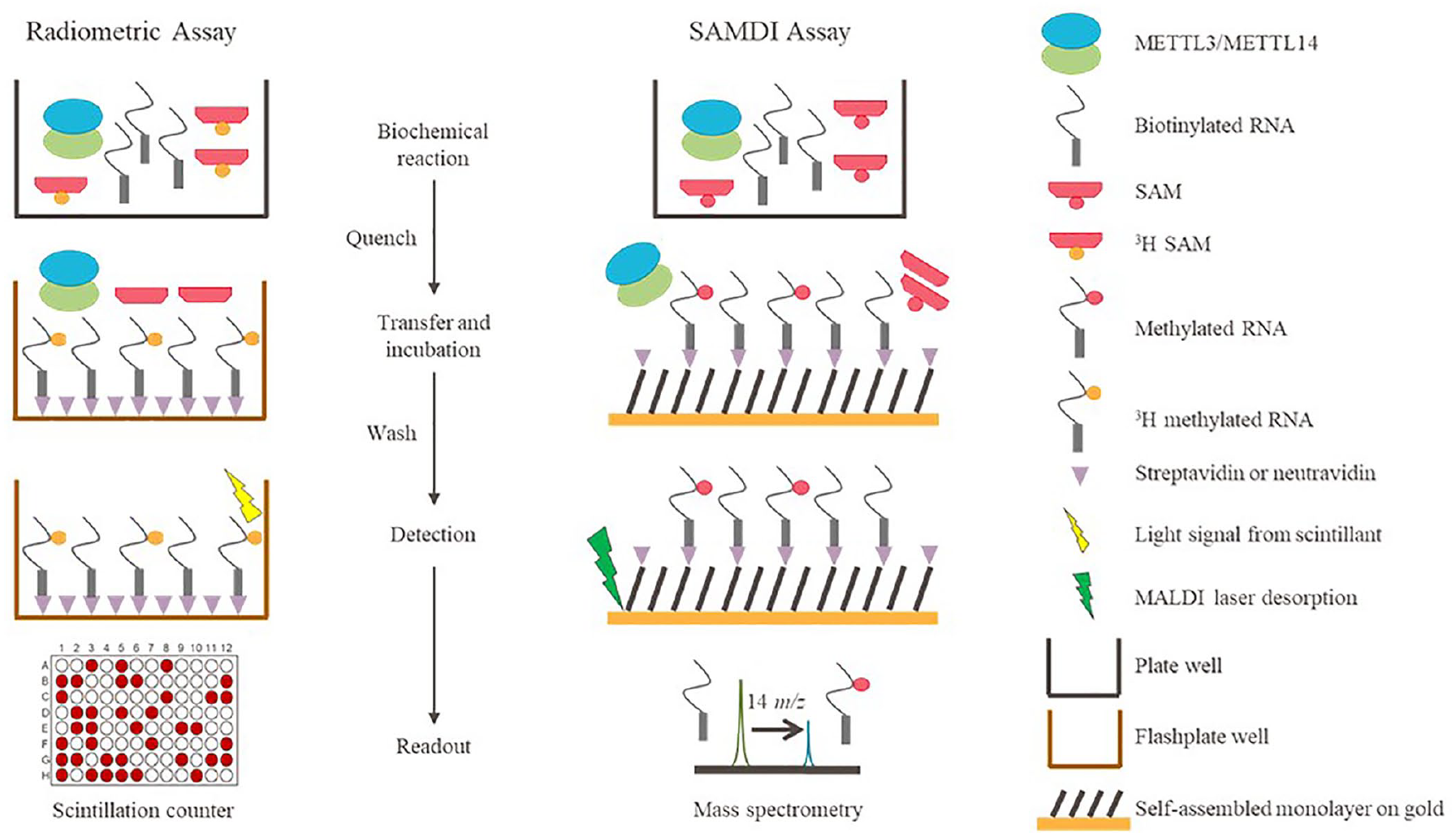
Schematic of the radiometric and self-assembled monolayer desorption/ionization (SAMDI) assay workflows.
Materials and Methods
Reagents
Biotinylated RNA oligomers were synthesized by Integrated DNA Technologies (Coralville, IA) and purified by high-performance liquid chromatography (HPLC) to greater than 95% purity. SAH, SFG, and SAM were purchased from Cayman Chemicals (Ann Arbor, MI). Tritiated SAM ( 3 H-SAM) was purchased from American Radiolabeled Chemicals (St. Louis, MO). 384-well streptavidin-coated flashplates (cat. no. SMP410A001PK) were purchased from PerkinElmer (Waltham, MA). All other reagents were purchased from Thermo Fisher Scientific (Waltham, MA) or MilliporeSigma (Burlington, MA).
Protein Production
Constructs for full-length, wild-type human METTL3 (UniProtKB accession no. Q86U44-1) and full-length human METTL14 (UniProtKB accession no. Q9HCE5-1) containing an N-terminal 6-His affinity tag and TEV protease cleavage site (His-TEV-METTL14) were each cloned into the pFastBac1 baculovirus expression plasmid (Thermo Fisher Scientific). Viral amplification was performed using standard procedures. For protein production, P2 virus was used to infect a Spodoptera frugiperdo (Sf9) cell culture using a ratio of 1:100 (METTL3) and 1:200 (METTL14) to maximize formation of the METTL3/His-TEV-METTL14 complex. Cells were harvested after 72 h by centrifugation. Protein was purified by resuspending the cells in buffer containing 20 mM Tris-HCl, pH 8.0, 300 mM NaCl with cOmplete Protease Inhibitor Cocktail (MilliporeSigma), and lysed using sonication. After centrifugation to remove cell debris, the METTL3/His-TEV-METTL14 complex was purified by nickel affinity (GE Healthcare, Chicago, IL) with 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 20 mM imidazole as the wash buffer, and 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 250 mM imizazole as the elution buffer. After cleavage of the affinity tag by recombinant TEV protease, nickel affinity chromatography was used to further purify the cleaved METTL3/METTL14 complex using 20 mM Tris-HCl, pH 8.0, 300 mM NaCl as an elution buffer; and size exclusion chromatography was used as a final purification step and buffer exchange into 25 mM HEPES, 100 mM NaCl, pH 7.0. Protein was concentrated to ~3 mg/ml (Nanodrop 2000c, Thermo Fisher Scientific) and stored at −80 °C.
Radiometric METTL3/METTL14 Assay
The methyltransferase activity of the METTL3/METTL14 complex was measured using a radiometric flashplate-based assay similar to one previously described. 21 Assays were performed in 25 µl volume in 384-well V-bottomed polypropylene microplates (cat. no. 781280, Greiner Bio-One, Monroe, NC) at ambient temperature. Optimized 1× assay buffer was 20 mM HEPES pH 7.5, 50 mM KCl, 250 µM MgCl2, 1 mM DTT, 0.01% Tween-20, 0.01% bovine skin gelatin (BSG), and 0.004 U/µl RNAseOUT (cat. no. 10777019, Thermo Fisher Scientific). For compound screening, METTL3/METTL14 [final concentration (f.c.) = 2.5 nM] was added using a Multidrop Combi (Thermo Fisher Scientific) and preincubated for 5 min. Reactions were started by adding 3′ biotinylated RNA (f.c.= 100 nM) and 3H-SAM (f.c.= 100 nM) substrates. Reactions proceeded for 30 min and were quenched with excess non-radioactive SAM (f.c.= 15 µM). The reactions were then transferred to streptavidin-coated flashplates and incubated for 2 h at 25 °C. Following two washing cycles with 0.1% Tween-20, the plates were sealed and read on a TopCount (PerkinElmer) plate-based scintillation counter. For determination of kinetic parameters, reaction times were optimized so that measurements were taken during the initial velocity phase of the reaction.
Mass Spectrometry METTL3/METTL14 Assay
Methyltransferase reactions were performed in a similar manner to the radiometric assay described above with 10 nM METTL3/METTL14, 200 nM SAM, and 125 nM RNA substrate in the 1× optimized assay buffer. The reactions were run in 20 µl volume in 384-well V-bottomed polypropylene microplates (cat. no. 781280, Greiner Bio-One) at ambient temperature and quenched by the addition of 0.5% formic acid (final). At defined times, a 2 µL sample of the quenched methyltransferase reactions was then transferred using a 384-channel pipette station to SAMDI 384-biochip arrays functionalized with a neutravidin-presenting self-assembled monolayer. The SAMDI arrays were incubated for 1 h in a humidified chamber to allow the specific immobilization of the biotinylated RNA substrate and methylated product. The samples were then purified by washing SAMDI arrays with deionized ultrafiltered (DIUF) water and dried with compressed air. A matrix comprising 2-hydroxy-5-methoxybenzoic acid in acetonitrile (30 mg/mL) and ascorbic acid in aqueous ammonium citrate (500 mM) was applied by dispensing 300 nL on each spot in the array. SAMDI–mass spectrometry (MS) was performed using the reflector-negative mode on an AB Sciex TOF/TOF 5800 System, a matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF) mass spectrometer (AB Sciex, Framingham, MA). The conversion of substrate to product was calculated by using the ratio of product area under the curve (AUC) over the sum of the substrate and product AUC peaks.
Data Analysis
GraphPad (San Diego, CA) or Grafit (Erithacus Software, West Sussex, UK) was used to calculate enzyme kinetics and parameters such as KM and kcat. Michaelis–Menten fits of steady-state enzyme velocities were applied in calculations; IC50 values and Hill slopes were generated using four-parameter fits. The quality and robustness of the assay were determined by analysis of the Z’ factor. 35 Ki values for inhibitors were calculated from the midpoint values of concentration–response plots (i.e., IC50 values) using the appropriate Cheng–Prusoff equation, 36 as recently reviewed by Buker et al. 37
Results
Characterization of the METTL3/METTL14 Protein Complex
Production of high-quality protein is an important component of assay development efforts. Therefore, selection of the appropriate protein constructs for purification efforts is a critical first step. The METTL3/METTL14 heterodimer composed of both full-length proteins had previously been shown to be catalytically active in vitro.17–21 Although WTAP is important for cellular function, previous efforts to purify METTL3/METTL14/WTAP were unsuccessful, 20 because WTAP was prone to aggregation and a homogeneous ternary complex containing an equimolar ratio of each protein could not be obtained. 20 Therefore, these efforts focused on production of the dimeric METTL3/METTL14 complex. In these experiments, the affinity tag was attached to the noncatalytic subunit, METTL14, to minimize the potential of purifying uncomplexed METTL3. Size exclusion chromatography performed as the last step of purification showed the resulting protein complex eluted as a single peak ( Suppl. Fig. S1A ), and SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) analysis clearly showed the presence of both proteins ( Suppl. Fig. S1B ). The protein complex was 88% pure, as measured by a Bioanalyzer (Agilent, Santa Clara, CA), and calculations of the individual concentrations of METTL3 and METTL14 were 16 µM and 17 µM, respectively, indicating a 1:1 protein complex was obtained.
Optimization of METTL3/METTL14 Assay Conditions
A robust enzymatic assay measuring RNA methylation and capable of compound screening was desired for METTL3/METTL14. A fully characterized radiometric flashplate assay for the complex has previously been published using a 27-nucleotide sequence with a central DRACH sequence (5′-UACACUCGAUCU
Using these optimized conditions, assay linearity was tested at multiple enzyme complex concentrations and at multiple timepoints. Steady-state enzyme kinetic analysis was then performed by titrating either SAM or the 27-nucleotide RNA at two fixed concentrations of the other substrate ( Suppl. Fig. S2 ). Multiple timepoints at each of these conditions were collected, and initial velocities calculated, from the linear region of the progress curves. Fitting of the data to the Michaelis–Menten equation yielded substrate KM values that individually did not differ by more than twofold when calculated at the two concentrations of the other substrate. The calculated substrate KM values for the 27-nucleotide RNA and SAM were 43 ± 11 nM and 127 ± 24 nM, respectively, which are comparable to the previously reported values of 22 ± 2 nM and 102 ± 15 nM ( Table 1 ). Similarly, the calculated kcat in these experiments was measured to be 12 ± 1/h and is also comparable to the previously reported value of 18 ± 2/h ( Table 1 ).
Kinetic Parameters and Inhibitor Ki Values for METTL3/METTL14 Assays.
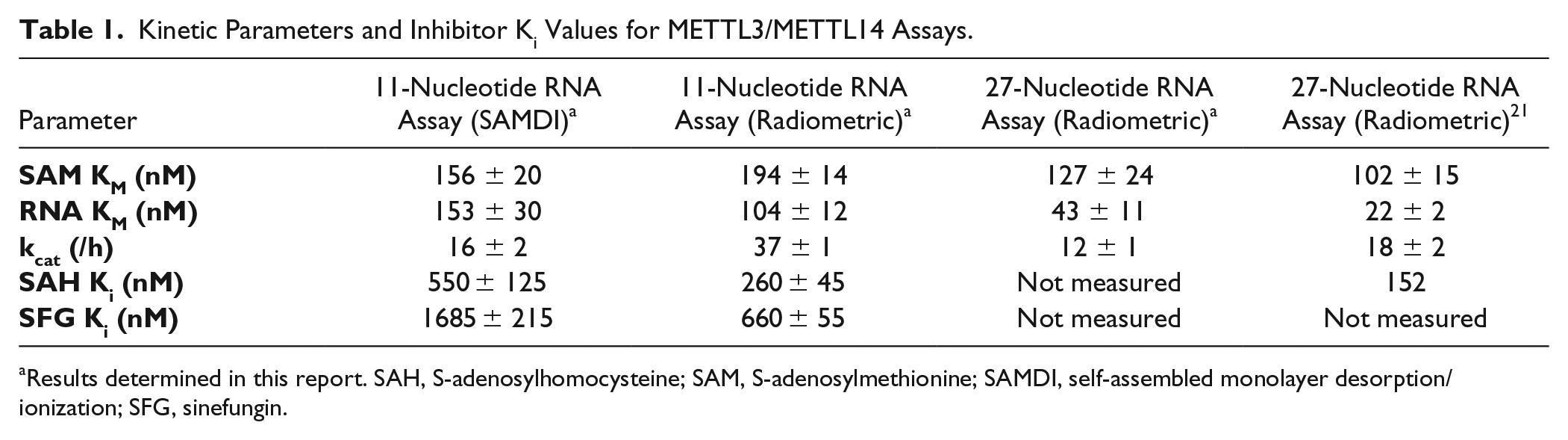
Results determined in this report. SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SAMDI, self-assembled monolayer desorption/ionization; SFG, sinefungin.
Development of the METTL3/METTL14 SAMDI Assay
To develop a SAMDI assay for RNA methylation, an 11-nucleotide single-stranded RNA substrate featuring the 5′-GGACU-3′ DRACH motif positioned centrally within the sequence (5′-UCU
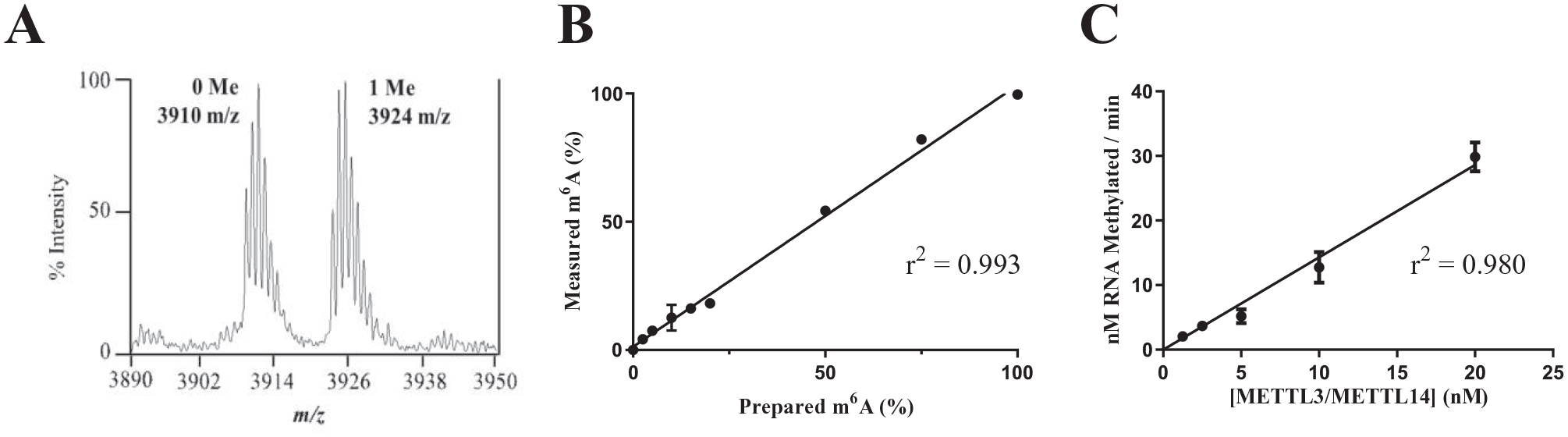
An 11-nucleotide RNA is an appropriate substrate for self-assembled monolayer desorption/ionization (SAMDI) and radiometric assay development. Samples of substrate and product for 9 different ratios (0, 2.5, 5, 10, 15, 20, 50, 75, and 100% product) of the 11-nucleotide sequence were generated, and the ratios of substrate and product peak areas were measured and plotted. Measurements were performed in triplicate. (
RNA molecules are susceptible to degradation by RNA nucleases, which can copurify with recombinant proteins. 38 This degradation has the potential to change the measured activity of the RNA-modifying enzyme and therefore negatively affect the quality of an assay when compared to a homogeneous RNA substrate. Many assay formats, including that of the radiometric flashplate assay, cannot directly assess the RNA integrity within the assay; therefore, addition of commercial RNase inhibitors is often included as protection against trace RNA nucleases. 38 In contrast, the SAMDI methodology can monitor substrate integrity within the assay by observing and measuring peaks corresponding to potential degradation products in addition to the substrate and product peaks. Therefore, the impact of RNA integrity on decreasing the concentration of the RNase inhibitor, RNaseOUT, was tested. In the absence of the RNase inhibitor, RNA degradation was seen with increasing protein concentrations ( Suppl. Fig. S3 ). RNaseOUT was titrated to 1/100th the recommended concentration (final concentration = 0.004 U/µL), with no change to assay quality, and this concentration was used for further assay characterization.
Using the assay conditions optimized for the 27-nucleotide substrate in the radiometric assay and the reduced concentration of RNaseOUT, plots of velocity as a function of enzyme concentration were generated using the 11-nucleotide RNA substrate with linear relationships observed over a range of enzyme concentrations (
Fig. 3A
). KM values for the 11-nucleotide RNA substrate and SAM cofactor (
Fig. 3B
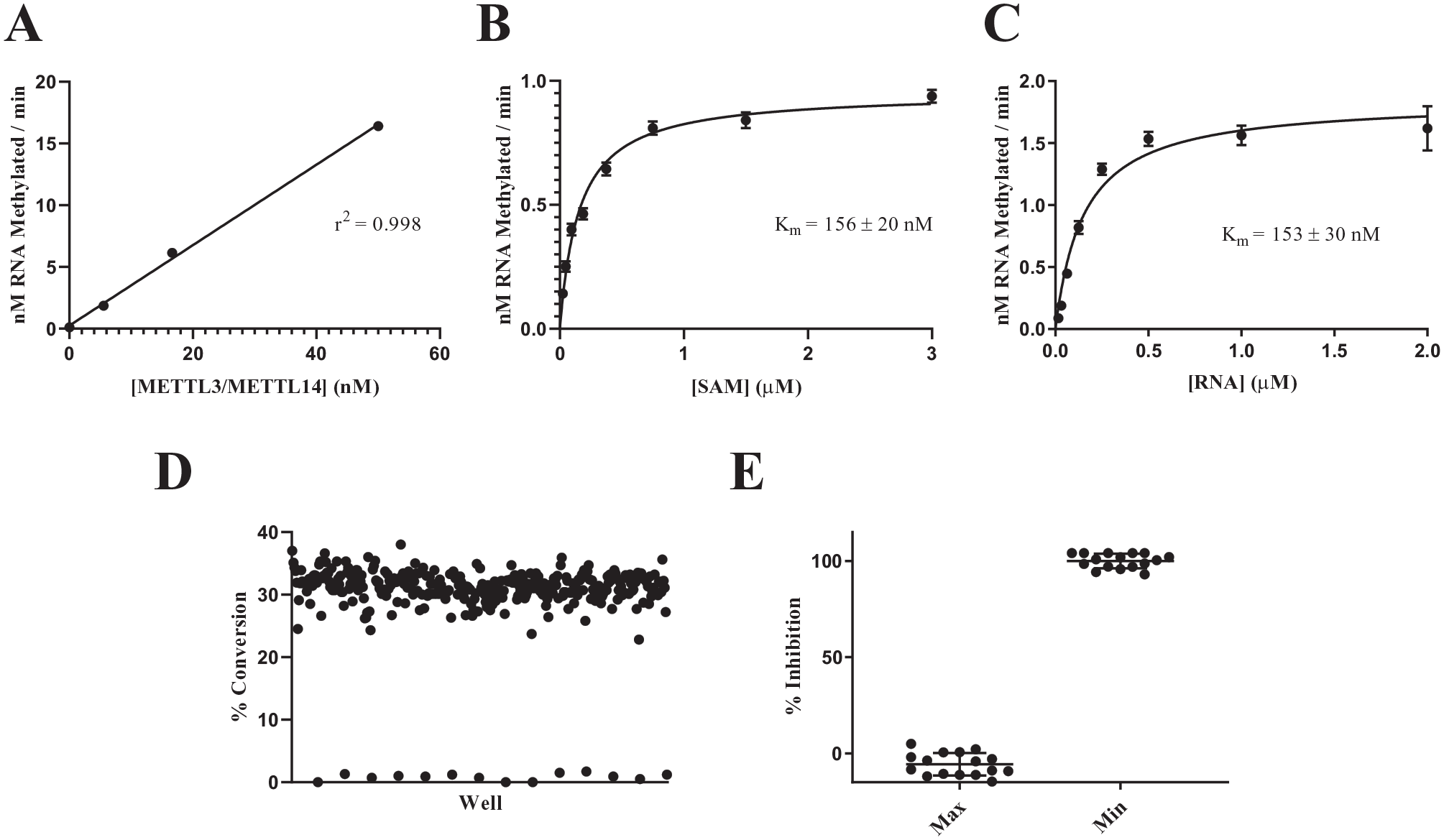
Characterization of the METTL3/METTL14 self-assembled monolayer desorption/ionization (SAMDI) assay using the 11-nucleotide substrate. (
Characterization of the METTL3/METTL14 Radiometric Assay
Because a linear relationship between enzyme concentration and initial velocity was observed radiometrically with the 11-nucleotide RNA substrate, radiometric assay development with this shorter substrate was completed. Kinetic parameters for the 11-nucleotide RNA substrate, using the final buffer conditions (see above), were determined (
Fig. 4A
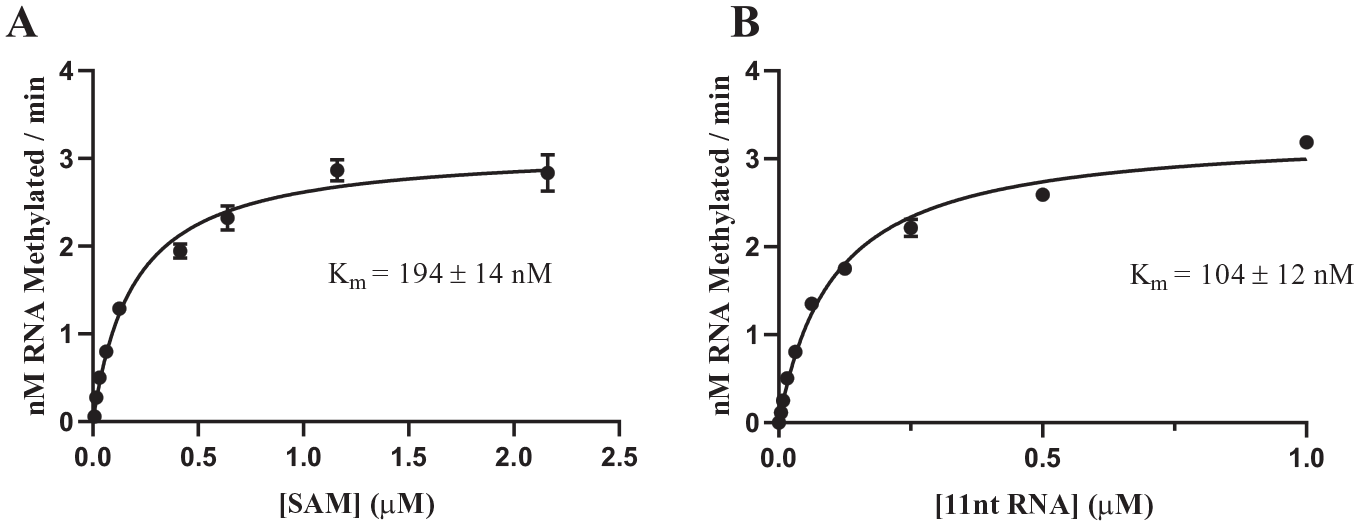
Characterization of the METTL3/METTL14 radiometric assay using the 11-nucleotide substrate. KM values were determined for (
The previously reported radiometric assay for METTL3/METTL14 differs from the present format in several ways; 21 in addition to the longer RNA construct used, there are also differences in solution conditions (e.g., buffer composition) between the present assay and that of Li et al. 21 Nevertheless, overall, the kinetic parameters for METTL3/METTL14 catalysis using these differing assays agree quite well. The KM of SAM in both assay formats agrees within a factor of twofold and is found to be independent of the length of the RNA substrate ( Table 1 ). Likewise, comparing equal-length RNA substrates, we find that the KM values are comparable (within a factor of threefold) between assays. A reduction in KM value (likely reflecting greater binding affinity) is observed for the 27-mer RNA substrate compared to that for the 11-mer RNA substrate. In the absence of crystallographic data for the METTL3/METTL14-RNA complex, a detailed structural rationale for this difference cannot be provided. The data are, however, consistent with the notion that exosite interactions between the enzyme complex and RNA recognition elements beyond the DRACH consensus sequence contribute to the binding affinity of the longer RNA substrate.
Comparison of IC50 Values of METTL3/METTL14 Inhibitors
The IC50 values of the universal methyltransferase inhibitors SAH and SFG were tested in both assays to compare measured potencies using the two different methodologies. SAH binds to METTL3/METTL14 as the reaction product, and its affinity has been previously measured. 21 In addition, high-affinity binding of SAH to the METTL3 active site has been corroborated by several cocrystal structures of SAH bound to METTL3/METTL1417–19 (PDB 5TEY, Protein Data Bank, http://www.rcsb.org/structure/5TEY). In stark contrast, no information on the potency of SFG for METTL3/METTL14 has been previously reported. The IC50s of SAH and SFG, as measured by the radiometric (SAH = 520 ± 90 nM; SFG = 1320 ± 110 nM) and SAMDI (SAH = 1110 ± 250 nM; SFG = 3370 ± 430 nM) methodologies, compared extremely well and were within threefold of each other ( Fig. 5 ), thus affirming the appropriateness of these assays for small-molecule screening.
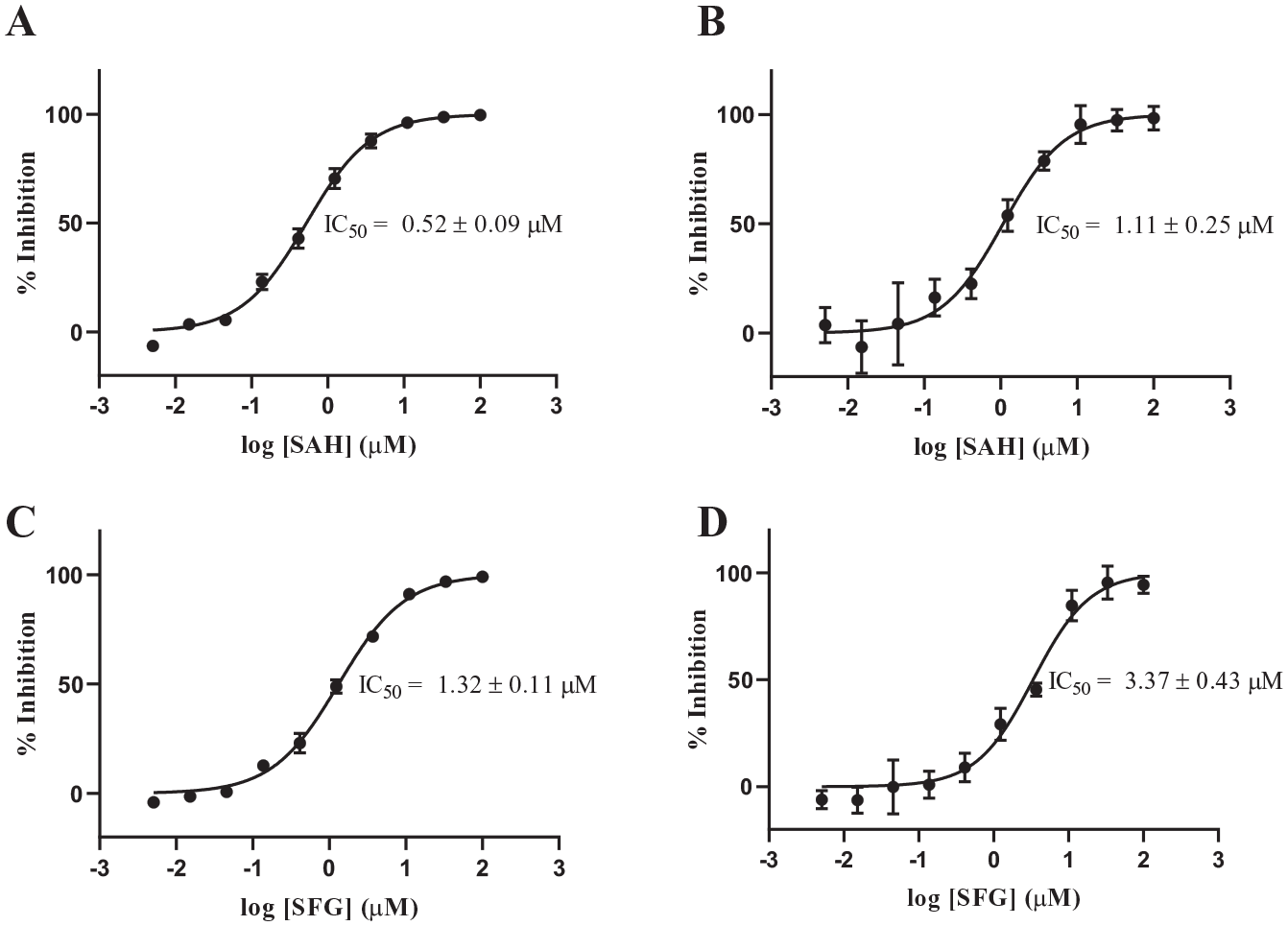
IC50 measurements of known METTL3/METTL14 inhibitors. IC50 values for the reaction product S-adenosylhomocysteine (SAH) were measured in the (
The radiometric IC50 values presented above for SAH were lower than the previously measured value of 900 nM measured by Li et al. 21 Because SAH and SFG are competitive inhibitors of the substrate SAM, measured IC50 values will be dependent on the concentration of SAM used in the assay relative to the measured SAM KM value. Calculation of the binding constant, Ki, however, can take these assay condition differences into account so that binding among assays can be compared. 39 Encouragingly, the SAM KM values measured in this report and in Li et al. 21 are in good agreement with each other, differing by less than twofold ( Table 1 ).
Using the model of SAM-competitive inhibition, the Ki values of SAH were determined for the radiometric (260 nM) and SAMDI (550 nM) assays ( Table 1 ). Li et al. reported a SAM concentration of 500 nM for their assay, a concentration that is higher than their SAM KM of 102 nM. Using the same model of competitive inhibition, the resulting Ki of SAH from the data reported by Li et al. is 152 nM ( Table 1 ). The SAH Ki values for the 11-mer nucleotide assays presented here agree within twofold of each other but are higher than the calculated Ki values derived from Li et al. by 1.7-fold and 3.5-fold for the radiometric and SAMDI formats, respectively. This variance may be attributed to the differences between the assays (buffers, substrates, etc.) outlined above. Substrate concentrations near the measured KM values were chosen for the present work, since assays run under these conditions are preferred for screening large diversity libraries, because such conditions afford the researcher an equal likelihood of identifying inhibitors with diverse modes of interaction with the target enzyme. 40
Discussion
The present study, describing the METTL3/METTL14 assay, represents the first report using SAMDI mass spectrometry to analyze RNA modification. This technology has previously been applied to enzymes that add or remove methyl groups from protein substrates using biotinylated peptides, such as the protein lysine methyltransferases WHSC1/NSD2/MMSET, 31 EZH2, 34 and SETDB1; 32 the protein arginine methyltransferase CARM1/PRMT4; 33 and the lysine demethylases KDM4C and KDM5A. 41 These previous applications used peptide substrates known to be stable under a variety of assay conditions. SAMDI has also been reported to analyze reactions on DNA.42,43 In contrast, RNA is much more susceptible to degradation, especially from RNA nucleases that are resistant to heat denaturation. 38 Researchers who regularly work with RNA are cautioned to work in a separate laboratory area, use RNase-free water and tips, and separate chemicals in addition to RNase inhibitors to minimize RNA degradation. 38 Trace RNases that copurify with added proteins are more difficult to remove, however, and must be managed with RNase inhibitors. The ability to monitor both substrate and product not only provides an understanding of progression of the enzymatic reaction but also allows substrate and product degradation to be monitored and experimental conditions (e.g., time, temperature, RNase inhibitor type and concentration, use of RNase-free materials, and protein concentration) to be optimized for generation of reproducible and high-quality assay data. In this assay, the ability to monitor RNA degradation allowed for the titration of the RNase inhibitor (RNaseOut) to 1/100th of the previously determined concentration with little impact on RNA stability or data quality; in the absence of the RNase inhibitor, RNA degradation was observed at all protein concentrations ( Suppl. Fig. S3 ). For other assays that use RNA as a substrate, the amount of RNase inhibitor required to manage RNA degradation may vary depending on the protein concentrations used and the stability and/or structure of the RNA substrate [single-stranded, double-stranded, and secondary and tertiary structures, e.g., transfer RNA (tRNA)].
The kcat value of METTL3/METTL14 (16–37/h) may be viewed as an unusually low rate of catalytic turnover. When compared to other Type I methyltransferase enzymes, however, this kinetic constant is consistent with that seen within the larger protein superfamily. The average kcat values for all protein arginine methyltransferases (PRMTs) reported in the literature ranged from 54/h (PRMT3) to 0.006/h (PRMT2), 44 while kcat for murine DNMT1 was measured at 0.66/h. 45 In contrast, METTL3/METTL14 appears to bind its substrates more tightly than PRMT enzymes. The average KM value for SAM and peptide substrates of the PRMTs is typically in the low micromolar range, 44 whereas the DNA and SAM KM values for murine DNMT1 are 250 nM and 680 nM, respectively. 45 Additional detailed kinetic characterization of other RNA methyltransferases within the Type I family will be necessary to determine if the METTL3/METTL14 SAM and RNA substrate KM values are characteristic of RNA methyltransferases generally.
Radiometric assays have been used in numerous hit-finding campaigns and inhibitor programs for protein methyltransferase enzymes (e.g., Refs. 46–50). These assays are desirable for screening and profiling methyltransferases based on their assay sensitivity, lack of interference from optically active compounds, and general utility throughout the target class. 51 In addition to requiring specific licenses from regulatory agencies for radioactivity, however, the use of tritiated SAM produces both solid and liquid radioactive waste, which can be a barrier or deterrent for some laboratories. The present SAMDI assay provides an additional assay method for methyltransferases with high sensitivity, high-throughput capabilities, no interference from optically active compounds, and no unusual waste products. These factors can be advantageous for high-throughput screening efforts involving large numbers of compounds with respect to solid and liquid waste management while maintaining high-quality data production. The SAMDI technology does, however, require dedicated and specialized mass spectrometry equipment and expertise that may be more difficult to procure for many general laboratories when compared to radiation capability.
The utility of the SAMDI assay for the methyltransferase enzyme family is dependent on identification of an enzymatically used substrate in which the shift in the mass of the substrate and product can be clearly resolved in the MALDI-TOF instrument. Therefore, SAMDI-based assays for protein methyltransferase and lysine demethylase enzymes used peptide substrates rather than full-length proteins or oligonucleosomes,31–34,41 because the 14 Dalton mass difference corresponding to the addition or removal of a single methyl group is challenging for MALDI to resolve with substrates larger than 5000 Daltons. The METTL3 assay presented here required the identification of an enzymatically used, smaller single-stranded RNA substrate (11 nucleotides, MW = 3911 Daltons) than had been previously published (27 nucleotides). 21 RNA methyltransferases covalently modify a large variety of RNA substrates and sequences, including larger molecules such as tRNA and ribosomal RNA (rRNA). 27 Development of SAMDI-compatible assays for specific enzymes in this target class may require directed efforts to identify shorter RNA sequences (e.g., tRNA loop regions) that are catalytically used as substrates. At the same time, orthogonal enzymatic assays for inhibitor profiling and validation are valuable assets for reducing false-positive results from compound screening and characterization, and additional efforts toward the development of multiple assays and assay formats are advisable, whenever technically feasible, for high-value targets of interest.
Development of an assay under conditions when substrate concentrations are close to KM values is preferred for screening large diversity libraries, because these conditions afford an equal likelihood of identifying inhibitors of diverse interaction modalities with the target enzyme.39,40 Inhibitors for protein methyltransferases have been found that bind in the SAM pocket, protein substrate pockets, and allosteric binding sites;25,52 and inhibitors for the protein methyltransferases DOT1L, EZH2, PRMT5, and PRMT1 that are competitive with either SAM (DOT1L, EZH2, and PRMT5) or protein (PRMT5 and PRMT1) substrates are currently in clinical trials (www.clinicaltrials.gov). No specific and selective inhibitors of human RNA methyltransferases have been published to date; 3 hence, it is currently unknown what mechanism of inhibition may be of greatest clinical value for RNA methyltransferase enzymes in general or METTL3/METTL14 specifically. It has been speculated that due to the nature of the RNA methyltransferase protein structure and conformational flexibility, bisubstrate inhibitors may have particular utility for this target class; 27 this is due to the relatively high levels of SAM substrate found in cells. Previous work indicated that SAM was found at concentration ranges of 10–100 µM in various rat organs, 53 while more recent experiments indicate levels of 100–150 nmoles/g of tissue in mouse liver and kidney. 54 The assays developed and presented here provide optimal conditions for identification of compounds that inhibit METTL3/METTL14 enzymatic activity regardless of the mechanism of inhibition. More generally, the results presented here portend the ability to develop similar assays that would be applicable to a broad spectrum of RNA methyltransferases, other RNA-modifying proteins, and other RNA-using enzyme systems.
Supplemental Material
Supplemental_Figures_Buker_etal_METTL3_Mass_Spec_Assay_20190701 – Supplemental material for A Mass Spectrometric Assay of METTL3/METTL14 Methyltransferase Activity
Supplemental material, Supplemental_Figures_Buker_etal_METTL3_Mass_Spec_Assay_20190701 for A Mass Spectrometric Assay of METTL3/METTL14 Methyltransferase Activity by Shane M. Buker, Zachary A. Gurard-Levin, Benjamin D. Wheeler, Michael D. Scholle, April W. Case, Jeffrey L. Hirsch, Scott Ribich, Robert A. Copeland and P. Ann Boriack-Sjodin in SLAS Discovery
Footnotes
Acknowledgements
The authors wish to thank Yu Xia for coordination of protein production efforts, Chatura Jayakody for sample management support, and Nicolas Holt for performing Bioanalyzer experiments.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: SMB, AWC, SR, RAC, and PAB-S are employees and stockholders of Accent Therapeutics, Inc.; ZAG-L and MDS are employees of SAMDI Tech; JLH is an employee of Confluence Discovery Technologies; and BDW was an employee of Confluence Discovery Technologies at the time of these investigations. The work contained in this report was performed at the request of Accent Therapeutics, Inc.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.