
Abstract
Compound pooling, or multiplexing more than one compound per well during primary high-throughput screening (HTS), is a controversial approach with a long history of limited success. Many issues with this approach likely arise from long-term storage of library plates containing complex mixtures of compounds at high concentrations. Due to the historical difficulties with using multiplexed library plates, primary HTS often uses a one-compound–one-well approach. However, as compound collections grow, innovative strategies are required to increase the capacity of primary screening campaigns. Toward this goal, we have developed a novel compound pooling method that increases screening capacity without compromising data quality. This method circumvents issues related to the long-term storage of complex compound mixtures by using acoustic dispensing to enable “just-in-time” compound pooling directly in the assay well immediately prior to assay. Using this method, we can pool two compounds per well, effectively doubling the capacity of a primary screen. Here, we present data from pilot studies using just-in-time pooling, as well as data from a large >2-million-compound screen using this approach. These data suggest that, for many targets, this method can be used to vastly increase screening capacity without significant reduction in the ability to detect screening hits.
Introduction
The past decade has seen revolutionary growth in the capacity of high-throughput screening (HTS). Miniaturized assay formats, together with advancements in assay, reader, and liquid-handling technologies, have enabled rapid screening of compound libraries consisting of millions of compounds. However, as compound collections continue to grow, innovative strategies will be required to further increase the capacity of primary screening campaigns.
Compound pooling has been explored in many permutations during the past two decades as one way to increase capacity and reduce the time and costs associated with screening.1,2 Historically, however, this approach has met with variable and somewhat limited success.3–7 Pooling strategies typically use multiplexed library plates containing 10 or more compounds per well stored at high concentrations in DMSO over long periods of time. The difficulties with this approach likely arise from many factors, including the potential for reactivity between compounds, solubility issues due to high compound concentration and alterations in ionic strength, the negative effects of high organic load in the assay well, as well as the potential for synergistic and antagonistic interactions between compounds.5,8
In recent years, various strategies have been used to circumvent some of these potential difficulties. One such approach is the creation of strategically designed multiplexed libraries in which compound properties and potential interactions are considered in building pools of compounds to be stored in multiplexed library plates.2,5 This is an interesting approach, although it does require significant informatic investment and does not easily offer the flexibility needed to accommodate a dynamically growing collection. Self-deconvoluting pooling strategies, which screen each compound multiple times in different pools of compounds, also reduce the overall number of wells screened and offer the ability to identify testing errors and false readings due to compound interactions. 9 This method takes into account library size, as well as expected hit and error rates, to build a multiplexed library designed to detect all true hits during primary screening, without the need for retests. However, advances in compound management and delivery systems have already drastically reduced the cycle time between primary screening and retests, and it is expected that introduction of closed-loop screening enabled by on-system compound storage and retrieval will further decrease the cycle time. In addition, this method uses complex mathematical algorithms to develop appropriate mixing designs and deconvolution analysis, and it would therefore require significant investments in infrastructure and computational resources.
For the reasons cited above, HTS at Bristol-Myers Squibb (BMS) has typically used a one-compound–one-well approach. However, as compound collections grow and our ability for further miniaturization becomes limited, innovative strategies are required to increase the capacity of primary screening campaigns. 10 To implement compound pooling at BMS, we needed to identify a simple, flexible, and robust method of compound pooling that would not compromise screening quality. We reasoned that if many of the difficulties that arise during compound pooling result from long-term storage of complex compound mixtures at high concentrations, then we might circumvent these difficulties by implementing a just-in-time pooling method that pools two compounds per well in an assay plate immediately prior to the addition of assay reagents. Pools are limited to two compounds per well to keep DMSO concentrations at or below 1%, thereby minimizing the deleterious effects of high DMSO concentration. We hereafter refer to this process as compound duplexing to emphasize the fact that we are pooling only two compounds per well.
Central to this novel compound duplexing method is the routine integration of acoustic dispense technology into each automated screening system at BMS, which enables just-in-time compound delivery for all HTS campaigns.11–14 This technology uses sound waves to eject nanoliter-sized droplets of compound in DMSO from a 1536-well compound source plate into a 1536-well assay plate immediately prior to assay.11–14 With acoustic dispensing integrated into each screening system, the entire BMS screening collection can be contained in 1536-well compound source plates, which can be housed locally in a centralized, environmentally controlled plate storage system in the HTS lab. Plates are then retrieved on an as-needed basis, using an automated system that feeds all robotic systems for screening. Acoustic dispense technology integrated into each system is then used to transfer compound from each source plate directly into the assay well, immediately prior to assay. Just-in-time compound duplexing, therefore, becomes an extension of our usual process: Instead of compound addition from one compound source plate into an assay plate, compound is added to an assay plate from two different compound source plates (an addition from one source plate, followed by a second addition of compound from a second compound source plate). As an added precaution, mixing of compound droplets can be minimized using the precise positional accuracy of acoustic dispense technology to dispense compound pairs in nonoverlapping droplets in the assay well. This method is technically simple, leverages existing infrastructure and informatics, and has the distinct advantage of enabling flexibility in selecting desired pairs of compound source plates for duplexing.
The primary goal of this study was to determine whether a just-in-time approach to compound duplexing would compromise screening quality and accuracy relative to the standard one-compound–one-well approach. We also wanted to understand the general applicability of this just-in-time duplexing approach across different assays and target classes. To do this, we needed to set up a general validation strategy for each assay to be considered for duplexing. Here we describe a validation strategy to evaluate this method of just-in-time compound pooling (duplexing), along with the results of this validation across several different assay and target types. We also present validation data and screening results for an enzyme target in which greater than 2 million compounds were screened using this just-in-time duplexing method.
Materials and Methods
Compound Storage and Retrieval
Compounds were selected for screening from the BMS repository of master source plates for small-molecule screening. Compound stocks (2 mM in DMSO) were stored in a single-compound-per-well format in heat-sealed 1536-well master plates compatible with acoustic dispense technology [Cyclic Olefin Copolymer (COC) Plates, product no. 3730; Corning, Corning, NY]. Master plates were stored and retrieved from temperature- and humidity-controlled environmental incubators (Liconic, Woburn, MA).
Automation and Duplexing Method
All studies were performed on automated robotic platforms that integrate instrumentation for noncontact acoustic dispensing of compound in DMSO solution (Labcyte ECHO) with instruments for assay reagent dispensing and assay readout. Although each validation study used very different assay technologies for diverse targets, the general pooling scheme was similar for each study. Acoustic dispense technology was used to transfer compound from source plates formatted in a single-compound-per-well format into assay plates immediately prior to assay. The actual volume of compound transferred differed depending on the final assay volume; generally, 10–30 nL of 2 mM compound in neat DMSO was transferred from the compound source to the destination plate, such that the final concentration of each compound was 10 µM and the final percentage of DMSO was ≤1% (in both single and duplexed assays). In single-compound-per well assays, compounds from a single source plate were transferred into a destination assay plate. For two-compound-per-well duplexing, compounds from two source plates formatted in a single-compound-per-well format were sequentially transferred into the assay plate using acoustic transfer just prior to assay. Two acoustic dispense programs were used for dispensing of each source plate into the destination plate, such that the dispense pattern was set at a slight offset (1 mm) relative to that of the second plate. This resulted in deposition of two droplets per well, in which each droplet was deposited into different areas of the same well, thereby minimizing interaction of the droplets ( Fig. 1 ). Although this dispense offset is an additional precautionary measure, this may not be strictly necessary, particularly if assays are initiated immediately after compound addition.
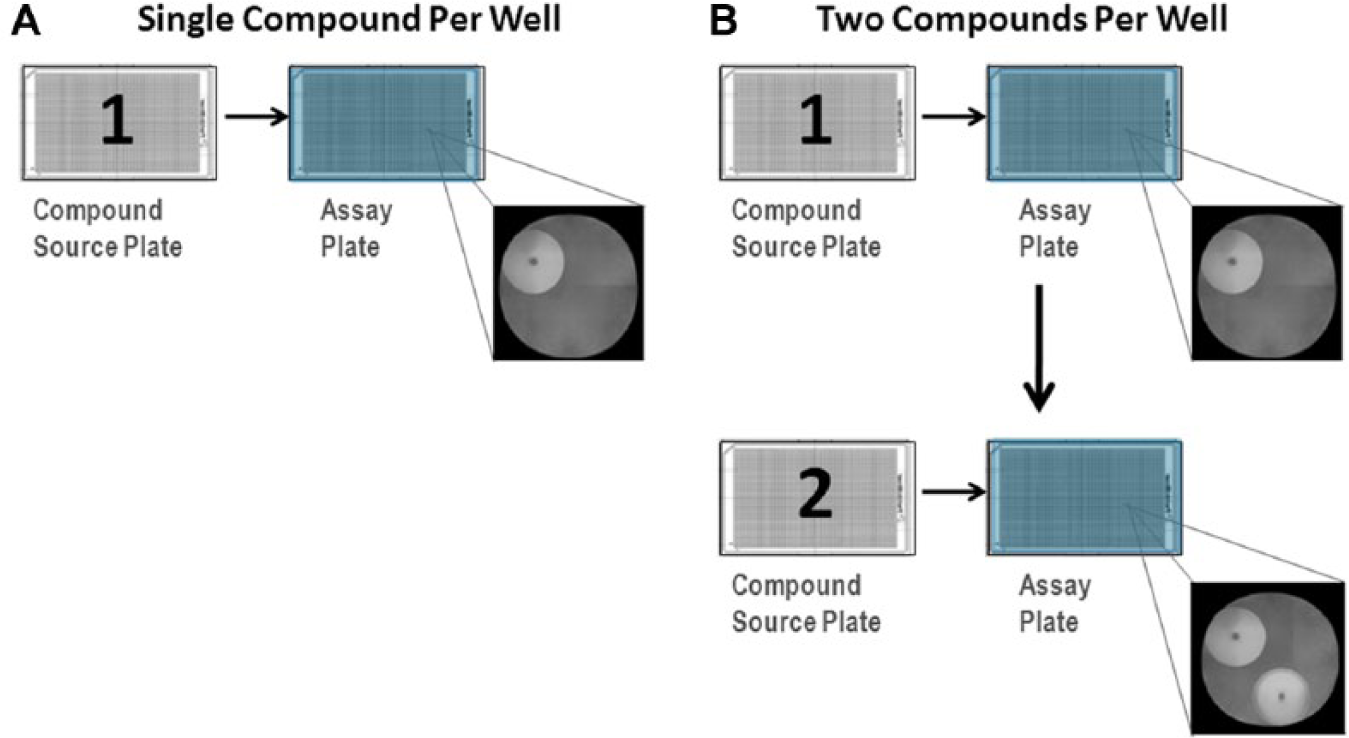
Schematic illustration of compound addition in the standard one-compound-per-well (
Biological Assay Methodologies
Pilot validation studies were conducted for a variety of target classes, including kinase, peroxidase, protease, G protein-coupled receptor (GPCR), and nuclear hormone receptor (NHR) targets. Both biochemical and cell-based assay formats were evaluated (biochemical assays: kinase, peroxidase, and protease; and cell-based assays: GPCR and NHR).
Biochemical kinase assays were performed by time-resolved fluorescence resonance energy transfer (TR-FRET; Cisbio Bioassays, Bagnols-sur-Cèze, France). Peroxidase and protease targets were screened using fluorescence and kinetic-fluorescence detection of fluorescent products. Fluorescence intensity was monitored on either an Envision plate reader (PerkinElmer, Waltham MA) for FRET-based and fluorescent probe displacement assays or a Viewlux (PerkinElmer) for kinetic-fluorescence assays. The GPCR target was screened in antagonist mode using an Aequorin cell line (PerkinElmer) and assayed as described previously 15 using a CyBi Lumax (CyBio USA, Woburn, MA) for cell addition and detection. The nuclear receptor assay used a cell line heterologously expressing a luciferase reporter driven by nuclear receptor activation; luciferase was detected by luminescence signal on substrate addition (Steady-Glo Luciferase Assay System; Promega, Madison, WI) using a Viewlux (PerkinElmer).
Each assay was conducted in 1536-well plates (Corning) with total assay volumes ≤8 µL. Test compounds were delivered to destination plates using an acoustic transfer via an Echo 550 (Labcyte, Sunnyvale, CA). For biochemical assay formats and the Aequorin assay, compounds were delivered to dry destination plates prior to assay. For biochemical assay formats, enzyme and substrate solutions were added using a Combi Multidrop (Thermo Scientific, Waltham MA) immediately after compound addition. In the Aequorin assay, cells were added to assay plates containing acoustically dispensed compound using the CyBi Lumax (CyBio USA). For the cell-based luciferase assay, cells were first plated into destination plates and test compounds were subsequently delivered to assay wells via acoustic transfer prior to assay.
Z’ Calculation, % Inhibition Calculation
For each assay, the high-signal (uninhibited) and low-signal (i.e., no enzyme or no activating stimulus) controls were used to calculate Z’ values 16 to monitor assay robustness. The percent inhibition for each assay was calculated according to the following equation:
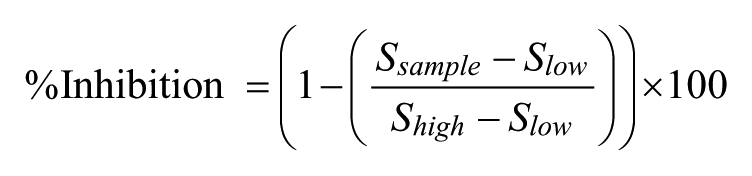
The values of S sample , S high , and S low in the equation refer to the fluorescence or luminescence of the sample, high-control, and low-control wells, respectively.
High-Throughput Screen Conducted by Just-in-Time Compound Duplexing
After completion of a number of pilot studies across multiple targets, a large-scale (>2 million compounds) HTS campaign was conducted using the just-in-time duplexing method against the protease target validated as part of our exploratory studies. All screening runs were performed on a fully automated robotic system equipped with an Echo 550 (Labcyte, Sunnyvale, CA), several Combi Multidrop instruments (Thermo Scientific, Waltham MA), and an Envision plate reader (PerkinElmer, Waltham MA), as well as carousels and stackers for compound source plates and assay plates. Compound plates (1536-well ECHO-compatible COC Plates; Corning) contained a single compound per well at 2 mM in DSMO; plates from the screening collection were randomized and paired on a carousel for just-in-time duplexing. Assay plates were 1536-well black polystyrene (product no. 3724BC; Corning).
Screening was performed by sequentially printing from two compound source plates into a single assay plate, with 20 nL acoustically dispensed from each source plate into the assay plate. The final assay volume was 4 μL, such that the final concentration of each test compound was 10 μM and the total DMSO was 1%. After compound dispense from both source plates was complete, 2 μL of 2× protease target (0.5 nM) or assay buffer (for low control wells) was added to the assay plates containing compound. Following a 10-min pre-incubation with compounds at room temperature, assay buffer containing 2× (800 μM) protease substrate H-Ala-Pro-AFC (7-amino-3-trifluoromethylcoumarine) was added. Plates were read immediately on an Envision plate reader (PerkinElmer) using excitation/emission wavelengths 405/535 using an on-the-fly protocol (10-point time course read every minute for 10 min). The reaction slope was determined for each well and was subsequently converted to percent inhibition relative to the reaction slopes of high-signal and low-signal (no-enzyme) controls as indicated above.
Establishing the Primary-Screening Hit Cutoff
After completion of the primary screening at a single compound concentration, active compounds meeting hit criteria were selected for follow-up in concentration response. A hit cutoff was determined by calculating the mean (µ) and standard deviation (σ) of percent inhibition of all wells screened. For validation studies, two different hit cutoffs were evaluated for comparison (µ+2σ, µ+3σ). Based on analysis of validation studies, a µ+2σ hit threshold was used for full-deck screens performed using compound duplexing.
Cluster Analysis
To identify compounds of interest for follow-up after HTS, several triage steps were performed on compounds above the hit cutoff (µ+2σ). First, undesirable compounds (including beta lactams, peptides, and molecules containing silicon and boron) were removed from further analyses by applying chemical filters. Initial clustering was performed using an internal BMS atom pair-based similarity algorithm based on the work of Carhart et al., 17 using a Tanimoto similarity coefficient of 0.65. With final goals of optimizing the clogP, maximizing the activity, and minimizing the surface area, a scoring function was applied to further reduce the total set of compounds:
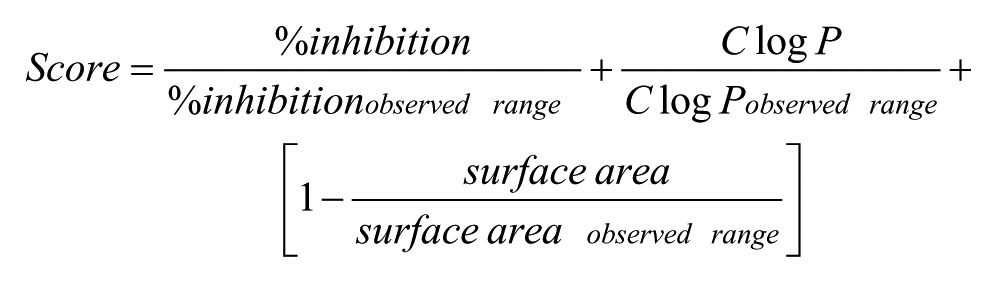
All singletons were retained for follow-up screening, as were the top scoring members of each cluster (20% of each cluster or best one for clusters with 2–7 compounds).
Results
Method Validation Strategy
High-throughput screening is a statistical process performed on a massive scale, with the ultimate goal of maximizing the probability of finding hits, or active compounds against the target or phenotype of interest. Although it is possible to optimize screen performance to reduce the occurrence of missed hits, or false negatives, even standard screening methods are associated with some rate of incorrectly categorized active and inactive compounds. For these reasons, prior to performing any HTS campaign, it is necessary to conduct validation studies to optimize the screen to maximize the probability of identifying hits. This is particularly true when comparing a new screening format with that of a more established format.
As a first step in comparing the just-in-time duplexing method with the standard one-compound-per-well method, a set of validation experiments were conducted using both methods across several different assay types and targets (biochemical assays: kinase, peroxidase, and protease assays conducted by fluorescence, kinetic fluorescence, or TR-FRET; and cell-based assays: GPCR and NHR by aequorin and luciferase reporter assay methods, respectively). This method uses a randomized set of screening compound source plates to evaluate each assay in triplicate on three separate test occasions, in both the one-compound–one-well method and the two-compounds-per-well (duplexing) method. In this way, screen performance, as well as the relative rates of false positives and false negatives, could be determined for both methods.
For each target and assay investigated, a set of 1536-well compound source plates (containing approximately 10,000–20,000 compounds) were randomly selected from the screening library. The compounds in these source plates were screened in both the one-compound–one-well method and pooled as two compounds per well (duplexing), with each compound screened in both methods in triplicate on at least three separate test occasions ( Fig. 2 ). In all cases, DMSO concentration was limited to ≤1% final assay concentration, in both the standard and duplexed conditions, to reduce the likelihood of interference from high DMSO in the assay. Although this is particularly important for cell-based assays, we almost always adhere to the same limits for biochemical assays as well. Statistical robustness for each assay was determined such that Z’ values for each test plate exceeded 0.5 for inclusion in the analysis. 16
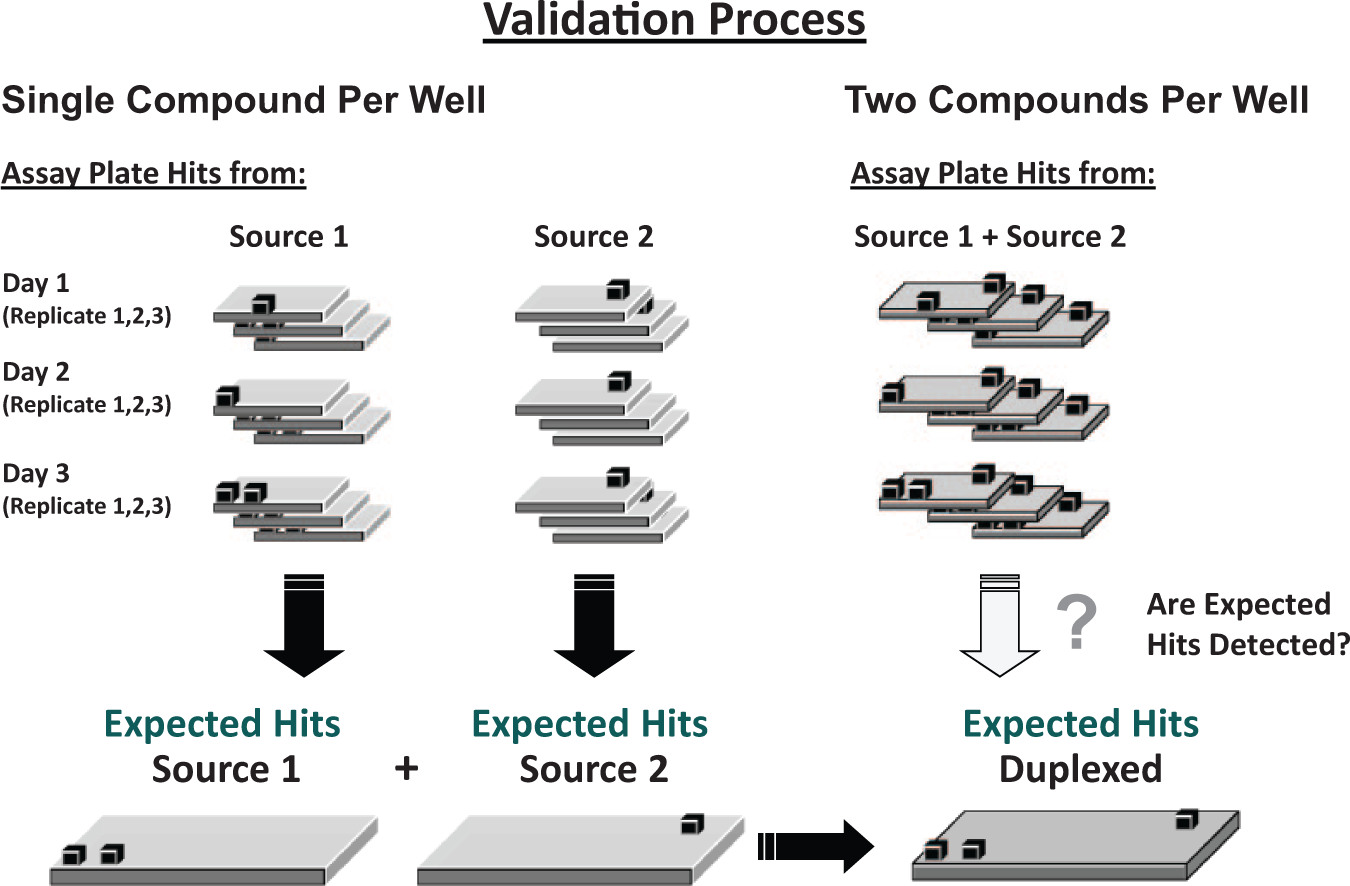
Validation procedure for pilot studies comparing a one-compound-per-well process to a duplexing process. A hit cutoff is generated based on the mean and standard deviation (2σ or 3σ) of the percent inhibition across all replicates from the one-compound-per-well validation tests. Data from multiple replicates of each test plate are used to generate a set of expected hits based on the number of compounds with an average percent inhibition above the hit cutoff. The same set of test plates are then paired and duplexed according to the process outlined in Figure 1 . Using the expected hit list, the rate of false negative (hits missed from the expected hit list) and false positives (hits not on the expected hit list) can be compared between the two processes.
For each study, hit identification was performed according to standard methods. Due to the number of compounds tested in primary HTS, this phase of screening typically tests each compound at a single concentration without replicates. Because primary screens conducted with a very large, diverse collection of molecules yield a high percentage of inactive compounds, the mean result (e.g., mean percent inhibition, µ) of the entire data set is typically near zero. Therefore, for unbiased collections, a hit cutoff is typically established at two or three standard deviations from the mean (µ + 2σ, or µ + 3σ) of the entire data set.
To understand the relevance of the false-negative and false-positive rates of the duplex assay, it is critical to also understand these parameters in the standard one-compound-per-well method. This evaluation was not necessarily focused on the ability to detect true hits verified by concentration response. Rather, we wanted to understand whether the duplexing method could identify all of the same hits identified with the standard method. Therefore, this analysis focused on the ability to detect the expected hits based on our standard one-compound-per-well primary screening process.
Because each compound was tested in multiple replicates during the validation process, a hit cutoff for the standard one-compound-per-well method was determined by averaging all of the replicates for this method and determining the mean and standard deviation of the averaged data. A compound was considered a hit if the average percent inhibition was greater than the mean plus three standard deviations (µ + 3σ) of the averaged data. This approach minimizes the impact of assay variability and gives a best approximation of the expected hits for the standard condition.
To estimate the relative rates of “false negatives” for the standard condition, the data from each replicate of the standard condition were compared to the list of expected hits derived from the average of all replicates of the standard condition. Likewise, the relative rate of “false positives” in a given replicate of the standard condition was determined by identifying the number of compounds that were incorrectly identified as active in that replicate (compounds not on the expected hit list).
To analyze the two-compound-per-well (duplex) format, a list of wells expected to be above the hit cutoff in the pooled condition could be generated based on the list of expected hits in the standard condition. A well was expected to be a hit in the pooled condition if either or both of the two compounds in that well were determined to be a hit in the standard condition (greater than three standard deviations plus the mean). Multiple hit cutoffs were evaluated for the multiplexed data set to determine the best method for maximizing the probability of detecting expected hits using the duplex method. Three hit cutoffs were evaluated for each validation set: the mean plus one, two, or three standard deviations of all results for each replicate data set. In each case, the positive wells (hits greater than the hit cutoff) were identified in the duplex condition and the well was identified as either a correctly identified positive or negative well, a missed hit (false negative), or an incorrectly identified positive (false positive). The rates of false negatives and false positives were then identified for each replicate of the duplex condition, for each of the three cutoffs established.
Pilot Validation Studies Evaluating Just-in-Time Compound Duplexing
An example of the validation results for a TR-FRET kinase assay (time-resolved Förster resonance energy transfer assay) is shown in Figure 3 . In this validation, approximately 5800 compounds were screened in both the single-compound-per-well and the two-compound-per-well format. The screen validation was completed in triplicate for both formats on each day of three separate test occasions. Using a hit cutoff of three standard deviations from the mean (µ + 3σ) for the single-compound-per-well format, the cutoff was established at 50% inhibition based on the average results from each of 9 replicates ( Fig. 3A ). Compounds with an average percent inhibition above 50% in the single-compound-per well format were designated as “expected hits” (closed circles in Fig. 3 ). Figure 3B shows the average inhibitory activity of all validation compounds screened as two compounds per well (duplex format). Each replicate was then evaluated individually, to determine the number of expected hits that were not identified in the single or duplex condition for that replicate ( Fig. 3C ). For each replicate, the number of expected hits falling below hit cutoffs of 1, 2, or 3 standard deviations from the mean were determined for the single and duplex conditions. Using a 50% inhibition cutoff (µ + 3σ), any given replicate of the single-compound-per-well format missed an average of 6% of the expected hits ( Fig. 3D ). The mean plus three standard deviations of the duplex data set was significantly higher than that of the standard condition (µ + 3σ = 60% inhibition). If a three-standard-deviation cutoff was used for the duplex data set, the percentage of missed hits (18%) was significantly higher than that of the standard process ( Fig. 3D ). However, if a two-standard-deviation cutoff was used (41% inhibition), the rate of missed hits, or false negatives, for the duplex condition was not significantly different from that of the standard process using a three-standard-deviation cutoff (4 ± 2% duplex vs. 6 ± 2% single). Note the expected hits that were missed in one or more replicates of the single or duplex conditions (red closed circles; Fig. 3D ) tended to be weaker hits close to the hit cutoff, further suggesting that missed hits are more likely the result of day-to-day variability rather than masking of activity due to duplexing. Taken together, these results suggest that if a large-scale screen were performed with this assay in duplex format using a cutoff of two standard deviations from the mean, then the risk of missing hits would not be statistically different from the risk with the standard screening process.
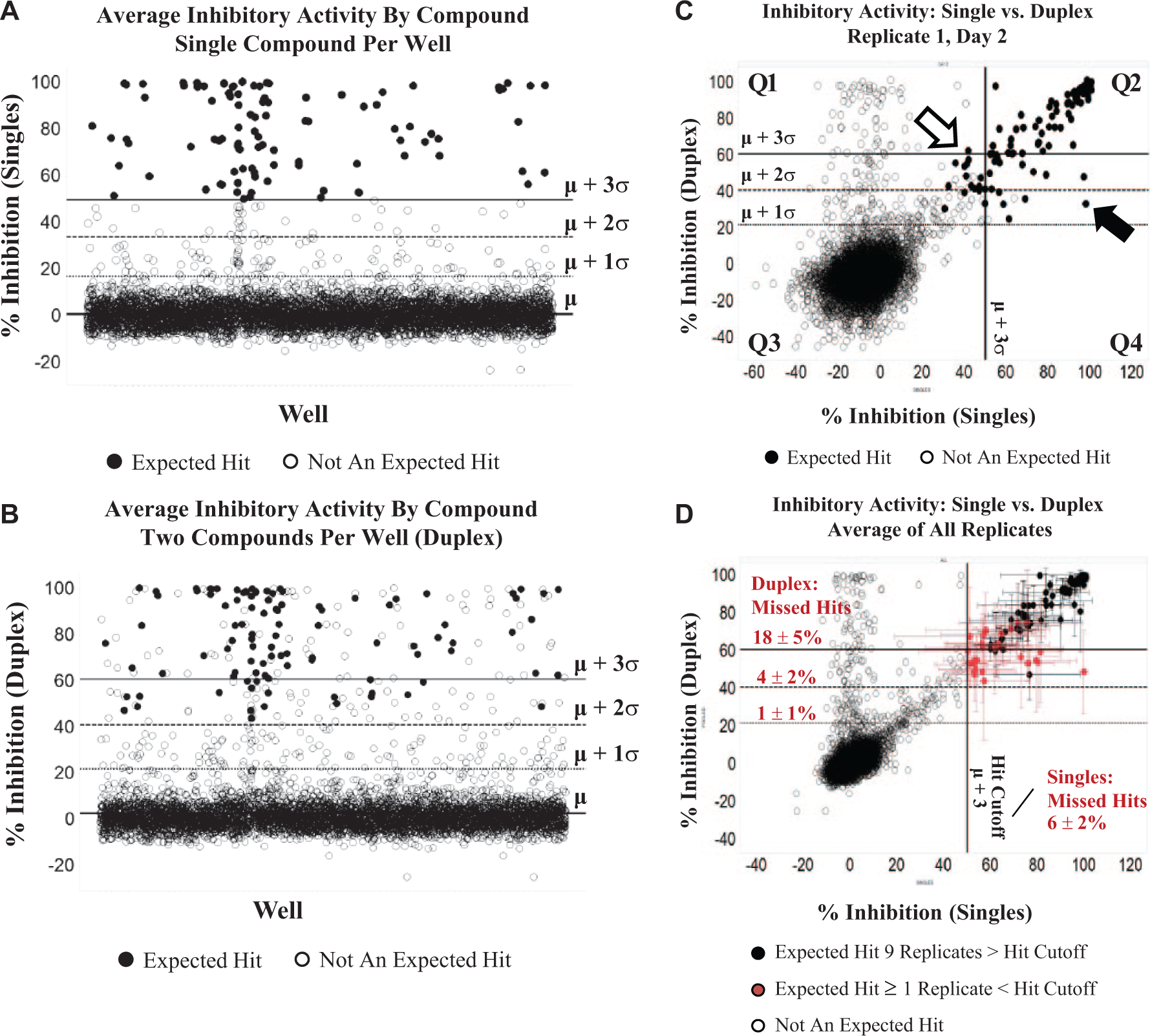
Effect of hit cutoff on false-negative rates. Scatterplots of percent inhibition in a pilot validation study for a time-resolved fluorescence resonance energy transfer (TR-FRET) kinase assay, comparing single (
Similar results have been obtained for several different target classes and assay types, including both biochemical assays as well as cell-based formats (
Fig. 4
). The rates of false negatives have not differed between the duplex process using a two-standard-deviation cutoff and the standard process using a three-standard-deviation (σ) cutoff (
Fig. 4A
), suggesting that if a 2σ cutoff is used for a duplex screen, there is a low risk of missing hits that would have been detected using the standard process with a 3σ cutoff. Note that the percent inhibition of the hit cutoff at the mean plus three standard deviations (µ + 3σ) in the single-compound-per-well format was roughly similar to the value for the hit cutoff at the mean plus two standard deviations (µ + 2σ) in the duplex condition (
Fig. 4
,
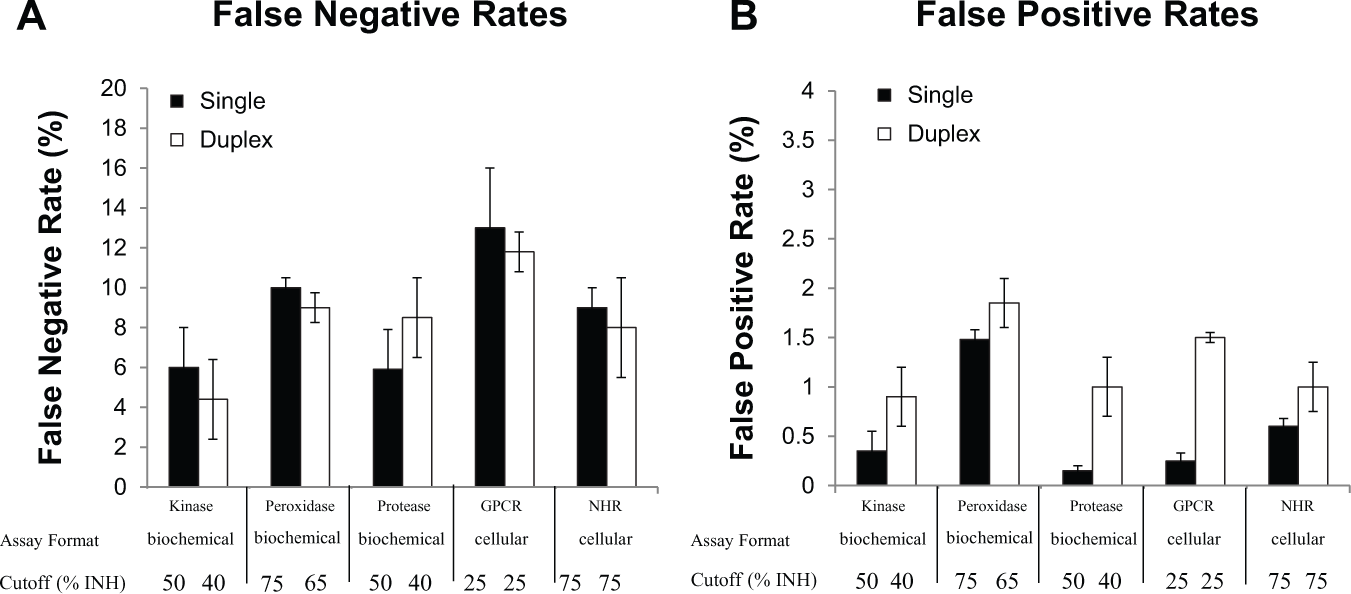
Analysis of percentage of false-negative and false-positive rates across a series of target classes and assay formats, in which the hit cutoff for the standard single-compound-per-well format was set at µ + 3σ, and the hit cutoff for the duplex condition was set at µ + 2σ. Data are presented as mean ± standard deviation of false-negative (
Large-Scale (>2 Million Compounds) HTS Campaign by Just-in-Time Compound Duplexing
For each new screening campaign, pilot validation studies as described above can be used to assess the risk of conducting a large HTS using the just-in-time pooling method. For screens in which the risk of missed hits is not significantly higher than that using the standard method, screening capacity can be significantly increased without a significant increase in screen cost.
After completion of a number of pilot studies across multiple targets, a large-scale HTS campaign was conducted using the just-in-time duplexing method. Given the chemical diversity of our screening library, it was hypothesized that screening additional compounds by the duplexing method could enable identification of additional chemically viable lead chemotypes (without significantly increasing screening costs). Pilot validation studies for the protease target in Fig. 4 indicated that the risk of false negatives associated with just-in-time compound duplexing in this assay was similar to the risk using our standard one-compound-per-well screening method, as long as a two-standard-deviation cutoff is used for the duplex condition. Therefore, the decision was made to screen >2 million compounds against this target using just-in-time compound duplexing.
As with any screening campaign, a number of different methods were used to monitor screen quality throughout the screening process. In addition to monitoring plate statistics, such as Z’ and change in overall signal throughout a screening run, quality control plates are typically introduced at periodic intervals throughout a screening run. These quality control plates may include compound source plates containing only DMSO and compound plates containing known inhibitors. Reference inhibitor plates may include compound serial dilutions, to allow for tracking reference compound potency (IC50) throughout the screen, or may consist of reference compounds at a single concentration randomly dispersed throughout a source plate. In both cases, a screener can use these reference plates to monitor detection of expected hits or compound potency relative to historical performance. In many cases, at the outset of a screening project, very few reference inhibitors are known. Thus, one additional advantage of a rigorous pilot screen (as described above) is the identification of reference inhibitors that can be used for quality control of subsequent large-scale screening campaigns. All of the above quality control measures can be used with a screen conducted by just-in-time duplexing.
In addition to these measures, quality controls to monitor duplexing are useful. In duplexing screens, each compound is randomly pooled with another compound in the screening collection by sequentially transferring compounds from two compound source plates into the same assay plate. Validation studies for each screening assay are essential to build confidence that compound duplexing does not mask detection of hits. In addition, quality control measures can be put in place to further enhance confidence in detection of true hits. For example, reference plates with known inhibitors can be randomly pooled with different compound source plates throughout a screen conducted by duplexing. In this case, monitoring detection of known inhibitors can help provide additional assurance that compound duplexing has not masked the ability to detect hits. In this screen, references of varying potencies that were known prior to HTS were used to further expand quality control. As part of the quality control process, a source plate containing reference compounds was randomly paired with different compound source plates from the screening collection, creating unique duplex pools with the reference compounds throughout the screen. As seen in this case, multiple compound pools were tolerated with little to no change in percent inhibition ranking of the reference compounds plated at the screening concentration, indicating the ability to continually detect reference compounds pooled with random compound source plates throughout this screen ( Fig. 5 ).
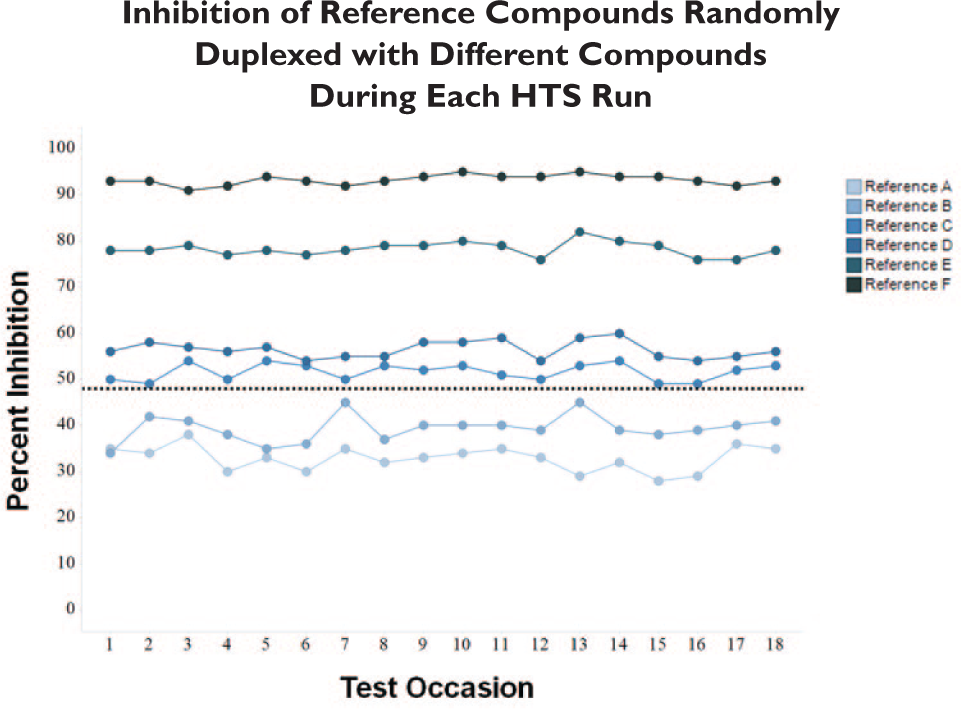
Analysis of a series of known reference inhibitors tested in different duplex pools throughout a primary screening campaign. Reference compounds were plated at a single concentration on a compound source plate, which was randomly paired with a test plate from the screening library to create a different pool on each of 18 separate test occasions. None of the compound pairings significantly altered the inhibition of any of the references tested during this screening campaign.
Figure 6 summarizes the results of the 2-million-compound primary screen performed by just-in-time compound duplexing. In this screen, the mean percent inhibition of all compounds tested was 10% inhibition. The mean plus three standard deviations for this screen was 88% inhibition, and the mean plus two standard deviations was 62%. Based on the pilot validation, an approximately two-standard-deviation distance from the mean cutoff was used. All wells showing greater than 60% inhibition of protease activity were selected for hit confirmation. In this screen, there were approximately 23,000 wells meeting the hit criteria. Given that each well contained two compounds in the primary screen, approximately 46,000 compounds were received for hit confirmation. At the stage of hit confirmation, each compound is tested in triplicate as a single compound per well to confirm activity and identify the active compound(s) in each pool.
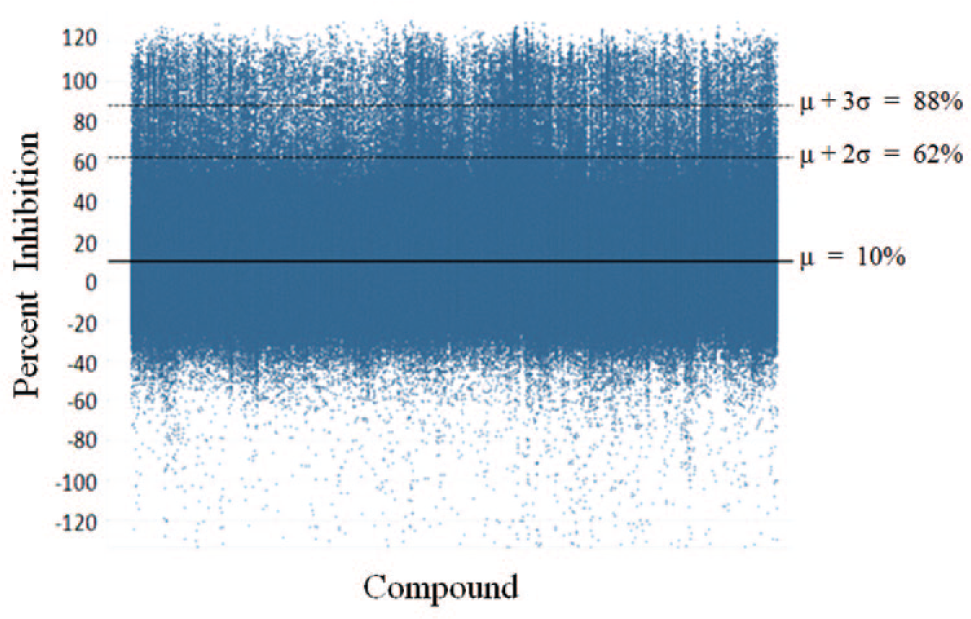
(
Based on pilot studies, it was estimated that approximately 25% (11,000) of the retested compounds would be active. Two factors are considered in this estimation. Approximately half of the compounds (23,000) were expected to be inactive based on the fact that most pools are likely to contain one inactive compound, and another 12,000 compounds (1% of 1.2 million wells tested) were estimated to be false positives based on the 1% false-positive rate determined in the pilot screen ( Fig. 4 ). The actual number of compounds that confirmed at retest in triplicate was 11,482. This is consistent with our estimations predicted based on the pilot study results.
It is important to note that increasing screening capacity and the number of hits in a particular screening campaign does not necessarily improve the initial success of a screen. Rather, initial success depends on the number of different chemotypes and viable chemical starting points identified. Therefore, it was of interest whether increasing our screening capacity using just-in-time compound duplexing identified chemotypes that we would not have identified using our standard screening process.
After further biological and chemical triage of the active compounds from this screen, approximately 4000 compounds were selected for further evaluation concentration response. Clustering analysis of these hits indicated that 2396 were represented in 568 clusters of 2 or more compounds. In addition, 1541 compounds were not related closely enough to each other to be assigned to any cluster. In a screen of approximately half the size, the number of hits taken through to concentration response determination is typically fewer than 2000 compounds. Often, to partially compensate for a smaller screen size, screen hits are used as the basis for similarity searching to identify additional compounds for screening. However, because this expansion is based on similarity searching, very different chemotypes are not usually identified based on this method. Given the number of clusters and the large number of singletons in this screen, it is likely that even if this screen had been conducted using a diverse subset of compounds selected from the larger collection, a smaller screen could not have identified all of the chemotypes identified in this screen.
Because this screen was performed only by the just-in-time duplexing method, it is not strictly possible to determine whether this screen identified compounds and chemotypes that would not have been identified using the standard screening method. Typically, the standard screen is composed of a diverse subset of approximately 1 million compounds from the entire compound collection. Because this screening campaign contained all of the compounds from which this subset is derived, it is possible to determine whether any of the screening hits are compounds from this 1-million-compound subset. Analysis of the hit chemotypes revealed that 473 out of the 570 clusters with 2 or more compounds had at least one member represented in the 1M compound diversity pick. However, 97 clusters and 582 singletons did not contain any compounds from the diversity pick. Furthermore, of the 10 chemotypes that were of most interest based on follow-up studies, half of these were not represented in the 1-million-compound diversity pick, suggesting that the expanded screening capacity offered by the just-in-time compound duplexing method had a significant impact on the success of the screen.
Discussion
As compound collections grow, it becomes increasingly important to maintain an adequate sampling of library collections in HTS campaigns. Toward this goal, we have identified a novel compound pooling method that uses just-in-time compound duplexing to increase screening capacity without compromising data quality. This method circumvents issues related to the long-term storage of complex compound mixtures by using acoustic dispensing to enable pooling directly in the assay well immediately prior to assay. Using this simplified duplexing method, we can effectively double the capacity of a primary screen.
As with any HTS, a thorough validation data set is essential for a successful screen. The validation strategy detailed here is simply an extension of a typical screen validation strategy, which evaluates replicate-to-replicate and day-to-day variability, the hit rates, and the rates of false negatives and positives using a diverse sampling of the screening collection. Using this strategy, the relative risk of screening with the duplex format can be determined and a go/no-go decision for each screen can be made. These validation methods could also be used to extend the concept of just-in-time compound pooling. For example, a higher-density compound mixture could be evaluated. The one caveat to this technique is that DMSO load increases with each compound added to the pool; limiting to two compounds per pool allows us to maintain a low DMSO concentration in the assay. However, given the right assay with good DMSO tolerance, this concept could be further explored using the validation methods presented here. Similar validation could also be used by laboratories without in-line compound dispense automation to explore alternative methods for compound dispense and timing (pre-printing droplets in advance, freeze/thaw of droplets, and/or extended time between printing and assay).
In the large-scale duplex screen presented here, more than 2 million compounds were screened in a two-compound-per-well format using a biochemical kinetic fluorescence assay. In this example, validation with representative screening deck plates appears to have adequately predicted the number of hits as well as the rates of false positives for this screen. As expected based on the validation, there were a large number of compounds that required retest confirmation and follow-up concentration response potency determination. Depending on processes and automation for compound management at a given facility, this is a significant factor to consider when evaluating the pros and cons of using such a technique. The biggest challenge at our facility has been in prioritizing hits for follow-up in groups outside of the HTS department. However, hit clustering and high-throughput hit assessment for selectivity and/or cellular activity in the case of biochemical targets have significantly facilitated following up the most relevant hits. Although delivering more hits is typically helpful in establishing early structure–activity relationships (SAR), the compelling need for screening more compounds is the potential to sample additional chemical space and expand the pool of active chemotypes for further evaluation. Indeed, from the analysis of the samples of interest from the first four targets to use this method, the just-in-time duplexing approach clearly provides for more opportunities for lead series selection. Although this is not the only method to assure that screens adequately sample the diversity of a screening collection, this method offers one method to both establish early SAR and deliver a diverse pool of hits for early-discovery efforts.
To date, we have completed 20 large-scale >2 million-compound HTS campaigns using duplexing; 70% were biochemical screens, and 30% were cellular screens. We have yet to observe a case in which the risk of false negatives is significantly increased in the duplex format. To improve chances for success, cell-based duplex screens have been limited to antagonist screens in which compound incubations with cells are <6 h and DMSO concentrations are kept to ≤1%. Cell-based screens for activators (agonists and positive allosteric modulators) are screened using a one-compound-per-well approach, to reduce the potential risk of masking weak activators by pooling with an antagonist. We use duplexing only for short-term cellular assays to minimize effects due to cytotoxicity. DMSO concentrations are kept low to minimize effects on assay signal. These precautionary guidelines, together with thorough validation studies, have helped alleviate concerns due to historical experiences with pooling. However, with appropriate validation, it is not out of the question to further explore a broader range of cellular screens in the future.
A primary goal for the HTS laboratory at BMS is to develop a toolkit of flexible screening strategies for driving drug discovery. Introduction of this just-in-time duplexing method has enabled the screening of larger and more diverse compound collections against targets of interest using less resources than would previously have been required using our standard one-compound-per well format. In some cases, such as in assays with precious or expensive reagents, this process can enable screens that would otherwise not be possible. Implementing duplex screening for suitable assays and targets may significantly affect our ability to continue to maintain a strong early-discovery pipeline, enabling screening of more targets and reducing cycle time by allowing a more adequate sampling of a large diverse compound collection earlier in the lifetime of each discovery program.
Footnotes
Acknowledgements
The authors would like to thank Drs. Deborah Loughney, Stephen Johnson, and Brian Claus for their useful suggestions and support of this work. We also thank Robert Bertekap, Kim Esposito, Kim Nowak, Monique Anthony, Ramesh Padmanabha, and Kristen Pike for their contributions to discussions and experimental work.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.