
Abstract
Biologics represent a fast-growing class of therapeutics in the pharmaceutical sector. Discovery of therapeutic antibodies and characterization of peptides can necessitate high expression of the target gene requiring the generation of clonal stably transfected cell lines. Traditional challenges of stable cell line transfection include gene silencing and cell-to-cell variability. Our inability to control these can present challenges in lead isolation. Recent progress in site-specific targeting of transgene to specific genomic loci has transformed the ability to generate stably transfected mammalian cell lines. In this article, we describe how the use of the Jump-In platform (Life Technologies, Carlsbad, CA) has been applied to drug discovery projects. It can easily and rapidly generate homogeneous high-expressing cell pools with a high degree of reproducibility. Their use in cell-based screening to identify specific binders, identify binding to relevant species variants, or detect functionally relevant therapeutic antibodies is central in driving drug discovery.
Introduction
Since the first approval of a monoclonal antibody in 1986 (Muromonab-CD3, 1 Murine IgG2a Anti-CD3 antibody) for use in humans, the subsequent three decades have seen the development of chimeric, humanized, and fully human antibodies. Antibodies are now the fastest growing class of therapeutic agents. Similarly, therapeutic peptides have experienced considerable success since the development of drugs such as somatostatin 2 and Humulin, the first approved genetically engineered human insulin therapeutic3,4 in the late 1970s. Many peptide drugs have been approved by US Food and Drug Administration and European Medicines Agency, with many targeting glucose metabolism but also antibiotics and human immunodeficiency virus fusion inhibitors. 5
Therapeutic antibody development has become an established process in the biopharmaceutical industry. Different methodologies are routinely used to access the vast sequence potential of the antibody repertoire. These include immunizing mice (wild-type or humanized) combined with hybridoma technology, phage and ribosome display technologies presenting antibody variable regions as Fab or single-chain fragments, and antibody cell surface display using yeast or mammalian cells. All these methods for therapeutic antibody generation require a process to enrich for a subset of relevant antibodies that specifically bind the target of interest, followed by high-throughput screening (HTS) to identify potential leads and subsequent characterization of a smaller number of antibodies to identify potential therapeutic candidates. Typically, this pipeline approach of antibody discovery is supported by the use of recombinant protein reagents. 6
There are a number of challenges involved in developing successful candidate antibody and peptide biologic drugs. Two of these are generation of antibodies to complex membrane targets, including G protein–coupled receptors (GPCRs) and ion channels, and ensuring the adequate demonstration of efficacy and safety in pharmacology and toxicology species such as mouse or cynomolgus monkey. To this end, the ability to easily and rapidly generate stably transfected cell lines is important. The isolation and identification of antibody therapeutics requires a high level of the target protein to be displayed on the cell surface, which can be achieved by the generation of stably transfected cell lines and isolation of the clones with the desired expression levels. The most commonly used method for generating stable cell lines relies on random integration of plasmid DNA or transduction with viruses, with subsequent antibiotic selection of a stable pool followed by screening to identify clones that have the required a high expression level. Olivares et al. 7 first described the use of phage R4 integrase to target the integration of a plasmid-bearing attB and attP sequence at att sites previously integrated randomly or using the phiC31 integrase into the host cell genome. Also, Lieu et al. 8 have demonstrated the potential of this technology to generate cell lines that would be suitable for small-molecule functional screening and how the use of pretargeted cell lines reduced the effort and time taken to generate a homogeneous pool stably expressing the gene of interest. Briefly, so-called platform cell lines are generated that bear a single R4 attP sequence and a promoterless antibiotic resistance gene that has been stably integrated into a single locus in the host genome. Cotransfection of this platform cell line with an R4 integrase-expressing plasmid and a plasmid consisting of the gene of interest and an additional promoter flanked by R4 attB sites allows the locus to be retargeted. The R4 integrase activity and the presence of the att sequences enable the promoter to be positioned upstream of the promoterless antibiotic resistance gene so that cells containing the gene of interest specifically inserted at the predefined locus can be selected using the appropriate antibiotic. This method enables the rapid generation of a pool of cells with a gene of interest integrated at a single genomic locus and avoids the need to generate clonal cell lines via laborious single-cell cloning. Here we demonstrate the suitability of this platform to rapidly generate high-expressing cell pools to support biologic discovery projects. We focus specifically on the potential use of the cell pools as a source of antigen for species cross-reactivity determination, and we demonstrate a correlation between functional data from Jump-In and the relevant primary cell line.
Materials and Methods
Vectors
The following plasmid vectors were purchased from Life Technologies (Carlsbad, CA): pJTI R4 DEST CMV EmGFP pA (transfection control vector), pJTI R4 DEST CMV pA (expression vector), and pJTI R4 Int (R4 integrase required to catalyze recombination reaction).
Target genes were obtained from GeneArt (Life Techno-logies) as synthetic genes representing the coding region for the entire protein sequence cloned into the Gateway entry vector pDONR221 (Life Technologies). Target gene sequences used mouse TROP2 (GenBank accession NP_064431.2), mouse glucagon (GCGR, GenBank accession NP_032127.2), human FPR1 most common allele (differing from GenBank accession AAA16863 by 1 coding single-nucleotide polymorphism SNP I11T), and then two C-terminal recombinant fusions with glycine serine linker (GGS4) or either Avitag (GLNDIFEAQKIEWHE with AA linker) and ten polyhistidine (His10) or His10 alone, named mTROP2, mGCGR, hFPR1AviHis10, and hFPR1His10, respectively.
A plasmid construct pGEN IRESneo human FPR was provided by AstraZeneca (Charnwood, UK) containing the human FPR1 complementary DNA (cDNA) that encodes the natural nucleotide sequence encoding the same human FPR1 amino acids as in hFPR1AviHis10 and hFPR1His10 described above.
Cloning into Jump-In Destination Vector
The entire coding region for human FPR1was amplified from pGEN IRESneo human FPR by PCR using gene-specific primers (forward CACCATGGAGACAAATTCCTCTCTCC and reverse TCACTTTGCCTGTAACGCCACCTCTG sequences). The PCR product was cloned into pENTR/D-TOPO (Life Technologies) following the manufacturer’s instructions and then nucleotide sequence confirmed by Sanger sequencing.
All inserts representing the genes of interest were subcloned from pDONR221 or pENTR/D-TOPO into pJTI R4 DEST CMV pA using Gateway LR cloning reaction following the manufacturer’s instructions and nucleotide sequence confirmed using Sanger sequencing. The three human FPR1 destination plasmids were designated pJTI R4 DEST-hFPR1, pJTI R4 DEST-hFPR1AviHis10, and pJTI R4 DEST-hFPR1His10. The mouse TROP2 and mouse GCGR destination vectors were designated pJTI R4 DEST-mTROP2 and pJTI R4 DEST-mGCGR, respectively.
Generating Targeted Stable Transfected CHO-K1 Cell Pools
The Jump-In CHO-K1 (JI CHO-K1) platform cell line was acquired from Life Technologies for evaluation. Culturing of the cells was performed as described in the Jump-In CHO-K1 Retargeting Kit user guide (www.lifetechnologies.com). The transfection of JI CHO-KI with destination vectors was carried out as outlined in the Jump-In CHO-K1 Retargeting Kit user guide.
Expression confirmation and assessment of the relative expression levels were performed by flow cytometry. Cells expressing human FPR, mouse TROP2, and mouse GCGR were detected with the appropriate fluorescently labeled ligand for mouse GCGR (Alexa Fluor 647 glucagon; Cambridge Research Biochemicals, Billingham, UK) or labeled antibodies phycoerythrin-conjugated mouse anti–human FPR clone 350418 (R&D Systems, Minneapolis, MN) or APC-conjugated goat polyclonal anti–mouse TROP-2 (R&D Systems) using the following method. Cells were harvested with StemPro Accutase (Life Technologies) resuspended in culture media and centrifuged at 200 × g for 5 min to pellet the cells. The cells were then resuspended in ice-cold FACS buffer (0.01% sodium azide, 1% fetal bovine serum, 0.5M EDTA in phosphate-buffered saline) and washed twice by centrifugation and resuspension in FACS buffer. Then, cells were incubated with the primary antibodies at 10 µg/mL or glucagon–Alexa Fluor 647 (20 nM) for 60 min on ice. The cells were washed twice with FACS buffer before being fixed with BD CellFix (Becton Dickinson, Franklin Lakes, NJ). The samples were analyzed using a FACSCanto II instrument (Becton Dickinson).
Fluorometric Microvolume Assay Technology Cell-Binding Assays
Dylight 649–labeled goat anti–mouse IgG Fc (γ) fragment specific was purchased from Jackson Immuno-Research Labs (West Grove, PA). Thirty percent bovine serum albumin (BSA) and Costar nonbinding surface 384-well plates were both from Sigma-Aldrich (St. Louis, MO). PhyNexus Protein A tips were purchased from PhyNexus (San Jose, CA).
Anti–human TROP2 mouse IgG monoclonal antibodies were purified from hybridoma supernatant using 20 µL Protein A tips (PhyNexus) on a PerkinElmer (Waltham, MA) MiniTrak robotic liquid handling system. The concentrations of purified antibodies were determined by absorbance at 280 nm using an UV-Star 384-well plate (Greiner Bio-One, Monroe, NC). Cell-based fluorometric microvolume assay technology (FMAT) assays were performed on this panel of antibodies to identify clones with cross-reactivity to mouse TROP2. In brief, JI CHO-K1 mTROP2 cells and JI CHO-K1 parental cells were lifted from tissue culture flasks using StemPro Accutase (Life Technologies) and washed with phosphate-buffered saline (PBS) containing 1% BSA. The cell pellets were resuspended in the PBS/1% BSA at a concentration of 1.33 × 105 cells/mL. Dylight 649–labeled goat anti–mouse IgG Fc (γ) fragment-specific polyclonal antibody was diluted to 3.3 nM in PBS/1% BSA. The purified anti–human TROP2 mAbs were diluted to 5 µg/mL in the same buffer, and 5 µL of the diluted antibodies was added into each well of 384-well nonbinding surface plates followed by 15 µL Dylight 649–labeled anti–mouse antibody and 30 µL JI CHO-K1 mTROP2 cell suspension. The final 50-µL volume in the well contained 4000 cells and 1 nM Dylight 649–labeled detection antibody. After 10 h of incubation at room temperature, plates were read using Applied Biosystems Cellular Detection 8200 System (Life Technologies). An FMAT screen using JI CHO-K1 parental cells was performed in parallel. Positive binding was set arbitrarily at a count of over 50 events with JI CHO-K1 mTROP2 cells and 0 events in JI CHO-K1 parental cells.
Glucagon Receptor cAMP Accumulation Assay
Cell-based cAMP accumulation assays were used to screen peptides for agonist activity at endogenous and transfected mouse glucagon receptor. Peptides were solubilized in DMSO or PBS and serial dilutions prepared in black shallow-well u-bottom 384-well plates (Corning, New York, NY) in 5 µL assay buffer (HBSS; 25 mM HEPES, IBMX, 0.1% BSA, pH 7.4) using an ECHO acoustic noncontact dispenser (Labcyte, Europe, Dublin, Ireland). A frozen cryo-vial of JI CHO-K1 mGCGR or mouse hepatocytes (Life Technologies) was thawed rapidly in a water bath, transferred to prewarmed assay media, and centrifuged at 240 × g for 5 min. Cells were resuspended in assay buffer and 5 µL cell suspension was added and plates incubated at room temperature for 30 min.
cAMP levels were measured using a commercially available cAMP dynamic 2 HTRF kit (Cisbio, Codolet, France), following the two-step protocol as per the manufacturer’s recommendations. In brief, anti-cAMP cryptate and cAMP-d2 were made up separately in conjugate and lysis buffer provided. Then, 5 µL anti-cAMP cryptate was added to all wells of the assay plate, and 5 µL cAMP-d2 was added to all wells except nonspecific binding (NSB) wells, to which conjugate and lysis buffer was added. Plates were incubated at room temperature for 1 h and then read on an EnVision (PerkinElmer) plate reader. Data were transformed to % Delta F as described in the manufacturer’s guidelines and analyzed by four-parameter logistic fit to determine EC50 values.
Results and Discussion
Jump-In Platform Cell Lines Have Potential to Support Antibody Discovery
A fully human anti–formyl peptide receptor (FPR1) antibody with cross-reactivity to the cynomolgus FPR1 homologue has previously been generated (Douthwaite et al.). 9 Antibody isolation was achieved by immunization of transgenic mice, with a high-expressing cell line (CHO-hFPR-STR-TREX) generated by a plasmid random integration process and clonal selection, then subsequent engineering of antibody variable regions using phage display selecting on cell-surface FPR1, improving both potency and species cross-reactivity.
The number of human FPR1 receptors on the cell surface of the cell line CHO-hFPR-STR-TREX was calculated to be between 5 and 9 × 105 per cell. Receptor copy number was determined using quantitation of antibody binding sites using FACS staining with the human FPR1-specific antibody Mab3744-PE (R&D Systems) in conjunction with a calibrated curve prepared using Quantum Simply Cellular anti-Mouse IgG beads (Bangs Laboratories, Fishers, IN) . The CHO-hFPR-STR-TREX cell line was used for immunization, HTS FMAT binding assays, and phage display selections. Having expression levels of greater than 5 × 105 receptors per cell is a key factor in successful isolation of antibodies to a GPCR target when immunizing with cells alone (unpublished data). To understand whether the Jump-In platform offers a potentially faster and easier method to generate high-expressing homogeneous stably transfected pools that would support antibody discovery activities, we generated JI CHO-K1 pools expressing human FPR1 and compared the relative FACS shift with estimate expression.
Three human FPR1 destination constructs—pJTI R4 DEST-hFPR1, pJTI R4 DEST-hFPR1AviHis10, and pJTI R4 DEST-hFPR1His10—were used to stably transfect the JI CHO-K1 cell line, resulting in three pools resistant to the selection agent blasticidin: JI CHO-K1-hFPR1, JI CHO-K1-hFPR1AviHis10, and JI CHO-K1-hFPR1His10, respectively. Staining with MAB3477-PE and FACS analysis of the CHO-hFPR-STR-TREX and JI CHO-K1 FPR pools revealed all three JI CHO-K1 pools had homogeneous expression levels assessed by the relatively tight symmetrical peaks. The expression levels, when related to the CHO-hFPR-STR-TREX ( Fig. 1A ) cell line, were comparable for JI CHO-K1 hFPR1AviHis10 ( Fig. 1B ) and JI CHO-K1 hFPR1His10 ( Fig. 1C ) with a similar shift of MAB3744-PE stained populations, whereas the hFPR1 pool had a smaller shift by FACS ( Fig. 1D ), indicating a lower expression level at the cell surface.
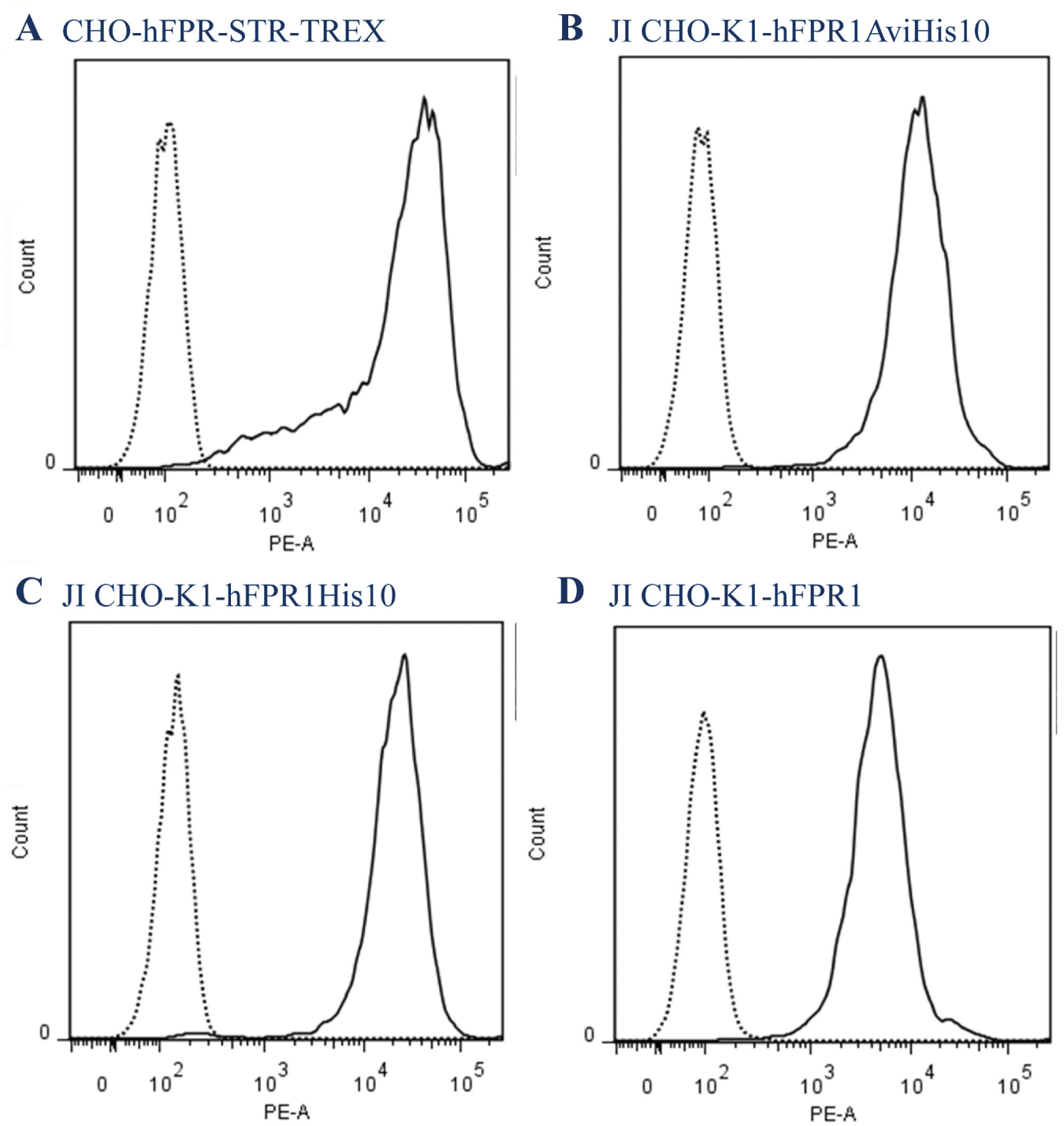
Flow cytometry plots for human FPR1 expressing cell lines compared with parental cell line stained with Mab3744-PE. (
As the JI CHO-K1 platform cell line contains a single integration site (data from Life Technologies) that is capable of high expression, this rules out the possibility of a positional effect as a consequence of random integration. The lower expression seen in JI CHO-K1-hFPR1 could be explained by hFPR1 cDNA maintaining the native codon usage for humans, whereas the hFPR1AviHis 10 and hFPR1His 10 have been codon optimized.
The R4 recombination reaction is an inefficient process that we observed as a small number of transgenic cells growing through the antibiotic selection (data not shown). As CHO cells are prone to phenotypic drift, this is a potential issue as any phenotype that results in lower expression would positively select those clones, leading to a bias in the final pool offering an alternative explanation to the relative codon usage.
With the FACS shifts comparable to the CHO-hFPR-STR-TREX cell line and inferred high expression level seen for two of the JI CHO-K1 pools expressing human FPR1, this would indicate that Jump-In pools are suitable for generating specific antibodies in an immunization strategy and selecting engineered antibodies.
Characterization of Biologics for Species Cross-Reactivity
The ability to generate and characterize biologic therapeutics that, in addition to their activity against the human target, bind and functionally modulate nonhuman pharmacology and toxicology species is critical to demonstrating in vivo efficacy and evaluating safety. Preclinical in vivo demonstration of efficacy and the maximum tolerated dose is critical to progressing therapeutics to the clinic. In an example shown here, anti-human TROP2 hybridoma monoclonal antibodies were successfully isolated from the strategy shown in Figure 2A . To enable the functional testing of lead antibodies in tumor inhibition in vivo models, it was essential to identify antibodies that cross-react with mouse TROP2. To that end, a JI CHO-K1 cell pool expressing mouse TROP2 was generated with high expression, as demonstrated by the large FACS shift compared with parent ( Fig. 2B ). A panel of 71 anti–human TROP2 antibodies were tested for their ability to bind mouse TROP2 by comparing the binding signal on the JI CHO-K1 mTROP2 cell pool with the background signal on the parental JI CHO-K1 cell line. Positive binding was set arbitrarily at a count of over 50 events for JI CHO-K1 mTROP2 cells and zero events on JI CHO-K1 parental cells. Three different antibodies that exhibit mouse TROP2 cross-reactivity in the FMAT binding assay were successfully identified ( Fig. 2C ).
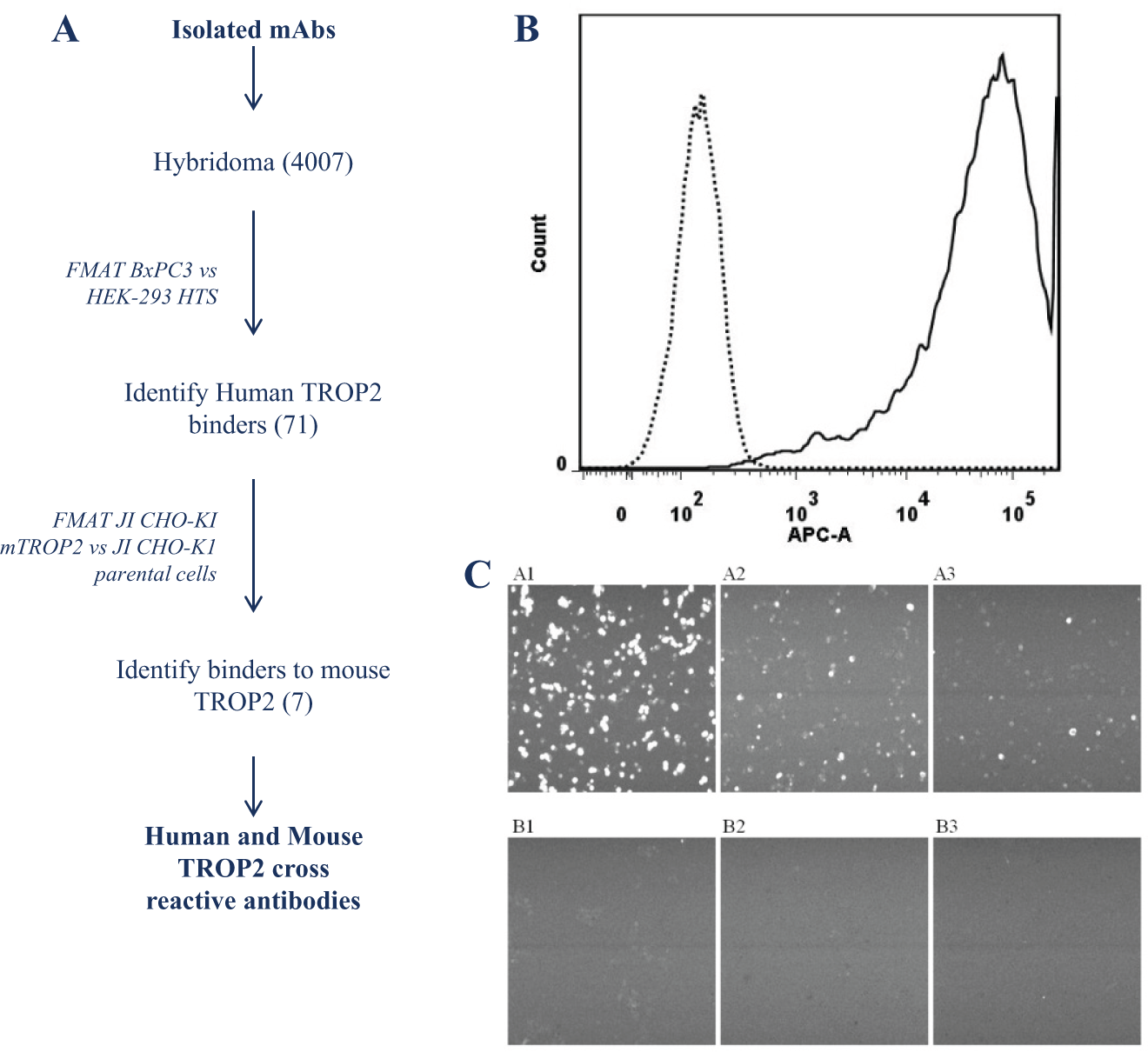
(
Optimized variants of the glucagon peptide have been developed as a potential therapeutic peptide following the screening cascade outlined in Figure 3A . Similarly to the anti-TROP2 antibody described above, it is necessary to evaluate in vivo efficacy and safety preclinically. Again, the JI CHO-K1 platform cell line was used to rapidly generate a mouse glucagon receptor (GCGR) JI CHO-K1 mGCGR. The absence of an anti–mouse GCGR antibody to perform FACS analysis meant fluorescently labeled glucagon was used instead to determine expression level compared with the parental cell line, as shown in Figure 3B . Using this JI CHO-K1 mGCGR cell pool, we were then able to successfully identify engineered glucagon peptides ( Fig. 3C , samples 1–7) with functional cross-reactivity to the mouse receptor, enabling the selection of leads that should be progressed for in vivo testing.
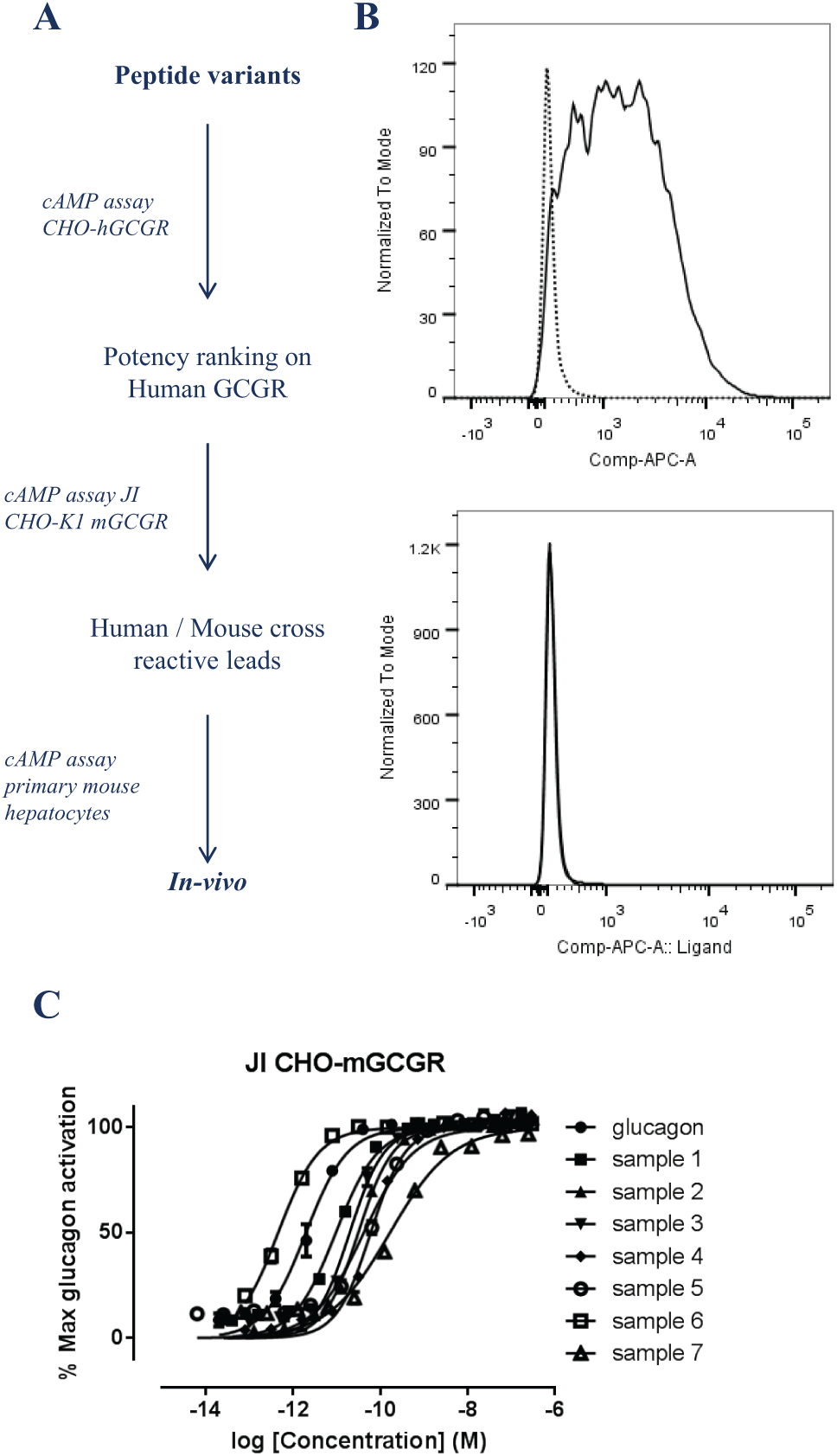
(
Using techniques such as sequence homology to predict cross-reactivity of a biologic to relevant nonhuman animal species is unreliable. Where sequence homology is of use is informing the selection of species that should be tested empirically, providing the highest chance of identifying cross-reactive leads. Consequently, it is essential to generate cell lines expressing species variants to enable HTS or characterization of lead molecules for their ability to cross-react with the target from pharmacology and toxicology species. The use of the Jump-In platform cells provides the ability to easily and rapidly generate cell lines with homogeneous high-level expression of different species variants to enable the empirical testing of leads. In the case shown, the anti–human TROP2 antibodies were identified using a high-throughput screening FMAT binding assay and subsequently characterized for their ability to bind to mouse TROP2. It would equally have been feasible to have performed parallel HTS to identify leads that bind human and mouse TROP2 while showing no binding to irrelevant or parental cell lines.
An alternative approach to stable cell line generation for profiling cross-reactivity species variants is transient transfection. This has the benefit of being rapid, taking only a few days once the expression construct has been generated. It is possible to then generate large assay-ready frozen cell banks. 10 The drawback is that transient transfection efficiency is less than 100%, expression levels are very heterogeneous, and it is challenging to generate large consistent batches. In agonist activity assays, potency is a function of receptor expression levels and thus requires controlling for consistency between batches. The Jump-In platform can overcome the challenges of transient transfection by generating high-level homogeneous expression. Although the time to generate Jump-In pools is longer than transient transfection (approximately 3 weeks from transfection), it is considerably shorter than generating clonal cell lines, which typically takes around 3 months to perform the transfection, antibiotic selection of the stably transfected pool, and subsequent isolation of clonal cell lines. In addition, it is feasible to generate multiple Jump-In cell lines in parallel as the most labor-intensive activity of isolating a high-expressing clone is removed.
Characterization of the Modified Human Glucagon Peptides for Mouse Glucagon Signaling and Correlation to Primary Hepatocytes
When generating a transgenic cell line, there is a general concern that the stably transfected cell line will not recapitulate the endogenous target function. Receptor function can be affected by a number of factors, including the parent cell line lacking relevant signaling pathways or not being able to respond due to species differences. Overexpression can also cause issues of undesired pharmacology such as high basal activity. These issues typically require the use of cell lines with endogenous target expression or primary cells in the screening cascade to understand the true potency of lead molecules to inform dosing levels prior to in vivo experiments.
In the case of the engineered glucagon peptides, it was possible to use the same assay measuring the cAMP production in response to GCGR activation by the glucagon peptide in the JI CHO-K1 mGCGR cell pool and primary mouse hepatocytes ( Fig. 3C and Fig. 4A , respectively). Plotting the potency relative to native glucagon potency in the two different cell assays clearly shows a good correlation in potency of the glucagon peptide variants ( Fig. 4B ), making this a suitable and reliable assay.
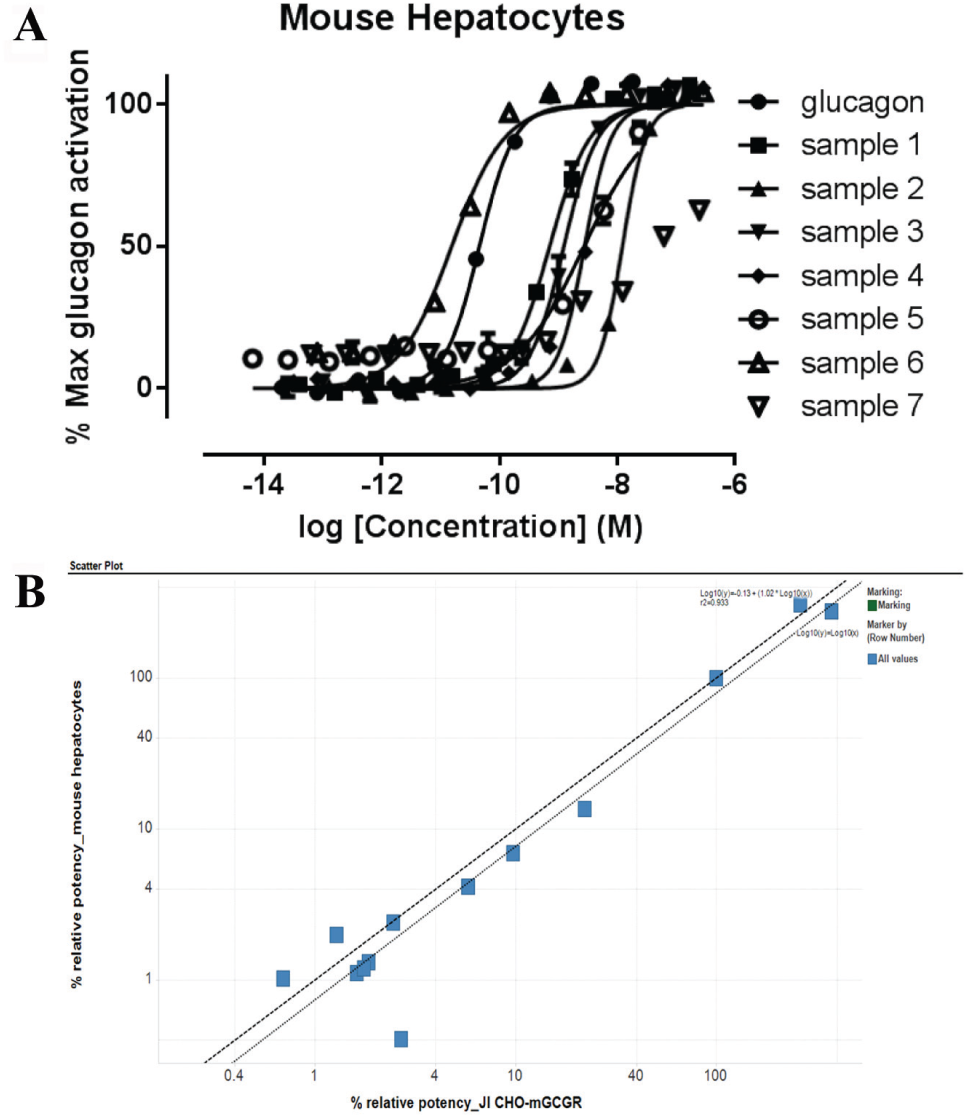
(
In summary, cell pools generated via the Jump-In platform can demonstrate homogeneous high expression of a target gene or of a gene of interest. This provides benefit to drug discovery by delivering expression levels required for immunization and “binding” assays. The ease of use of generating cell pools offers the potential to profile leads for binding to different species and sequence variants in parallel. Despite the single genomic insertion of the gene of interest in the Jump-In pool eliminating the problem of random integration affecting expression, it is important to bear in mind that there is potential for clonal variation within the population of cells. These variations caused by the polymorphic phenotype of the parental cell background could have effects on properties such as expression level and posttranslation modifications, which would need to be evaluated on an individual basis. Although not applicable for all receptors, high-level expression does not necessarily preclude the ability to develop a functional cell-based assay that will reflect endogenous receptor activity in primary cells. In this article, we have demonstrated a number of applications of the Jump-In platform relevant to biologics discovery, including the generation of cell lines suitable for generating monoclonal antibodies, identification of species cross-reactive antibodies, and cell-based functional testing of lead molecules. The studies we conducted here have used the Jump-In cells in short-term culture (e.g., to address a specific question or produce material for immunization). If the cell pools were to be maintained in longer term culture and used for extended assay campaigns, the potential for clonal variability and the impact of long-term storage would need to be determined empirically.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Financial support was through authors’ employment at MedImmune.