
Abstract
Identification and characterization of kinase inhibitor potency and selectivity is often an iterative process in which a library of compounds is first screened against a single kinase, and hits from that screen are then profiled against other kinases to determine specificity. By developing kinase assays that employ either a terbium- or a europium-based time-resolved fluorescence resonance energy transfer (TR-FRET) readout, one can take advantage of the distinct emission properties of these labels to develop assays for 2 kinases that can be performed simultaneously in the same well. This not only increases the information content provided per assay well but can immediately provide information on compound specificity. The authors have applied this strategy to the development of multiplexed assays for 2 examples systems: EGFR and IKKβ, as well as lipid kinase family members mTOR and PIK3C3. They demonstrate the ability of these multiplexed assays to characterize selective kinase inhibitors in a dose-response mode, with no difference in results obtained from traditional single kinase assays performed separately.
Keywords
Introduction
T
Tb-based LanthaScreen® TR-FRET kinase assays have been described previously. 6,7 These assays use either a fluorescein-labeled peptide substrate or a green fluorescent protein (GFP) fusion of a protein substrate that is paired with a Tb-labeled phosphorylation site-specific antibody that recognizes and binds to the phosphorylated substrate. Following phosphorylation of the substrate by a kinase, the Tb-labeled phosphorylation site-specific antibody (the FRET donor) brings the Tb and the fluorescein (or GFP) labels into proximity, resulting in an increase in FRET. Although other phosphorylation events may be occurring in the reaction mixture, only the site of modification that the antibody is directed toward results in a FRET signal. In the absence of the specifically recognized phosphorylation event, the antibody remains unbound and FRET does not occur. This approach is analogous to systems using Eu as the donor and acceptor dyes such as allophycocyannin 8 or Alexa Fluor® 647 but allows for the use of GFP fusion proteins to be used as substrates.
Because Tb and Eu, as well as their corresponding acceptor fluorophores, have distinct, nonoverlapping emission spectra ( Fig. 1 ), we reasoned that it should be possible to use these labels to develop multiplexed assays to measure the activity of 2 kinases simultaneously from within the same assay well. Such a strategy would allow additional information to be determined, such as a first-pass look at compound selectivity, without a corresponding doubling of many of the reagents and consumables necessary to perform the analysis separately. For proof-of-principal experiments, we chose to develop multiplexed assays for the protein kinases EGFR and IKKβ (IKBKB) using labeled peptide-based substrates that would be specifically recognized upon phosphorylation by either Tb- or Eu-labeled antibodies. To develop a multiplexed assay for mTOR (FRAP1) and PIK3C3 (hVPS34), we used 2 different assay formats, each using a different donor-acceptor pair in the readout. mTOR activity was measured using GFP-4E-BP1 and a Tb-labeled antibody that specifically recognizes phosphorylation of this substrate. 7 To measure PIK3C3 activity, we used the Adapta® Universal Kinase assay format, which measures kinase-mediated adenosine diphosphate (ADP) formation by monitoring displacement of an Alexa Fluor® 647–labeled ADP analog from a Eu-labeled anti-ADP antibody. In this format, displacement of the Alexa Fluor® 647–labeled ADP analog is detected by a decrease in TR-FRET signal. For each of the 2 sets of kinases, we compared a set of inhibitors both separately and in the multiplexed format and found an excellent correlation between the multiplexed and individual assay formats.
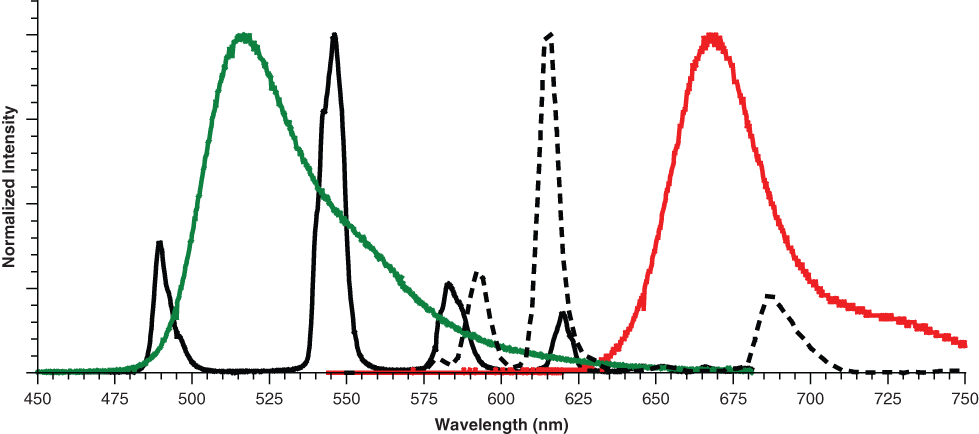
Emission spectra of terbium (Tb; solid black), europium (Eu; dashed black), fluorescein (green line), and Alexa Fluor® 647 (red line). The emission spectrum of green fluorescent protein (GFP; not shown) is similar to that of fluorescein. Tb→fluorescein/GFP fluorescence resonance energy transfer (FRET) is measured with a filter centered at 520 nm (25 nm bandwidth), and Eu→Alexa Fluor® 647 FRET is measured with a filter centered at 665 nm (10 nm bandwidth).
Materials and Methods
Purified recombinant proteins, assay reagents, and assay buffers were from Invitrogen, a part of Life Technologies Corporation (Carlsbad, CA). General chemicals and small-molecule inhibitors were obtained from Sigma-Aldrich (St. Louis, MO) or Calbiochem/EMD Biosciences (San Diego, CA). The EGFR and IKKβ assays were performed in 384-well low-volume nonbinding surface assay plates, part number 3676, from Corning (Corning, NY). The mTOR and PIK3C3 assays were preformed in a 384-well low-volume non-treated assay plate, part number 3674, from Corning. All experiments were repeated a minimum of 3 times in independent experiments.
Instrument settings
All assay plates were read on a Tecan Infinite F500 plate reader (Tecan, Foster City, CA) in time-resolved mode using a 100-µs lag time followed by a 200-µs integration time. For both Tb and Eu assays, an excitation filter at 340 nm with a 30-nm bandwidth was used. For the Tb readout, the fluorescein (or GFP) emission was collected with an emission filter centered at 520 nm with a 25-nm bandwidth, and the Tb emission was collected with an emission filter centered at 495 nm with a 10-nm bandwidth. Data were expressed as an emission ration of the fluorescein (or GFP) signal (520 nm) divided by the Tb signal (495 nm). For the Eu readout, the Alexa Fluor® 647 emission was collected with an emission filter centered at 665 nm with a 10-nm bandwidth, and the Eu emission was collected with an emission filter centered at 615 nm with a 10-nm bandwidth. All data were expressed as an emission ratio of the Alexa Fluor® 647 signal (665 nm) divided by the Eu emission (620 nm).
EGFR and IKKβ assays
All reactions were performed in a final volume of 10 µL at room temperature in kinase buffer A (Invitrogen part no. PV3189) consisting of 50 mM HEPES (pH 7.5), 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA. To determine the optimal kinase concentration for use in the assay, a dilution series of EGFR or IKKβ (IKBKB) was incubated with 200 nM Alexa Fluor® 647-poly GT, 200 nM fluorescein-IKK peptide, and 10 µM adenosine triphosphate (ATP). After a 1-h kinase reaction, a 10-µL solution of Eu-anti-PY20 antibody, Tb-anti-IκBα (pSer32) antibody, and EDTA, prepared in TR-FRET dilution buffer (20 mM Tris [pH 7.5], 0.02% NaN3, 0.01% NP-40), was added to each well for a final concentration of 2 nM for each antibody and 10 mM EDTA. After a 1-h equilibration period, the plate was read using the appropriate settings for LanthaScreen® Eu and LanthaScreen® Tb. For the inhibitor titrations, EGFR was used at a concentration of 8.8 ng/mL (0.1 nM), and IKKβ was used at a concentration of 24.4 ng/mL (0.2 nM).
mTOR and PIK3C3 assays
All reactions were performed in a final volume of 10 µL at room temperature in kinase buffer Q (Invitrogen part no. PV5125) consisting of 50 mM HEPES (pH 7.5), 1 mM EGTA, and 0.01% CHAPS supplemented with 2 mM dithiothreitol (DTT) and 2 mM MnCl2. To determine the optimal kinase concentration to use in the assays, a dilution series of mTOR (FRAP1) or PIK3C3 (hVPS34) was incubated with 300 nM GFP-4E-BP1, 100 µM PI:PS lipid substrate, and 10 µM ATP. After a 1-h kinase reaction, a 5-µL solution of Tb-anti-4E-BP1 (pThr46) antibody, Eu-anti-ADP antibody, Alexa Fluor® 647-ADP tracer, and EDTA prepared in TR-FRET dilution buffer was added to each well for a final concentration of 2 nM for each antibody, 3 nM Alexa Fluor® 647-ADP tracer, and 10 mM EDTA. After a 1-h equilibration period, the plate was read using the appropriate settings for Adapta® (identical to LanthaScreen® Eu) and LanthaScreen® Tb. For the inhibitor titrations, mTOR was used at a concentration of 360 ng/mL (2.1 nM), and PIK3C3 was used at a concentration of 800 ng/mL (6.2 nM). For the inhibitor dose-response curves using the Adapta® ADP detection readout, a “percent conversion” of ATP→ADP was calculated from the emission ratio using an ATP-ADP titration curve as per the manufacturer’s instructions.
Results and Discussion
Optimization of assay conditions for EGFR and IKKβ
The first step in developing a multiplexed kinase assay is to individually titrate each kinase in the presence of both the specific (intended) substrate and the substrate for the other kinase to be assayed and then to read out the assay using the detection reagents required for both assays. This is done to determine the appropriate amount of kinase required to achieve a suitable assay window (typically that which gives a 50%-80% change in the TR-FRET signal) as well as to ensure that the kinase of interest does not appreciably phosphorylate the substrate for the other kinase when present at this concentration. Because the TR-FRET-based assay signal is dependent on association of a phosphorylated peptide with an antibody that has a low-nM Kd (data not shown), an assay window of 50% to 80% represents <10% conversion of substrate to product, ensuring that the assay is performed within the linear range of the kinase reaction. Once appropriate kinase concentrations are determined individually, each titration can be repeated in the presence of the “optimal” amount of the other kinase to verify that there is no effect on the assay due to the presence of the second kinase.
EGFR activity was measured using an Alexa Fluor® 647–labeled poly-glutamate-tyrosine (poly-GT) substrate that is recognized upon phosphorylation using a Eu-labeled antiphosphotyrosine antibody (PY20), and IKKβ activity was measured using a fluorescein-IκBα-derived peptide substrate and a corresponding Tb-labeled anti-IκBα (pSer32) antibody. As shown in Figure 2A , titration of IKKβ resulted in a kinase-dependent increase in the Tb-dependent signal, with ~80% change in signal observed at 24.4 ng/mL IKKβ. Interestingly, an increase in the Eu-dependent signal (intended to be specific for the tyrosine kinase EGFR) was also seen at high concentrations of IKKβ ( Fig. 2B ), but this increase was negligible at the concentration of IKKβ that would be used in the assay (24.4 ng/mL). Whether this increase in signal was due to phosphorylation of the poly-GT substrate by IKKβ or by a contaminating tyrosine kinase was not explored. When the titration was repeated with EGFR, no increase in the Tb-dependent signal was observed at up to 500 ng/mL kinase ( Fig. 2A ), and an optimal concentration of 8.8 ng/mL EGFR was determined when reading the Eu-dependent signal ( Fig. 2B ). When the kinase titrations were repeated in the presence of the optimal concentration of the second kinase, there was essentially no change in either of the assay signals relative to that seen in the absence of the second kinase.
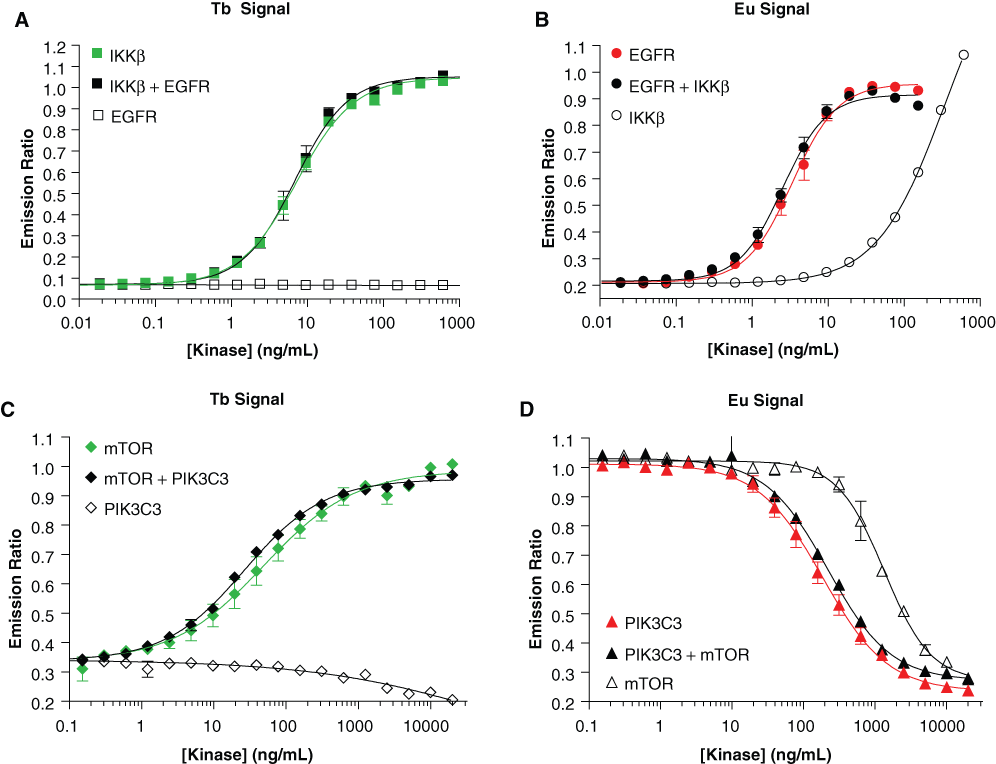
Optimization of kinase concentrations for multiplexed assays. Panels
Optimization of assay conditions for mTOR and PIK3C3
To develop a multiplexed assay to measure activity of mTOR and PIK3C3, we combined a traditional TR-FRET-based activity assay for mTOR (detection of acceptor-labeled product with donor-labeled, phosphospecific antibody) 7 with an assay for PIK3C3 that measures ADP formation through a competitive displacement of an ADP-derived tracer molecule from a Eu-labeled anti-ADP antibody. We reasoned that because the mTOR assay is expected to produce only nM amounts of ADP (because only nM amounts of phosphorylated product are necessary to saturate the 2 nM of anti-p-4E-BP1 antibody that is used), the amount of ADP formed in the mTOR catalyzed reaction should not affect the signal of the PIK3C3 catalyzed reaction, which would be optimized to produce larger amounts of ADP necessary to displace the tracer from the antibody. We also reasoned that because the Adapta® assay could be optimized such that <25% conversion of ATP to ADP would be required, changes in the amount of ATP present in the assay should have a negligible effect on the potency of mTOR inhibitors regardless of whether PIK3C3 was fully active or fully inhibited.
When mTOR was titrated into the assay system, a kinase-dependent increase in the Tb signal was observed, with an optimal assay concentration of 360 ng/mL ( Fig. 2C ). As was seen in the multiplexed EGFR-IKKβ assay, at high concentrations of mTOR, a signal change was observed in the Eu-dependent signal, likely due to substrate-independent, mTOR-catalyzed ATP hydrolysis that has been described previously ( Fig. 2D ). 7 However, this signal change was negligible at the concentration of mTOR to be used in the assay (360 ng/mL). Interestingly, when PIK3C3 was titrated into the assay system, there was a slight decrease in the Tb-dependent signal at high concentrations of PIK3C3 ( Fig. 2C ). However, this decrease was also negligible at the concentration of PIK3C3 that was to be used in the assay (800 ng/mL) as determined from the change in the Eu-dependent signal in the same reaction ( Fig. 2D ). As with the EGFR-IKKβ assay, when the kinase titrations were repeated in the presence of the optimal concentration of the second kinase, there was negligible effect seen on the changes in either assay signal relative to those seen in the absence of the second kinase.
Determination of compound potencies in multiplexed assays
Following the optimization of the kinase concentrations for the multiplexed experiments, each set of kinases was tested against a group of well-described inhibitors for that pair of kinases. For the IKKβ/EGFR set, the IKKβ-specific inhibitors BMS-345541 and IKK Inhibitor VII were used, as were the EGFR-specific inhibitors erlotinib and gefitinib. The broad-spectrum inhibitor staurosporine was tested as well. Inhibition of IKKβ activity with either of the IKK-selective small-molecule inhibitors or with staurosporine showed identical results when IKKβ was assayed alone or when multiplexed with the EGFR assay ( Fig. 3 and Table 1 ), and erlotinib and gefitinib showed no inhibition of IKKβ at up to 1 µM. Similar results were also observed when EGFR was inhibited with selective EGFR inhibitors or staurosporine: no significant difference was observed between the single kinase and the multiplexed formats. For the mTOR/PIK3C3 pairing, 3 inhibitors that had previously been shown to inhibit PI3K family kinases—PI-103, AS-605240, and LY249002—were tested. Again, no significant differences were observed between the single kinase and multiplexed formats ( Fig. 4 and Table 1 ). In separate experiments (not shown), no significant change in Z′ was observed between individual and multiplexed assays, with all assays exhibiting a Z′ of >0.85.
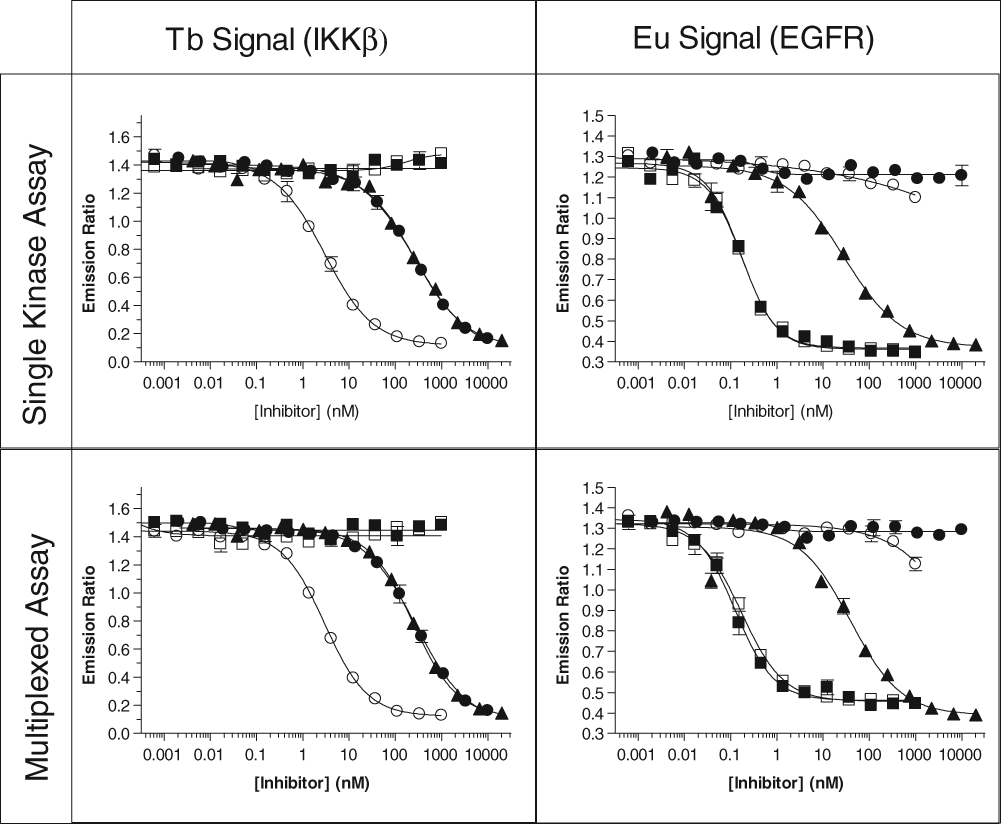
Representative dose-response curves for various inhibitors of IKKβ and EGFR alone, or multiplexed together, for both the terbium (Tb) and europium (Eu) LanthaScreen® assays. Legend: (■) erlotinib; (□) gefitinib; (▲) staurosporine; (●) BMS-345541; (○) IKK Inhibitor VII.
Comparison of IC50 Values for Selected Inhibitors as Determined in the Multiplexed or Single-Assay Formats for Both EGFR and IKKβ, as Well as the mTOR and PIK3C3 Example Systems
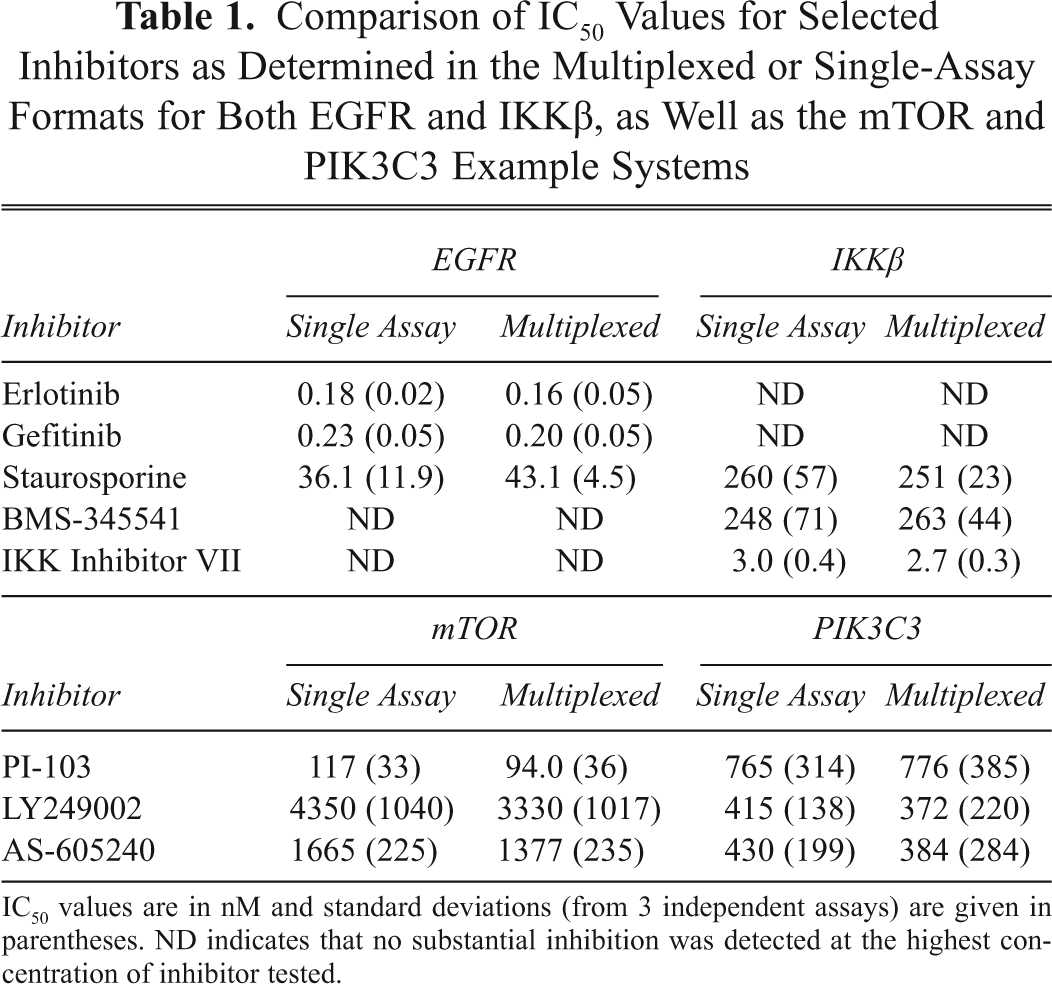
IC50 values are in nM and standard deviations (from 3 independent assays) are given in parentheses. ND indicates that no substantial inhibition was detected at the highest concentration of inhibitor tested.
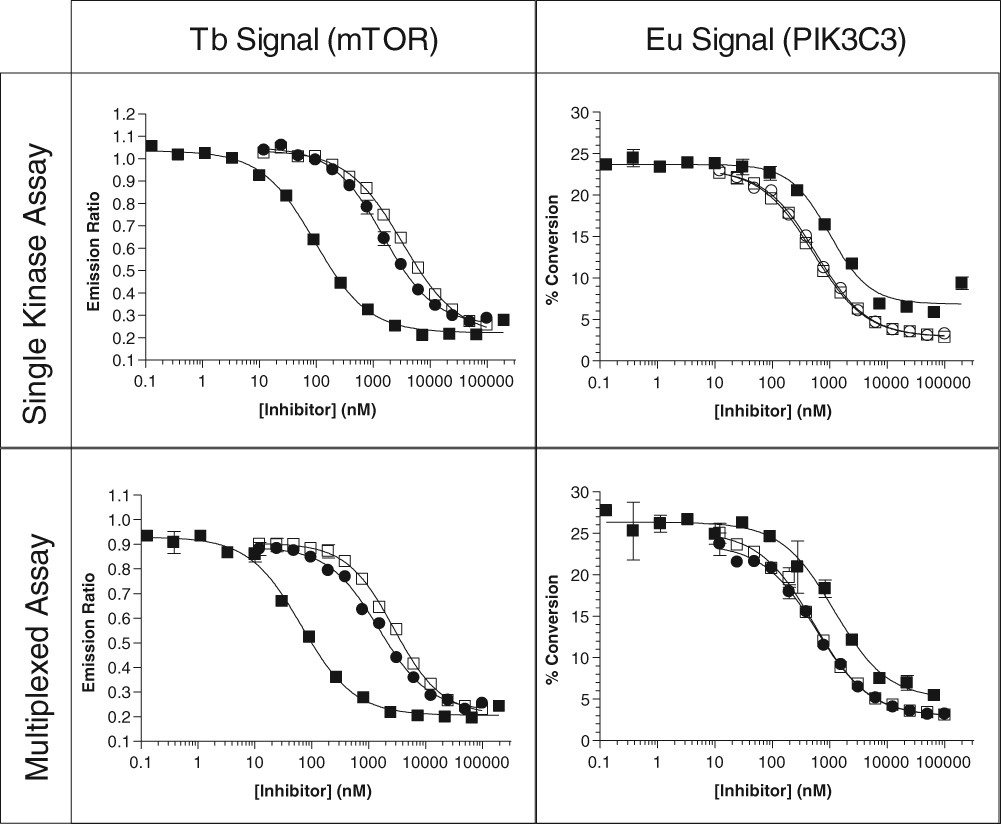
Representative dose-response curves for various inhibitors for mTOR and PIK3C3 alone, or multiplexed together, for both the terbium (Tb)–based LanthaScreen® and europium (Eu)–based Adapta® assays. Legend: (■) PI-103; (□) LY249002; (●) AS-605240.
Considerations in the development of multiplexed kinase assays
The ability to perform multiple kinase assays in the same well requires that both the reaction conditions and the detection conditions have a limited amount of cross-interference with one another. In the examples presented here, a small degree of cross-interference was observed: in the IKKβ—EGFR example, high concentrations of IKKβ showed an effect on what was intended to be an EGFR-specific signal that was dependent on phosphorylation of a “generic” tyrosine kinase substrate (whereas IKKβ is classified as a serine/threonine kinase). However, the concentration of IKKβ required for the assays was such that the amount of interference seen in the assay was negligible, as evidenced by the similarity in EGFR assay results in the presence or absence of IKKβ. Similarly, ADP formation by high concentrations of mTOR in the absence of its protein substrate 4E-BP1 interfered with what was intended to be a PIK3C3-specific signal.
In each of the examples presented here, substrates were chosen with the intention to minimize cross-phosphorylation by the other kinase present in the assay. Although a “generic” tyrosine kinase substrate was successfully used when multiplexing a tyrosine kinase assay with a serine/threonine kinase assay, it would be problematic to use such a substrate when assaying multiple tyrosine kinases in the same well. In addition, the quality and specificity of the antibodies are critical, as any cross-reactivity with the substrate for the second kinase would be expected to affect assay results. The PY20 antibody used here shows broad specificity for phosphorylated tyrosine within the context of a wide range of sequences, so when multiplexing tyrosine kinases with “specific” substrates for those kinases, it is likely that 2 different sequence-specific antiphosphotyrosine antibodies would be necessary (unless there was a fortuitous lack of affinity of PY20 for one of the phosphorylated substrates). The requirement for the use of “orthogonal” substrates in the multiplexed assay presented here may limit the utility of this strategy, especially for closely related kinases that may have overlapping substrate specificities.
An additional important consideration is the composition of the other assay components such as buffer and ATP concentrations. Because the IC50 of an ATP competitive inhibitor is dependent on the concentration of ATP present (relative to the ATP Km for the kinase), as defined by the Cheng-Prusoff equation (IC50 = Ki*(1 + [ATP]/Km), where Ki is a constant that describes the binding affinity of the inhibitor and Km is the Km for ATP), 9 inhibitor potency will be weakened when the ATP concentration is increased relative to Km. Although a given inhibitor will display a potency (IC50 value) that varies by less than 2-fold when assayed against a kinase in the presence of an ATP concentration that is at or below the ATP Km value, the observed potency rapidly shifts (weakens) according to 1 + [ATP]/Km. In the 2 multiplexed assays presented here, an ATP concentration of 10 µM was used, which was within 2-fold of the ATP Km determined in separate experiments (data not shown). In most screening settings that are designed to identify all candidate molecules (even weak inhibitors), the concentration of ATP used is at or below the ATP Km value for the targeted kinase. Thus, if the desire is to identify even weak inhibitors of the second kinase in an assay, the ability to do so will be limited if the ATP concentration used is too high relative to the second kinase’s ATP Km value. Although the ATP concentration used in the assay may be lowered, other practical factors such as a decreased reaction rate will then come into effect.
A final consideration that may come into play when multiplexing kinase assays is buffer composition. Although we have found that nearly all protein kinases perform well in a standard buffer containing HEPES, BRIJ-35, MgCl2, and EGTA, PI3K family members (including mTOR) can be rather sensitive to buffer components, including the buffer salt, the detergent, and the divalent metal. In the course of developing individual activity assays for mTOR and PIK3C3 (data not shown), we observed that both kinases were substantially more active using Mn2+ rather than Mg2+ as the divalent metal and that BRIJ-35 negatively affected activity, with mTOR performing best using polysorbate 20 and PIK3C3 performing best using CHAPS as the detergent. When the 2 kinases were assayed simultaneously, overall assay performance was better using CHAPS rather than polysorbate 20 as the detergent (data not shown), thereby influencing our choice of buffer in the assay. Although a suitable assay buffer could be readily determined for each set of kinases examined here, it is likely that there are some kinase combinations, especially those involving lipid kinases, that development of a suitable assay buffer could be a complex undertaking.
Conclusions
The unique spectral properties of lanthanide chelates, including their large Stoke’s shift and sharp emission peaks that span a broad range of wavelengths, with clear “silent” regions between those peaks, allow them to serve as FRET donors to a wide variety of common acceptor fluorophores that span the spectrum from “green” to near-infrared emitters. These properties have previously been exploited to develop assays in which a single donor molecule, either Tb or Eu, has been used to donate to more than one acceptor fluorophore. Here we have taken advantage of these properties to develop assays in which multiple lanthanides are used as FRET donors in a single well, with independent readouts. We have used this strategy to independently monitor multiple kinase reactions from within a single assay well, which may be useful in generating a “first-pass” look at compound specificity when screening compound libraries and also may be compatible with other commonly used kinase assay formats. 10,11 Because both Tb- and Eu-based TR-FRET assays across many target classes have been well described in the literature, we expect that this strategy can be easily adapted to a range of targets. In addition, applications can be envisioned in which the second assay performed in the well may be a control to improve confidence in the results from the primary assay, and studies toward such applications are under way in our laboratories.