
Abstract
African trypanosomiasis, leishmaniasis, and Chagas disease are 3 neglected tropical diseases for which current therapeutic interventions are inadequate or toxic. There is an urgent need to find new lead compounds against these diseases. Most drug discovery strategies rely on high-throughput screening (HTS) of synthetic chemical libraries using phenotypic and target-based approaches. Combinatorial chemistry libraries contain hundreds of thousands of compounds; however, they lack the structural diversity required to find entirely novel chemotypes. Natural products, in contrast, are a highly underexplored pool of unique chemical diversity that can serve as excellent templates for the synthesis of novel, biologically active molecules. We report here a validated HTS platform for the screening of microbial extracts against the 3 diseases. We have used this platform in a pilot project to screen a subset (5976) of microbial extracts from the MEDINA Natural Products library. Tandem liquid chromatography–mass spectrometry showed that 48 extracts contain potentially new compounds that are currently undergoing de-replication for future isolation and characterization. Known active components included actinomycin D, bafilomycin B1, chromomycin A3, echinomycin, hygrolidin, and nonactins, among others. The report here is, to our knowledge, the first HTS of microbial natural product extracts against the above-mentioned kinetoplastid parasites.
Keywords
Introduction
Human African trypanosomiasis, leishmaniasis, and American trypanosomiasis (Chagas disease) are just 3 of the many neglected tropical diseases that are responsible for causing thousands of deaths and underproduction leading to poverty in the world’s poorest continents. Human African trypanosomiasis occurs in 2 clinical forms: a chronic form caused by Trypanosoma brucei gambiense (mostly found in West and Central Africa), which accounts for more than 98% of reported cases, and an acute form caused by Trypanosoma brucei rhodesiense (mainly found in East and South Central Africa). Without treatment, both types of parasites penetrate the blood–brain barrier and invade the CNS, which is manifested in complex symptoms that lead to the death of patients. 1 Trypanosoma cruzi is the parasite responsible for Chagas disease (mostly in Latin American countries), which is characterized by 2 clinical phases: a short, acute phase defined by patent parasitemia, and a long, progressive chronic phase that can manifest symptoms after several years. 2 Leishmaniasis is caused by different species of the genus Leishmania. Three major clinical forms of the disease are visceral, cutaneous, and mucocutaneous leishmaniasis, which differ in immunopathologies and degree of morbidity and mortality. 3 Importantly, visceral leishmaniasis caused by Leishmania donovani is fatal if untreated. 3 Millions of people living in areas where these diseases are endemic are in desperate need of novel, safe, and affordable drugs to help alleviate their suffering.
In the drug discovery and development process, it is highly critical to have access to pure, highly potent, and low-toxicity compounds to be screened within the shortest possible time using the most cost-effective process. Most drug development methods rely on phenotypic and target-based HTS of synthetic chemical libraries to search for active compounds for further development into potent drugs, 4 but such synthetic libraries are often limited in structural diversity and novelty. Natural products offer an alternative source of highly underexplored chemical entities with privileged bioactive molecules that could be used as templates for the synthesis of novel drugs. The possibility of obtaining them by scaling up the fermentations of the producing microbial strain is an additional advantage that microbial natural products offer as compared to those from other sources, such as plants or marine invertebrates. After elucidation of the molecular structures of many natural products, chemists are often able to propose a total synthesis, rather than isolate them from their natural sources, thereby markedly reducing the cost of mass drug production. It has been estimated that about 60% of the drugs available now, including household names such as artemisinin, camptothecin, lovastatin, maytansine, paclitaxel, penicillin, reserpine, and silibinin, were directly or indirectly derived from natural products.5–7 Traditionally, plants have been excellent sources of antimalarial compounds. Artemisinin, the current World Health Organization–recommended sesquiterpene endoperoxide, which is used in combination therapies as a first-line treatment of malaria, is a natural product isolated from Artemisia annua (Asteraceae), a medicinal plant that has been used for more than a millennium in China. There are also numerous natural products in the drug discovery pipeline with promising anti-kinetoplastid activities. 8
The search, however, for active compounds from whole crude extracts, as is the case with natural product drug research, is both time consuming and capital intensive. For that reason, it is very critical to develop quick, efficient, and high-quality high-throughput primary screening assays integrated with chemistry platforms for early de-replication of known compounds to guarantee that only extracts with relevant pharmacological properties are selected for further development downstream. We report here on a very robust and validated pilot HTS platform for the screening of microbial extracts against Trypanosoma brucei brucei, L. donovani, and T. cruzi, the kinetoplastid parasites respectively responsible for human African trypanosomiasis, leishmaniasis, and Chagas disease. The purpose of this ongoing project is to identify, characterize, and develop potent natural product–based drug leads against these neglected tropical diseases.
Materials and Methods
Reagents
Fetal bovine serum (FBS), L-glutamine, sodium pyruvate, MEM nonessential amino acids, penicillin–streptomycin, and TrypLE Express were purchased from Invitrogen Gibco, Inc. (Life Technologies, Carlsbad, CA). NP40 was purchased from Thermo Scientific (Rockford, IL). Chlorophenol red-β-D-galactopyranoside (CPRG), phorbol 12-myristate 13-acetate (PMA), resazurin, amphotericin B, pentamidine, benznidazole, chromomycin A3 and bafilomycin B1, actinomycin D, diphenyl tetrazolium bromide (MTT), and methyl methane sulfonate (MMS) were obtained from Sigma-Aldrich (St. Louis, MO). Echinomycin and hygrolidin were purchased from Tebu Bio (Smithfield, NSW, Australia), and nonactin was provided by Santa Cruz Biotechnology (Dallas, TX).
Parasite Culture
T. cruzi Tulahuen C4 strain, expressing the β-galactosidase gene (LacZ),9,10 was cultured in RPMI-1640 supplemented with 10% inactivated FBS (iFBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37 °C and 5% CO2. L6 rat skeletal muscle cells were used as host cells for infection with transgenic T. cruzi trypomastigotes. T. b. brucei Lister 427 bloodstream form parasites were routinely grown in HMI9 medium 11 supplemented with 10% iFBS. Axenic L. donovani MHOM/ET/67/HU3 amastigote parasites (provided by Dr. L. Maes from LMPH, University of Antwerp, Belgium) were grown in Schneider medium supplemented with 20% iFBS, pH 5.4, at 37 °C and 5% CO2. For the Leishmania intracellular assay, L. donovani MHOM/ET/67/HU3 parasites stably expressing luciferase activity in THP-1 host cells were used. The THP-1 cells were grown at 37 °C and 5% CO2 in RPMI-1640 supplemented with 10% iFBS, 2 mM glutamate, 100 U/mL penicillin, and 100 μg/mL streptomycin.
β-D-Galactosidase Transgenic T. cruzi Assay
A Thermo Scientific Multidrop Combi dispenser (MTX Lab Systems, Vienna, VA) was used to dispense 55 µL of T. cruzi amastigote–infected L6 cell culture (2×103 infected L6 cells per well) into the 384-well Corning assay plates (Corning Inc., Corning, NY) already containing 5 µL of the microbial extracts to be screened [final extract concentration of 1/60× whole broth equivalent (WBE)] and controls. The plates were incubated at 37 °C for 96 h. Then, 15 µL of 100 µM CPRG and 0.1% NP40 diluted in PBS were added to each well, and the plates were incubated for 4 h at 37 °C in the dark. Absorbance at 585 nm was measured in an Envision plate reader (PerkinElmer, Waltham, MA). Extract activities were normalized using the in-plate negative (benznidazole at 10 µg/mL) and positive (0.167% DMSO) growth controls.
Resazurin-Based T. b. brucei Assay
Forty-five microliters (45 μL) of parasite culture containing 5×102 bloodstream form T. b. brucei parasites per well were added to 384-well Corning assay plates already containing 5 µL of the microbial extracts (final concentration of 1/1500 × WBE) and controls, and incubated for 72 h at 37 °C. Ten microliters (10 μL) of resazurin at working concentration [0.02% by mass prepared in Milli-Q water (Millipore, Billerica, MA)] were added per well, and the plates were further incubated for 6 h at 37 °C. The final fluorescence was determined at 550–590 nm. Pentamidine at 40 nM and 0.0067% DMSO were used as negative and positive growth controls, respectively.
Resazurin-Based L. donovani Axenic Amastigotes Assay
Forty-five microliters (45 μL) of parasite culture containing 1×104 axenic amastigote parasites per well were dispensed into 384-well Corning assay plates already containing 5 µL of the microbial extracts (final concentration of extract at 1/200 × WBE) and controls. The plates were incubated for 72 h at 37 °C. Ten microliters (10 μL) of resazurin at working concentration (0.02% by mass prepared in Milli-Q water) were dispensed per well, and fluorescence at 550–590 nm was monitored after 24 h of incubation at 37 °C. Plate normalizing was done using in-plate negative (amphotericin B at 10 µM) and positive (0.05% DMSO) growth controls.
Secondary Intracellular Amastigotes Screening Assay for L. donovani
THP-1 cells were differentiated into macrophages with 20 ng/mL of PMA in a 96-well plate at 3×104 cells per well for 48 h, followed by 24 h of culture in fresh medium. Afterward, these cells were infected with transgenic stationary L. donovani MHOM/ET/67/HU3 promastigotes expressing the luciferase gene. A volume of 100 µL of crude extracts (final concentration of 1/200 × WBE) was added to each well, and the plates were incubated at 37 °C for 72 h. Luminiscence was measured using the Promega kit luciferase assay system (Promega, Madison, WI).
Data Analysis
Pure compounds and extract activities were calculated automatically using the Genedata Screener software (Genedata AG, Basel, Switzerland), and the percentage inhibition of each extract (or compound) was determined by Equation 1, integrated in the Genedata Screener software:
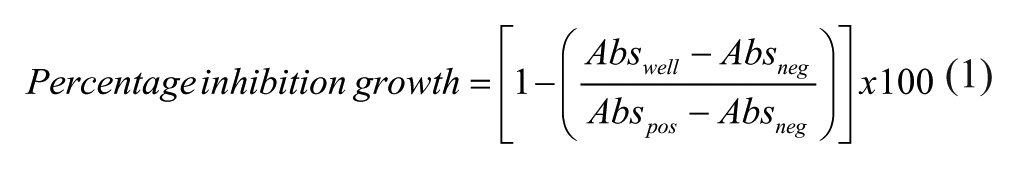
where Abswell is the absorbance measured per specific well; and Abspos and Absneg are the average absorbances measured for the positive and negative controls, respectively.
The RZ’ factors obtained for the T. cruzi, T. b. brucei, and L. donovani assays were 0.83, 0.74, and 0.70, respectively.
Determination of IC50 Values and Cytotoxicity for Pure Known Compounds
IC50 values of commercial known compounds were obtained from dose–response curves in the assays targeting the 3 parasites. Compound concentrations were started at 50 µM, and twofold serial dilutions were performed until nanomolar concentrations were attained (DMSO concentration lower than 0.5%). The IC50 values were calculated as the compound concentration that inhibits the growth of parasites by 50% using the online Genedata Screener software (Genedata AG, Basel, Switzerland). Cytotoxicity was assessed using the HepG2 liver cell line and the MTT assay. HepG2 cells (human liver carcinoma, CCL-8065) were grown in American Type Culture Collection (ATCC)-formulated Eagle’s Modified Essential Medium (MEM) with 10% qualified iFBS, 2 mM L-glutamine, 1 mM sodium pyruvate, and 100 µM MEM nonessential amino acids at 37 °C under a humidified atmosphere of 5% CO2.
Ten thousand cells per well were plated in 96-well plates with a cell culture robotic system, SelecT (TAP Biosystems, Royston, UK). After 24 h of incubation, compounds were added with the automated liquid-handling system Biomek FX (Beckman Coulter, Pasadena, CA), and plates were incubated for an additional 72 h. The MTT assay was performed, and plates were read in a Victor2TM Wallac spectrofluorometer (PerkinElmer). 12
Microbial Extracts Collection
For the primary screening campaign, a subset of the MEDINA microbial extracts collection consisting of 5976 microbial extracts (3568 from actinomycetes and 2408 from fungi) was used. This subset of extracts was previously found to inhibit less than 50% growth of HepG2 cells when tested at 1/40 × WBE final concentration for 24 h. The microbial extracts were obtained from a selection of producing strains that were cultivated for 7 and 14 days at 28 °C in the case of actinomycetes and for 21 and 28 days at 22 °C for fungi. The secondary metabolites in the broths were extracted with acetone (1:1) by mixing 10 mL of culture broth with 10 mL acetone and shaking in an orbital shaker (Adolf Kuhner AG, Birsfelden, Switzerland) for 1 h. The extracts were then centrifuged at 1500 × g for 15 min, and the supernatant was transferred to 16 mm glass tubes (EPA vials) and evaporated until half of the original volumes remained. DMSO was then added to each extract to a final crude concentration of 2 × WBE and 20% DMSO. Five hundred microliters (500 µL) of these crude extracts were stored at −20 °C in 96-well ABgene plates until needed.
Primary Screening and Dose Response Experiments
All of the extracts were first screened against each parasite, and those exhibiting more than 70% inhibition of growth were selected as hits and confirmed in 3 independent assays. Plates included the reference compounds benznidazole, amphotericin B, and pentamidine for T. cruzi, L. donovani, and T brucei, respectively. The extracts selected from this stage were then tested in a 5-point dose response assay; the first point was the initial dilution previously described for each assay, and the 4 subsequent points were twofold serial dilutions from each point. Extracts that exhibited a minimum of 70% inhibition at a fourfold dilution in dose response experiments were selected for de-replication by tandem liquid chromatography mass spectrometry (LC-MS).
LC-MS and Database Matching of Known Secondary Metabolites
Two microliters (2 μL) of the selected extracts were analyzed by LC-MS. Analysis was performed on an Agilent (Santa Clara, CA) 1100 single Quadrupole LC-MS, using a Zorbax SB-C8 column (2.1×30 mm), maintained at 40 °C and with a flow rate of 300 µL/min. Solvent A consisted of 10% acetronitrile and 90% water with 1.3 mM trifluoroacetic acid and 1.3 mM ammonium formate, whereas solvent B was 90% acetronitrile and 10% water with 1.3 mM trifluoroacetic acid and 1.3 mM ammonium formate. The gradient was started at 10% B and went to 100% B in 6 min, was kept at 100% B for 2 min, and was returned to 10% B for 2 min to initialize the system. Full diode array ultraviolet (UV) scans from 100 to 900 nm were collected in 4 nm steps at 0.25 s/scan. The eluting solvent was ionized using the standard Agilent 1100 ESI source adjusted to a drying gas flow of 11 L/min at 325 °C and a nebulizer pressure of 40 psig. The capillary voltage was set to 3500 V. Mass spectra were collected as full scans from m/z 150 to 1500, with 1 scan every 0.77 s, in both positive and negative modes. Database matching was performed using an in-house developed application in which the diode array detection, retention time, and positive and negative mass spectra of the active samples were compared to the UV-LC-MS data of known metabolites stored in a proprietary database when metabolite standard data were obtained using the exact same LC-MS conditions as the samples under analysis.
Bioassay-Guided Extract Fractionation
Extracts with LC-MS profiles suggestive of containing active novel compounds were regrown in 100 mL volume. An equal volume of acetone was added to each 100 mL fermentation, and the mixture was homogenized and shaken in a Kühner shaker at 220 rpm for 3 h. Each extract was split into 4 EPA vials, and 13 mL volumes were transferred from each vial into separate glass tubes and evaporated under a nitrogen stream until only 3.2 mL remained (extract concentration 2 × WBE). From each of these glass tubes, 5×520 µL extract volumes were transferred into 5 ABgene plates using a Multiprobe II liquid handler (PerkinElmer). Aliquots from 1 of these 5 ABgene plates were bio-assayed to confirm activity before storing the plate at −20 °C as a reference plate. The remaining 4 ABgene plates of each extract identified as active were centrifuged, each of the extracts was pooled together in an EPA vial and loaded onto a 10 g SP207ss charged column, and the flow-through was collected and stored at −20 °C. The column was then washed with an equal volume of Milli-Q water, and the aqueous flow-through also collected and saved in a new EPA vial at −20 °C. The column was finally eluted to dryness with 20 mL of acetone. The collected solutions were evaporated under a nitrogen gas to give a dry crude extract.
Each crude was then reconstituted in 400 µL DMSO, and half of this volume was injected and separated into 80 fractions using a linear gradient of 5–100% acetonitrile–H2O with 0.1% trifluoroacetic acid in reverse-phase, semipreparative, high-performance liquid chromatography (HPLC) using Agilent column Zorbax RX-C8, 5 µm, 9.4×250 mm, with UV detection at 210 nm (Gilson, Middleton, WI). The 80 fractions collected were evaporated to dryness in an ABgene plate using a Genevac evaporator (Genevac, Ipswich, UK). These fractions were then reconstituted in 30 µL of DMSO and 150 µL of Milli-Q water to make a final solution at 20% DMSO concentration. The fractions were then tested for activity. Active fractions from this stage were then analyzed by LC-MS.
Results
Optimization of Assays in a 384-Well Format
Our first goal was to optimize simple and reliable assays for HTS in a 384-well format to quantify the growth pattern of the 3 parasites used in the screening campaign, namely, T. cruzi, T. b. brucei, and L. donovani. These parasites have complex life cycles, with several stages that occur between the insect vector and vertebrate host. Our assays were developed using the mammalian stage of the parasites, in particular the intracellular amastigote form of T. cruzi, the bloodstream form of T. b. brucei, and both axenic and intracellular amastigote forms of L. donovani. For this study, we used T. cruzi parasites genetically engineered to express the E. coli β-galactosidase gene, lacZ.
9
L6 cells were infected with the trypomastigote forms of T. cruzi before exposure to the microbial extracts. In this way, we ensured that the extracts target the intracellular dividing amastigotes. The 384-well HTS assay was optimized starting from the previously reported 96-well HTS assay.
13
Two different parameters were considered for optimizing the primary screening assay, namely, the number of L6 host cells and the number of intracellular parasites. Three different amounts of L6 host cells and parasites per well were tested (2×103, 5×103, and 15×103), and 2×103 L6 cells with 2×103 intracellular parasites per well (1:1 host-to-parasite ratio) were chosen and used as the most suitable combination (
On the other hand, Leishmania has 2 major life cycle stages: promastigotes, which are easily cultured in suspension, and amastigotes, which are more difficult to maintain in vitro because they require macrophages as host cells. It has been previously shown that HTS screening using either axenic amastigotes or intracellular amastigotes of L. donovani identifies a similar number of hits.
14
Bearing in mind these considerations, we chose to use adapted axenic amastigotes of L. donovani strain MHOM/ET/67/HU3 for the HTS. We adapted the assay to 384-well microtiter plates from a 96-well assay described previously.
15
We performed the assay using a different number of axenic parasites per well (5×103, 104, 2.5×104, and 5×104) and selected 104 axenic parasites per well because that gave the optimal fluorescence level (
The bloodstream form of T. b. brucei strain Lister 427 was used for the HTS because it is better adapted to in vitro culturing than the human pathogenic T. b. rhodesiense or gambiense subspecies. In addition, any compound active against T. b. brucei will likely show a similar effect in T. b. gambiense, which is the main etiological agent of human African trypanosomiasis. Indeed, the drugs available for animal treatment of T. b. brucei, such as diminazene aceturate (Berenil), effectively kill T. b. gambiense. Eflornithine, used as a second-stage treatment for T. b. gambiense infection, was also shown to clear T. brucei parasitemia in animal experimental models. Pentamidine, the default chemotherapy for T. b. gambiense in the first clinical stage, is routinely used as a control against T. b. brucei both in vivo and in vitro.16,17 The genomes of both subspecies share more than 99% homology in their coding regions.
18
We used parasites in the logarithmic phase of growth (105–106 cells/mL) in all of our optimization steps. The initial culture was diluted to different starting concentrations (102, 2.5×102, 5×102, and 103), and a concentration of 5×102 cells per well was chosen because it gave a better signal-to-background ratio (
To determine the optimal extract concentration to obtain a reasonable hit rate, different extract dilutions were tested for each parasite. The 2 main factors considered when establishing the optimum extract concentrations were that (1) the crude extracts to be tested were prepared in 20% DMSO and that cytotoxic concentrations of this solvent should be avoided; and (2) a hit rate of about 2–5% is desirable, a hit being defined as an extract that, at a given dilution, exhibits a growth inhibition of ≥ 70%. Using the stated guidelines, dilutions of 1/60, 1/200, and 1/1500 were chosen for the assays of T. cruzi, L. donovani, and T. b. brucei, respectively (
Screening Campaigns and Hit Identification
From the primary screening of 5976 noncytotoxic microbial extracts from the MEDINA microbial extract collection (general workflow shown in
Fig. 1
), 228 extracts (3.82%) were initially identified as anti–T. cruzi hits, 134 (2.24%) as anti–L. donovani hits, and 91 (1.52%) as anti–T. b. brucei hits. We assessed the activities of the selected extracts again in triplicate by cherry-picking and confirmed 119, 64, and 51 extracts as anti–T. cruzi, anti–L. donovani, and anti–T. b. brucei hits, respectively (
Table 1
). Next, we used dose response experiments to investigate extract potency and selected only those exhibiting minimally 70% growth inhibition at a fourfold extract dilution (
Fig. 2
). Using this criterion, 50 anti–T. cruzi, 30 anti–L. donovani, and 21 anti–T. b. brucei extracts were selected as hits (
Table 1
) for LC-MS de-replication. Extracts showing higher dose–response curve slopes are preferred because these are expected to contain components that severely suppress parasite growth. For L. donovani, we used the disease-relevant intracellular amastigote assay (96-well format) as a secondary method to select 26 extracts out of the 30 initially selected after dose response for further processing. Taking into account the fact that there were several extracts with overlapping activities among the different parasites, the total number of active extracts that were further processed from this stage came to 80. Details on the number of hits identified through the screening procedure and those that are common to the different parasites are shown in
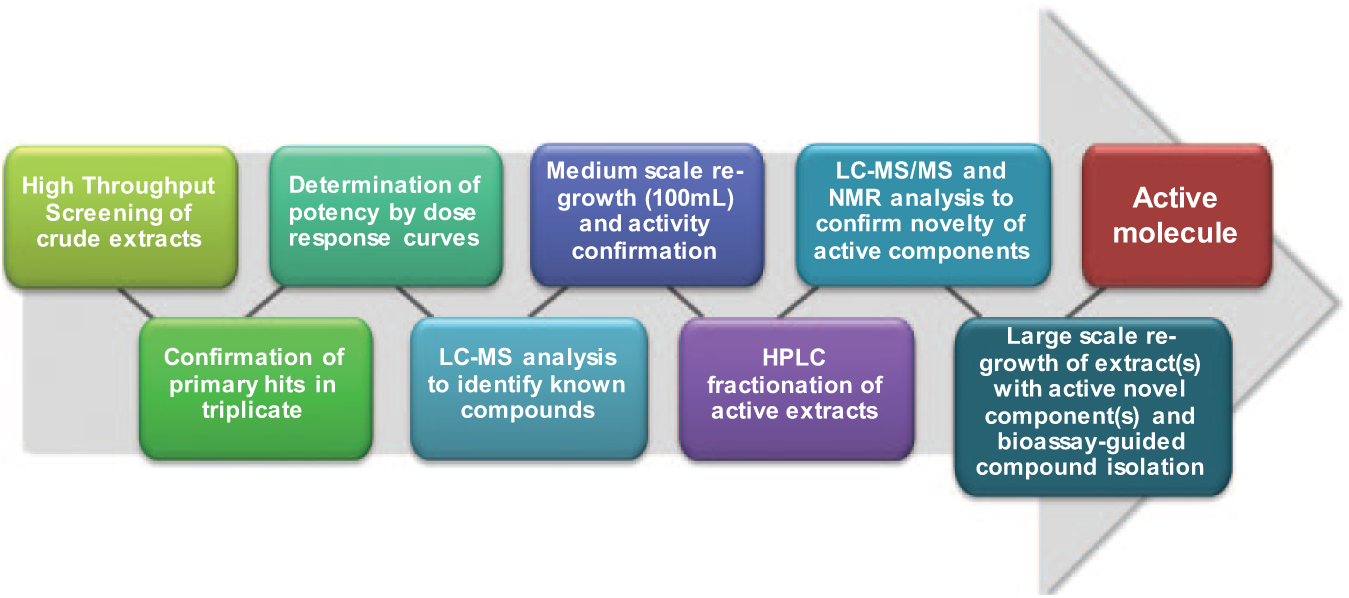
Schematic representation of the high-throughput screening (HTS) and integrated de-replicating chemistry platform used in discovering drug leads from microbial extracts.
Number of Extracts Identified as Hits at Different Stages of the Screening Process.
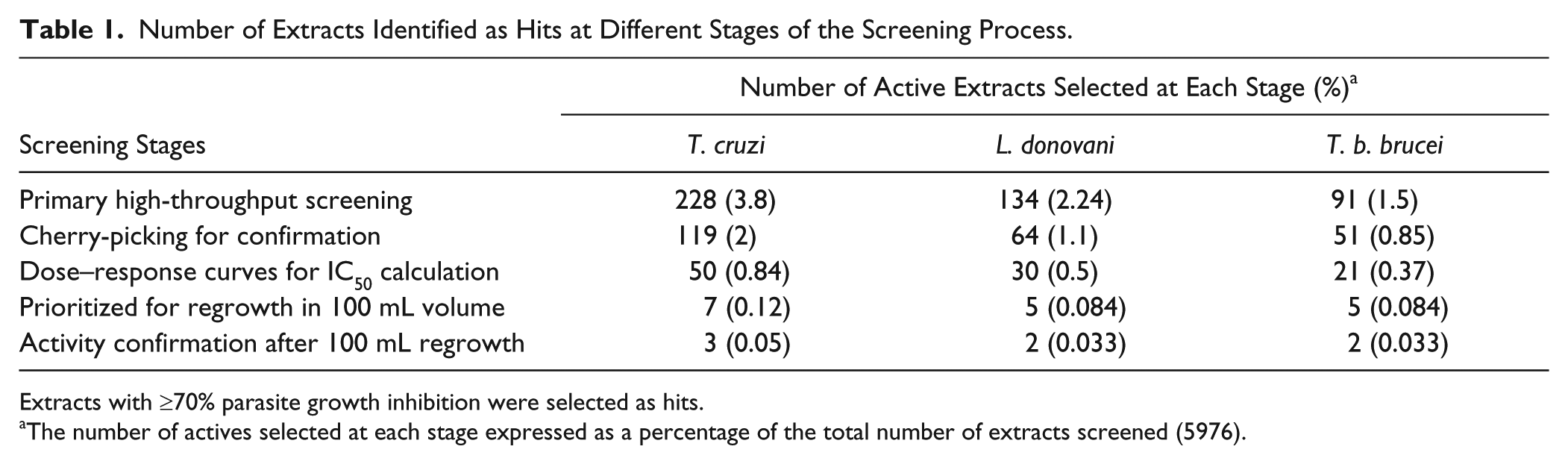
Extracts with ≥70% parasite growth inhibition were selected as hits.
The number of actives selected at each stage expressed as a percentage of the total number of extracts screened (5976).
(
Early LC-MS De-replication
A database search performed using our in-house application, which matches UV-LC-MS data of the metabolites in the active extracts to UV-LC-MS data of known metabolites stored in our proprietary database obtained using the exact same LC-MS conditions, did not identify any of the metabolites in 48 active extracts, and therefore these extracts were classified as potentially containing novel active components. The remaining 32 active extracts from the dose response stage gave profiles associated with known secondary metabolites, such as actinomycin C2/D, azalomycin F3/F4, bafilomycin B1/C/E, chromomycin A3, cordyol C, diorcinol, echinomycin, eupenifeldin, grisorixin, hygrolidin, lobophorin A/B, lysolipin I, nigericin, nonactins, polyenes (N = 7), quinomycin C, roridin H, rugulosin, skyrin, norcardamine, and violaceol I/II. Data indicating the distribution of known and unknown metabolites in the active extracts against the 3 different parasites are shown in
Figure 3
. Six of these metabolites (structures indicated in
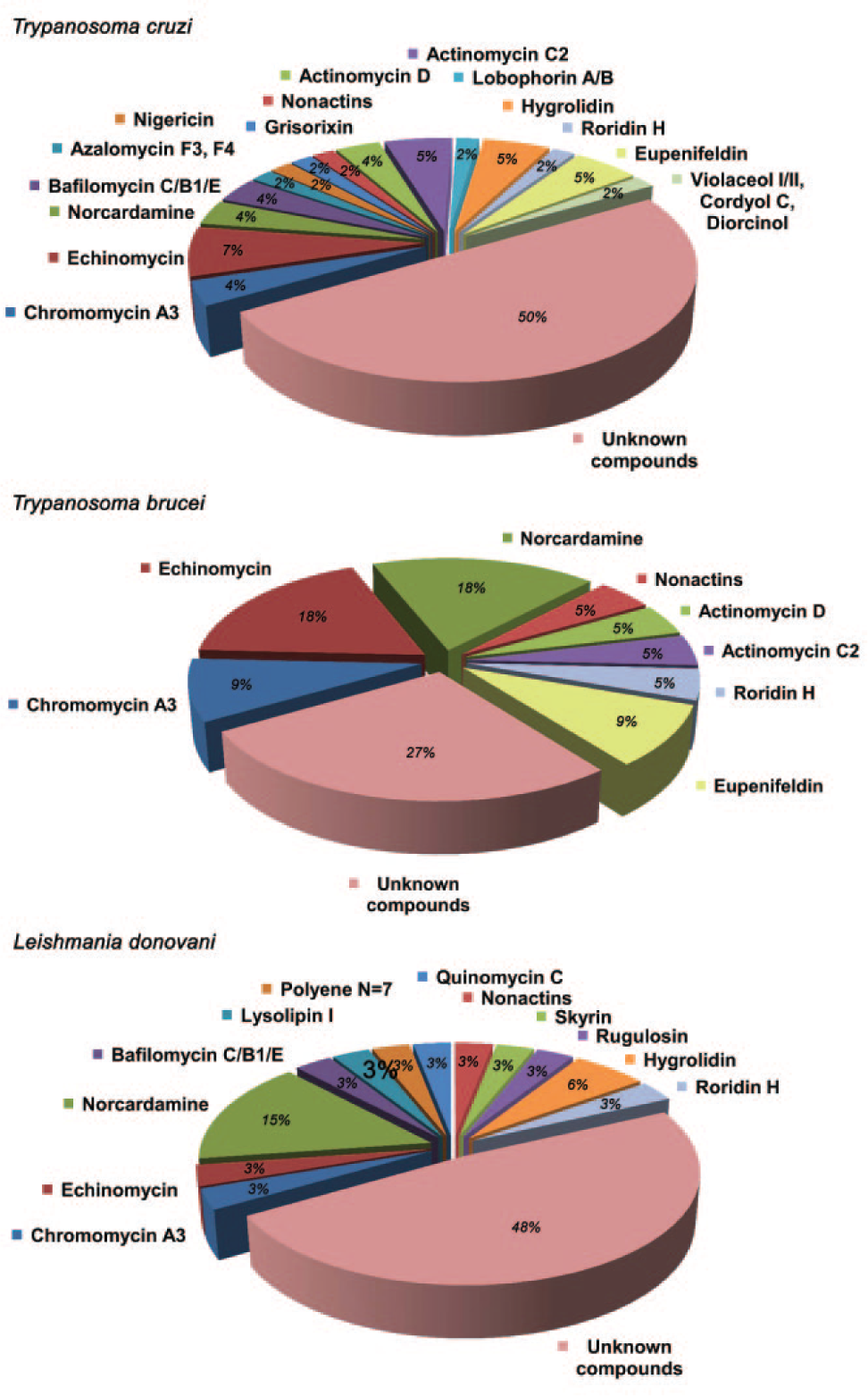
Frequency of appearance of known compounds after liquid chromatography mass spectrometry (LC-MS) identification in hit extracts active against Trypanosoma cruzi, Trypanosoma brucei brucei, and Leishmania donovani.
IC50s of 6 Known Compounds Identified by Liquid Chromatography Mass Spectrometry (LC-MS) Obtained in the 3 Antiparasitic Screens and Cytotoxicity Assays.a
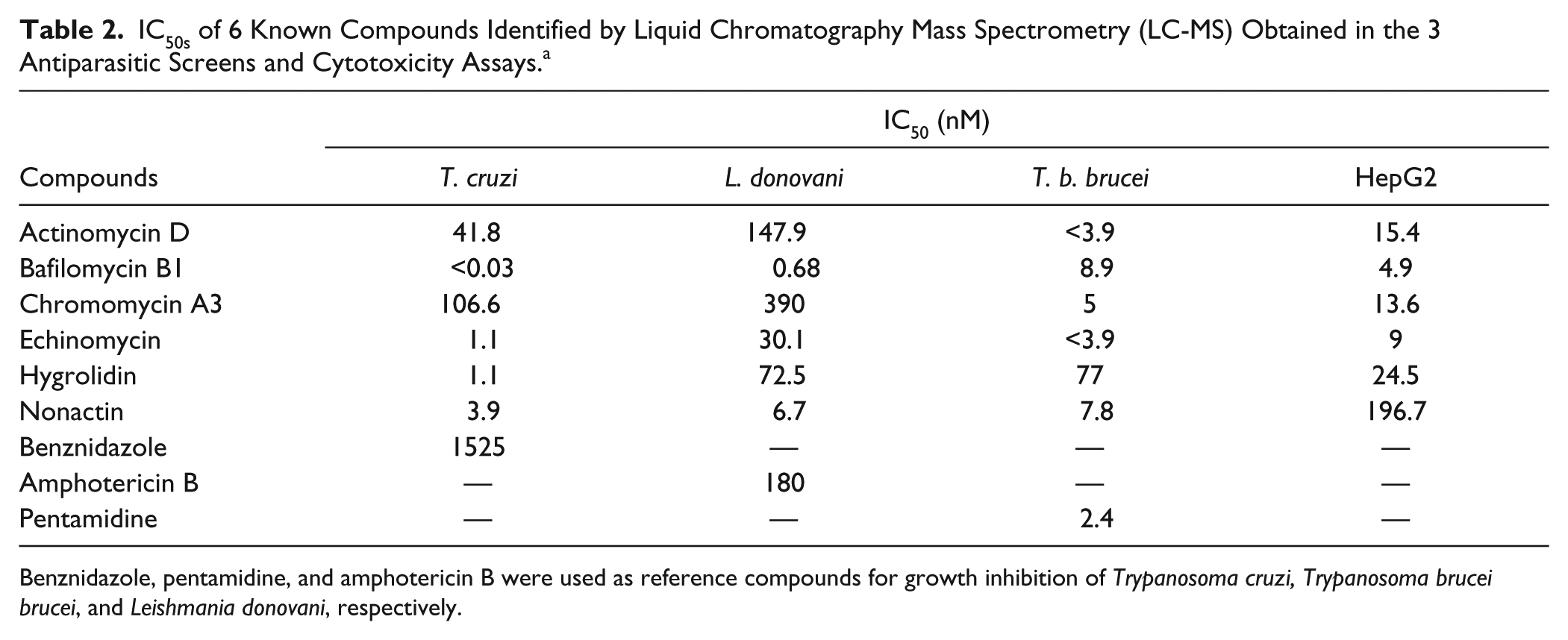
Benznidazole, pentamidine, and amphotericin B were used as reference compounds for growth inhibition of Trypanosoma cruzi, Trypanosoma brucei brucei, and Leishmania donovani, respectively.
Small-Scale Regrowths for Bioassay-Guided Fractionation
At this stage, 17 extracts (7 in T. cruzi, 5 in L. donovani, and 5 in T. b. brucei) identified as containing potentially novel active components have been prioritized, because they show well-resolved metabolic profiles, and they have been regrown in 100 mL volumes. The specific antiparasitic activities have been confirmed in 7 of them: 3 anti–T. cruzi, 2 anti–L. donovani, and 2 anti–T. b. brucei. Different culture conditions are being tested in an attempt to recover the activities in the 10 extracts that have not yet reproduced activities in 100 mL regrowths. Currently, we are running tandem LC–high-resolution MS (LC-HRMS) and nuclear magnetic resonance analyses of the previously generated 80 semipreparative HPLC fractions of each of the 7 active extracts to confirm the novelty of the active components. The extracts in which novel components are confirmed will be regrown in 1 L volumes for the purification of the active compounds.
Discussion
The main objective of this work was to establish a robust HTS platform for the discovery of natural product–based drugs against tropical diseases. In this pilot scheme, we screened a subset (5976 microbial extracts) of our natural products collection, which is one of the world’s most diverse and productive collection of filamentous fungi, actinomycetes, and unicellular bacteria for drug leads. This subset of extracts was previously found to inhibit less than 50% growth of HepG2 cells when tested at 1/40 × WBE final concentration for 24 h, hence this subset is termed “noncytotoxic.” Overall, the collection contains more than 110,000 microbial strains used to generate 130,000 microbial extracts in diverse conditions to ensure a rich metabolite-producing spectrum. In the present platform, a good assay plate layout, valid in-plate negative or positive controls, and a robust RZ’ score have been used, thus ensuring that requirements are met for robust and efficient HTS. For this screening campaign, the RZ’ was always required to be higher than 0.7.
End point readouts, buffers, equipment, and analytical methods used were optimized in trial runs before being used in the screening program. This is especially critical when dealing with parasites with complex life cycles. Routine procedures like parasite handling, dilutions, and equipment manipulation were standardized in trial runs to ensure reproducibility and accuracy.
The general workflow used ( Fig. 1 ) ensures that only extracts that have been tested and proven through rigorous methodology to consistently contain active and potentially novel metabolites against the screened parasites are selected for further development. Our efficiently integrated LC-MS de-replication system also enables us to pick up on active extracts containing compounds already known but for which that activity has not been previously reported.
After LC-MS de-replication of the active extracts, 32 of them were found to contain known compounds. In the case of T. cruzi and L. donovani, the number of known compounds found was higher than in T. b. brucei, but this difference could be a consequence of the higher extract dilution used in the screen and the slightly lower hit rate obtained for the latter. Extracts containing readily identifiable components were not developed any further. Six compounds (actinomycin D, chromomycin A3, nonactin, hygrolidin, echinomycin, and bafilomycin B1;
Moreover, the confirmation of these potentially bioactive known compounds further validates the strategy used in the present screening approach. We can confidently infer from the LC-MS de-replication data that the screening platform is able to effectively determine known active anti-kinetoplastid compounds from crude extracts, thus serving as proof that we have a working system that can be used to de-replicate extracts that contain either (1) generally cytotoxic metabolites or (2) known compounds that have hitherto not been identified as having the specific anti-kinetoplastid activities reported here. The downside of this de-replication step is that potentially active metabolites that may be present at undetectable levels in these crude extracts may be overshadowed by more abundant cytotoxic metabolites and consequently be missed.
As shown by the data above, we have established a robust HTS platform for the identification of potentially novel natural product–based drug leads against 3 neglected tropical diseases, namely, human African trypanosomiasis, leishmaniasis, and American trypanosomiasis. This platform has been used in a pilot project to screen 5976 microbial extracts and has confirmed 48 extracts (at this stage) as having novel potent anti-kinetoplastid metabolites. These are currently under study for the identification of novel hit components. In addition, further HTS of selected extracts is planned. We have also shown that our platform can be used both to validate already-known drugs for which anti-kinetoplastid activities have been previously described and to discover novel therapeutic applications of commercially available compounds for which those activities were unknown. Indeed, the depth of chemical diversity of natural products is unfathomable; there are as diverse chemotypes here as there is diversity of species and strains in this realm. It is our hope that, through this work, we will generate more interest in natural products as a source for novel chemotypes against global medical challenges, such as malaria and neglected tropical diseases.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Junta de Andalucía [BIO-199, P09-CVI- 5367, CTS-7282], the VI PN de I+D+I 2008–2011, ISCIII-Subdirección General de Redes y Centros de Investigación Cooperativa (RICET FIS Network: RD12/0018/0017, RD12/ 0018/0015, and RD12/0018/0005) SAF2012-34267 and SAF2010-20059, and FEDER funds from the European Union. F. Annang and M. Valente are funded by the European Commission FP7 Marie Curie Initial Training Network “ParaMet” [grant number 290080].
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.