
Abstract
Chagas disease affects 8 million people worldwide and remains a main cause of death due to heart failure in Latin America. The number of cases in the United States is now estimated to be 300,000, but there are currently no Food and Drug Administration (FDA)–approved drugs available for patients with Chagas disease. To fill this gap, we have established a public-private partnership between the University of California, San Francisco and the Genomics Institute of the Novartis Research Foundation (GNF) with the goal of delivering clinical candidates to treat Chagas disease. The discovery phase, based on the screening of more than 160,000 compounds from the GNF Academic Collaboration Library, led to the identification of new anti-Chagas scaffolds. Part of the screening campaign used and compared two screening methods, including a colorimetric-based assay using Trypanosoma cruzi expressing β-galactosidase and an image-based, high-content screening (HCS) assay using the CA-I/72 strain of T. cruzi. Comparing molecules tested in both assays, we found that ergosterol biosynthesis inhibitors had greater potency in the colorimetric assay than in the HCS assay. Both assays were used to inform structure-activity relationships for antiparasitic efficacy and pharmacokinetics. A new anti–T. cruzi scaffold derived from xanthine was identified, and we describe its development as lead series.
Keywords
Introduction
Chagas disease was first described by the Brazilian public health physician Carlos Chagas in 1909. 1 It is characterized by a generally asymptomatic acute phase, followed by an indeterminate chronic phase that can last for more than a decade. After this quiescent period, 30% to 40% of patients develop a life-threatening progressive inflammation of the heart and/or gastrointestinal tract. 2 Chagas disease is caused by parasites from Trypanosoma cruzi transmitted by an insect vector from the family Reduviidae, commonly known as the kissing bug or assassin bug. The disease is endemic in Latin America, from Argentina to the southern portion of the United States. In addition, human migration has increased the prevalence of Chagas disease in the United States, Europe, Japan, and Australia. 3 According to the World Health Organization, 8 million people are currently infected, with most cases in Latin America. 4 In the United States, the number of cases is estimated to be 300,000.5,6
The first anti-Chagas drugs, benznidazole and nifurtimox, were developed in the 1970s and are still the only available options for treatment. Neither compound is approved by the Food and Drug Administration (FDA). Drawbacks of these drugs include a prolonged treatment course (up to 180 days) and serious adverse effects.7–10 The recent results of CHAGASAZOL clinical trials confirmed efficacy of benznidazole for patients in the indeterminate chronic phase, but side effects of benznidazole remain an issue. Disappointing results in the same trial for posaconazole (Noxafil; Schering-Plough, Kenilworth, NJ) 11 and in another trial for E1224 (a prodrug of the antifungal agent ravuconazole; Eisai, Tokyo, Japan) 12 strengthen the urgent need for novel and better drugs.
The research and development (R&D) landscape for new drugs to treat neglected diseases has evolved in the past decade. The total annual R&D funding has increased from $2.6 billion in 2007 and $3.1 billion in 2010 to $3.2 billion in 2012. However, these figures represented less than 1% of all health R&D investments in those years, 13 while neglected diseases affect more than 15% of the world’s population. A current strategy for drug discovery and development targeting neglected tropical diseases relies on the partnership between public and private sectors—a public-private partnership (PPP)—pioneered by the Tropical Disease Research Unit at the University of California, San Francisco (UCSF) and Khepri Pharmaceuticals in 1995 and the Medicines for Malaria Venture (MMV) in 1999.14,15 In this model, the academic environment provides the biological or disease model expertise while the pharmaceutical company has compound libraries, the industrial scale, streamlined development process, resources, and experience to bring a discovery product to the patient, making it an ideal combination. This approach, combined with public and philanthropic funding, represents a great innovation for the development of drugs targeting diseases with a lack of commercial incentive. 16 The Drugs for Neglected Diseases Initiative (DNDi) was founded in 2003 following the MMV model and has been one of the major players in the field of Chagas disease drug development, acting in various stages from screening assay development to clinical trials. 17 DNDi was the sponsor of the recent E1224 clinical trial (ClinicalTrials.gov identifier NCT01489228). In 2012, Dundee University, GlaxoSmithKline, and the Wellcome Trust established a collaboration to deliver at least one treatment for Chagas disease, leishmaniasis, or human African trypanosomiasis. In 2013, Eisai and the Broad Institute entered into a global agreement to jointly discover and develop new therapeutic agents for the treatment of neglected diseases and tuberculosis. Chagas disease was the first selected project of the collaboration and was partially funded by the Global Health Innovative Technology Fund (GHIT Fund). 18 To our, knowledge, in addition to the examples mentioned above, including attempts to repurpose posaconazole and our own partnership described in this study, these are the major initiatives committed to bring new anti-Chagas chemotherapy to patients.
Our PPP, established between the UCSF and the Genomics Institute of the Novartis Research Foundation (GNF), is funded by the National Institutes of Health (NIH) of the United States. It is also an academic-industry partnership, and the goal is to identify at least one preclinical candidate to treat Chagas disease, preferentially with a new mechanism of action. One strength of our partnership is the systematic evaluation of antiparasitic activity of compounds in two different in vitro assays, using different parasite strains and readout methods to screen libraries of compounds. The assays were compared in this article, and results indicated that there might be distinct sensitivity toward a particular mechanism of action. Knowing also that T. cruzi strains may respond differently to a chemotherapy,19,20 we believe a drug discovery program should rely on more than one strain for efficacy assessment. We illustrate the hit optimization workflow using a series composed of xanthine analogues, which we were able to optimize for potency and pharmacokinetic properties, including in vivo efficacy proof of concept in a footpad mouse model. The xanthine substructure has not previously been described in the literature as being active against T. cruzi and has only recently been ascribed biological activity in the context of cystic fibrosis. 21 The collaboration flowchart with the decision-making points to identify a preclinical candidate is shown in Figure 1 . A lead compound would be characterized by a confirmed in vivo antiparasitic efficacy and adequate pharmacokinetics profile, as illustrated in the Figure 1 in the “Discovery” part. A preclinical candidate would be defined by a lead compound that passes toxicological tests, listed in the “Development” part of the figure, demonstrating a safety profile.
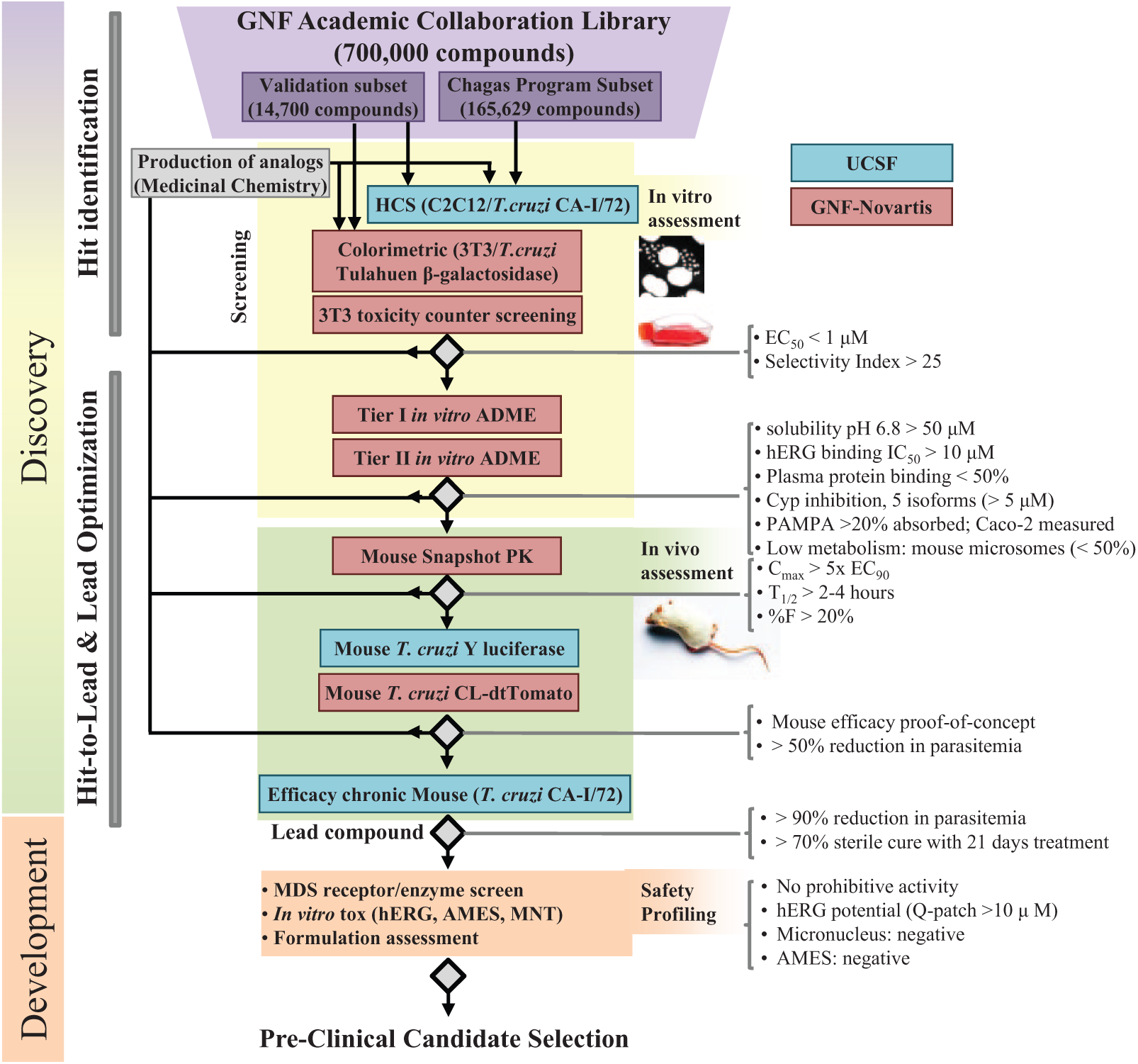
Chagas disease drug discovery and development decision cascade flowchart. The scheme describes the activities and decision points from the in vitro cell-based screenings of the compound libraries to the preclinical candidate selection.
Materials and Methods
Cell Culture
Mouse myoblast C2C12 cells (ATCC CRL-1772) and mouse fibroblast 3T3 (ATCC CL-173) were routinely cultured in RPMI 1640 medium supplemented with 5% to 10% heat-inactivated fetal calf serum (FCS) at 37 °C in 5% CO2. For the subculture, the culture media were removed, followed by washing the cells once with phosphate-buffered saline (PBS) and trypsinization with 0.25% Trypsin EDTA (25200-056; Gibco, Carlsbad, CA) for 2 min at room temperature. The cells were then counted and seeded at appropriate density in a new culture flask in the presence of RPMI 1640 medium. The T. cruzi CA-I/72 strain 22 came from a chronic chagasic patient. The T. cruzi CL strain expressing fluorescent tdTomato protein 23 was kindly provided by Rick Tarleton. Infectious trypomastigotes were obtained from the culture medium from infected host cells 4 to 7 days postinfection. T. cruzi Tulahuen strain expressing β-galactosidase 24 was kindly provided by Frederick Buckner. All the cells and parasites were kept in culture for no more than 20 passages. The genetically modified CL strain expressing the tdTomato was cultured in the presence of G418 antibiotic to prevent loss of the transgene.
Compound Sources
A library of 700,000 small molecules was assembled at GNF with a particular focus on compound drug-like properties and structural diversity (Academic Collaboration Library, ACL, San Diego, CA). This library consists of a collection of compounds that were selected after applying proprietary algorithms designed to select for optimal compound properties and eliminate undesirable functional groups. A subset of the library composed of 14,700 compounds was used to compare the two screening methods implemented for this work: the high-content screening (HCS) assay and the colorimetric screening assay. Another subset of the ACL collection comprising 165,629 compounds was also screened to identify T. cruzi growth inhibitors using an image-based HCS assay as the primary screening method.
Colorimetric Screening Assay
The Tulahuen strain of T. cruzi, constitutively expressing bacterial β-galactosidase reporter, was used in an infection/growth assay with 3T3 mouse fibroblast cells in 384-well plates. Compounds were tested up to 2 µM final concentration (in 0.2% DMSO) and incubated with the infected 3T3 cells for 6 days. To measure the amount of the parasite-expressed β-galactosidase, the cells were lysed by the addition of NP-40 detergent (0.1% final concentration), followed by the addition of the colorimetric β-galactosidase substrate chlorophenol red-β-D-galactopyranoside (CPRG). After a 2-h incubation at room temperature, the amount of CPRG hydrolyzed by β-galactosidase was quantified by measuring absorbance at 570 nM. The employed protocol was previously described in detail. 24
HCS Assay
For primary screening, 50 nL of compounds at 10 mM was dry-spotted in clear-bottom, 384-well plates (Greiner Bio-One, Monroe, NC). The compound-loaded plates were prepared at GNF and transferred to UCSF. A solution with trypsinized C2C12 cells at 1.0 × 104 cells/mL and T. cruzi CA-I/72 at 5.0 × 104 parasites/mL was dispensed in the plates using Matrix WellMate (Thermo Scientific, Waltham, MA), 50 µL/well, as a mixture/batch infection. The plates were incubated for 72 h at 37 °C and 5% CO2. After the incubation, plates were fixed for 2 h with 4% paraformaldehyde and rinsed with PBS to remove the fixative. Then, 50 µL/well of 5 µg/mL DNA fluorescent dye DAPI (4,6-diamidino-2-phenylindole) was then added. Plates were kept in the dark at 4 °C until image acquisition was performed.
For dose-response (EC50) determinations, compounds were serially diluted in DMSO from 10-mM stocks to give 10-point, 2-fold serial dilutions (compound concentrations from 10 mM to 19.5 µM). Then, 10 µL of PBS was added to each well of a clear-bottom 384-well plate (Greiner Bio-One), followed by transfer of 50 nL of serially diluted compounds (pin tool, VP Scientific, San Diego, CA; Biomek FxP, Beckman Coulter, Brea, CA). A solution with trypsinized C2C12 cells at 1.25 × 104 cells/mL and T. cruzi CA-I/72 at 6.25 × 104 parasites/mL was dispensed in the plates using Matrix WellMate, 40 µL/well, as a mixture/batch infection. The plates were incubated and fixed as described for primary screening.
Image Analysis
Plated cells and parasites were imaged using an IN Cell Analyzer 2000 (GE Healthcare, Piscataway, NJ). The excitation and emission filters used to detect DAPI were 350/50 nm and 460/40 nm, respectively. Four image fields were acquired per well, each with an exposure time of 150 ms. The IN Cell Developer Toolbox 1.9 was used for image analyses. Four types of objects were defined: nuclei, kinetoplasts and intranuclear foci, and cell body. Nuclei were segmented with object segmentation (kernel 21, sensitivity 1), which identifies objects based on size, and further reduced with clump breaking. Intranuclear foci and kinetoplasts were identified by binarization (whereby pixels are assigned a value of 1 if they are 33% brighter than the local average and 0 if they are not brighter than average) and intensity segmentation (using kernel size 17 to select small objects). To determine the cell body, we created an aggregate image that linked the kinetoplastids to a host-cell nucleus. First, host-cell nuclei and kinetoplastids were separated based on their size and intensity, using a granular segmentation algorithm (nuclei: kernel 21, sensitivity 1; kinetoplastids: kernel 3, sensitivity 25). Second, the image containing kinetoplastids was dilated, which tended to merge neighboring kinoplastids together into one body. Third, the nuclear and kinetoplastid images were summed into a new, aggregated image. Aggregate objects were then defined by intensity segmentation and clump breaking. Aggregate objects that did not contain intranuclear foci were considered out of focus and excluded from further analysis. The aggregate images represented cell bodies. Infected cells were defined as being >1.67-fold larger than the nucleus of that cell; uninfected cells had areas closer to the area of the nucleus itself. During assay development, both the fraction of infected cells (infected cells/total cells) and the area of infection (area of kinetoplastids/total nuclei) were evaluated. Only kinetoplastids linked to in-focus nuclei were included in the area calculation.
Ergosterol Depletion Assay
T. cruzi parasites were grown as epimastigotes in LIT medium at 27 °C, with 2 × 106 parasites seeded per well in a 96-well deep-well blocks (0.5 mL of media/well). Compounds were dissolved in DMSO and added to the wells as indicated (0.2% DMSO final concentration). Plates were sealed with airpore tape (Qiagen, Valencia, CA) and incubated for 1 h at 27 °C. Then, 2.4 µL of 14C mevalonate/well was added (PerkinElmer, Waltham, MA) and epimastigotes were labeled for 24 h at 27 °C. Plates were then centrifuged at 13,000 rpm for 3 min and washed 2× with Dulbecco’s Phosphate-Buffered Saline (DPBS) (0.5 mL/well). The plates with pelleted, labeled epimastigotes were resuspended in 100 µL of PBS, and lipids were extracted from the cell suspension with a 2:1 chloroform/methanol mixture. After concentrating the lipid extracts under a stream of N2 gas, the lipids were separated by thin-layer chromatography on silica plates using a toluene/diethyl ether (9:1) mixture as the mobile phase. After the separation, positions and identities of radioactively labeled lipid species on the thin layer were determined by autoradiography. Our protocol closely followed that described previously. 25
Pharmacokinetics Profiling
Mouse snapshot pharmacokinetics (PK) was performed as previously described to estimate compound oral bioavailability in the mouse. 26 Briefly, two mice were dosed by oral gavage with a compound, and four plasma samples were collected from each mouse over the period of 5 h postdose (0.5, 1, 3, and 5 h). Equivalent samples from two mice were pooled, and the compound plasma concentration at each time point was determined. Measured values were used to estimate compound oral bioavailability. More detailed characterization of compound pharmacokinetics in mouse was performed on compounds with a promising snapshot PK using the standard pharmacokinetic protocols. For permeability, we followed the protocol described by Skolnik and collaborators 27 using a robust 96-well Caco-2 assay that could predict intestinal absorption. Caco-2 cells are derived from colon carcinoma and can express tight junctions, microvilli, and transporters resembling enterocytes from the small intestine. The plasma stability of the compounds was tested according to the procedures established by Di and collaborators, 28 based on the following condition: 1 µM of tested compound, 2.5% DMSO, and 50% dilution of plasma in pH 7.4 buffer followed by the incubation at 37 °C for 3 h. After incubation, we measured how much of the compound remained intact.
HT-Equilibrium Solubility Assay
The assay tested compounds as 10-mM DMSO stock solutions, where a 20-µL aliquot of each compound was first dried in a 96-well plate before 200 µL buffer addition (67 mM potassium phosphate, pH 6.8) and shaking for 24 h on an orbital shaker. After shaking, samples were spun at 3700 rpm, and 150 µL of supernatant was transferred into clean 96-well plates. An 8-point dilution series was made for each compound in DMSO. Samples and standards were reformatted into a 384-well UV plate, with matrix matching by addition of assay buffer to the DMSO dilution series and addition of blank DMSO to test samples. The plates were read by a Spectramax Plus 384-well plate reader (Molecular Devices, Sunnyvale, CA), scanning from UV 250 to 420 nm with a step size of 5. Raw data were processed using a GNF in-house algorithm.
In Vivo Efficacy (Footpad Model)
In vivo activity of compounds was evaluated in the footpad model of acute Chagas disease as previously described in detail. 23 Briefly, BALB/c mice were infected in each pad of both hind feet with 2.5 × 105 of T. cruzi trypomastigotes (CL strain) expressing the tdTomato fluorescent protein. After allowing the infections to develop for 7 days, mice were dosed with experimental compounds for 5 days. At the end of treatment, the parasites in footpads were quantified by fluorescent imaging (IVIS 100; Xenogen, Alameda, CA).
Results and Discussion
Optimization of HCS Assay for T. cruzi Proliferation
An image-based HCS assay that measures proliferation of intracellular T. cruzi amastigotes was previously established in a 96-well plate format. The assay principle relied on the difference in size between mammalian nuclei (5–10 µm) and the parasite’s kinetoplast (0.5–1 µm each), which is more strongly stained by DAPI than by the parasite nucleus. The assay allowed quantification of the compound effect on both intracellular T. cruzi and host cells after 3 days of compound treatment. 29 In addition to measuring cytotoxicity and antiparasitic activity at the same time, this assay uses unengineered parasites and therefore works with multiple strains of parasite and multiple mammalian host cell lines.
To further increase the assay screening throughput, we adapted the published protocol 29 to a 384-well plate format. The published 96-well assay used bovine embryo skeletal muscle (BESM) cells, which are not immortal and are difficult to culture at the scale needed for high-throughput screening (HTS). 22 Therefore, we redesigned the assay with the mouse myoblast C2C12 cell line. The C2C12 cells formed monolayers in 384-well plates, a favorable property for high-quality image acquisition and analysis. Compared with the original protocol, we simplified sequential plating of host cells and parasites (two steps) and eliminated the postinfection wash and media replacement step. These three operations were replaced with a single step in which T. cruzi trypomastigotes and C2C12 host cells were preincubated for 10 to 30 min and then co-dispensed into the 384-well plate. By testing a matrix of serial dilutions of the C2C12 cells versus T. cruzi trypomastigotes, we determined that the optimal host cell density for seeding was 500 cells/well. This number took into account an expansion of C2C12 cells that occurs during the 72-h experiment, and it yielded the optimal host cell density for high-quality image acquisition at the end of the assay. The optimal parasite number for seeding was 2500 parasites per well (five parasites per host cell). This multiplicity of infection (MOI) allowed accurate identification of the intracellular amastigotes in the C2C12 cell cytoplasm after DAPI staining. We further streamlined the assay by prespotting the screening compounds dissolved in DMSO into the assay wells before adding the host cell/parasite mixture. By uncoupling compound addition and cell plating, we were able to add compounds to assay plates at the GNF and then ship the assay-ready plates to UCSF for screening. Typical assay throughput was forty-eight 384-well plates per run, corresponding to approximately 15,000 tested compounds per run.
For HCS, we collected images using a 20× objective, and the DAPI-stained DNA from both host cells and parasites was captured and the size of the objects measured. Compounds were considered a hit if (a) their antiparasitic activity was greater than two standard deviations from the untreated control (DMSO) and (b) did not reduce the number of host cells to less than 50% of the untreated control wells (coincides with the average number of cells from DMSO controls minus three standard deviations from the DMSO controls). Figure 2A–C provides examples of images of wells incubated with the reference drug benznidazole as a positive control ( Fig. 2A ) and DMSO as negative control ( Fig. 2B , C ).
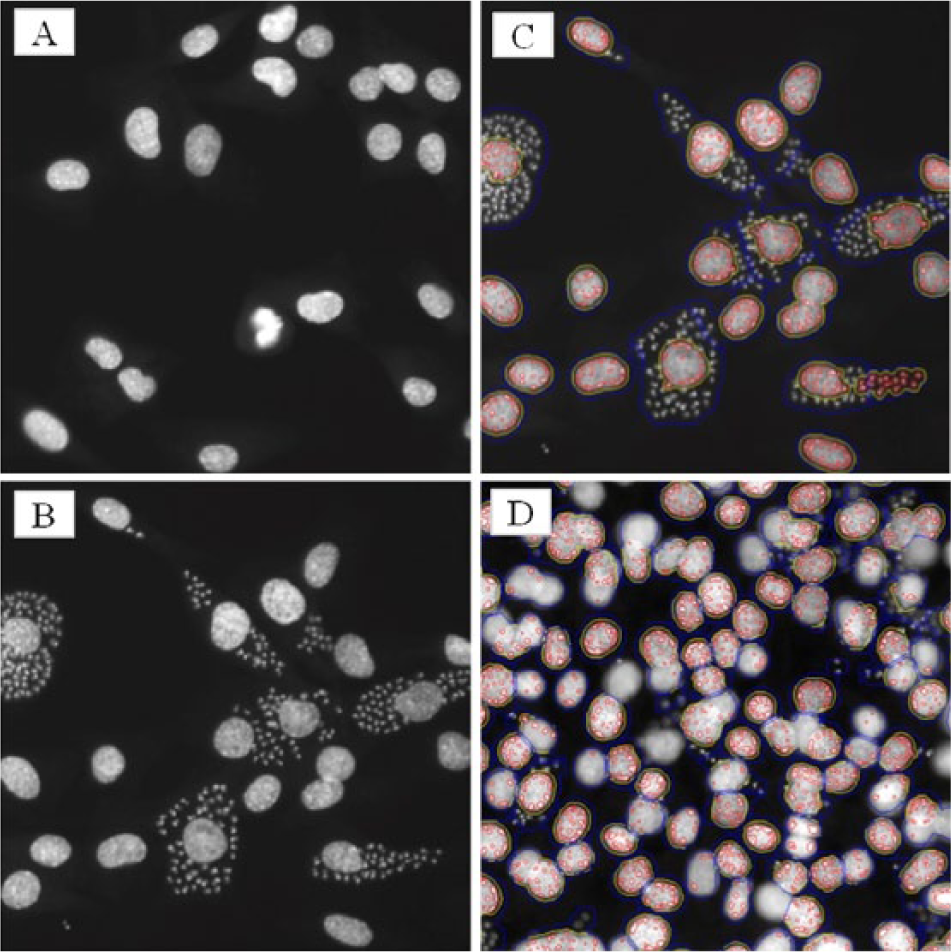
Image-based high-content screening (HCS) data analysis. Schematic representation of different phenotypes identified in the readings. (
Analysis of the HCS images was designed to address two experimental issues. First, some images contained cells that were out of focus, resulting from a high local cell density, edge-well effects, or lens artifacts. Due to their small size, we were unable to accurately count kinetoplasts in the poorly focused cells. We therefore defined out-of-focus cells by inspection of intranuclear foci, small granular structures within the cell nucleus that can be seen in well-focused images but not in blurred images. The cells with nuclei lacking these subnuclear structures were deemed out of focus and discarded from further analysis ( Fig. 2D ). Second, as the cell density increased, the cells became progressively smaller in size. Nuclei also varied in size. To address these issues, we defined an infected cell by the ratio of the area of kinetoplasts/nuclear area (rather than defining an infected cell as being larger than a certain size). By analyzing only in-focus cells and measuring the area of infection, we were able to extract data from images over a wide range of cell density.
Screening and Hit Selection
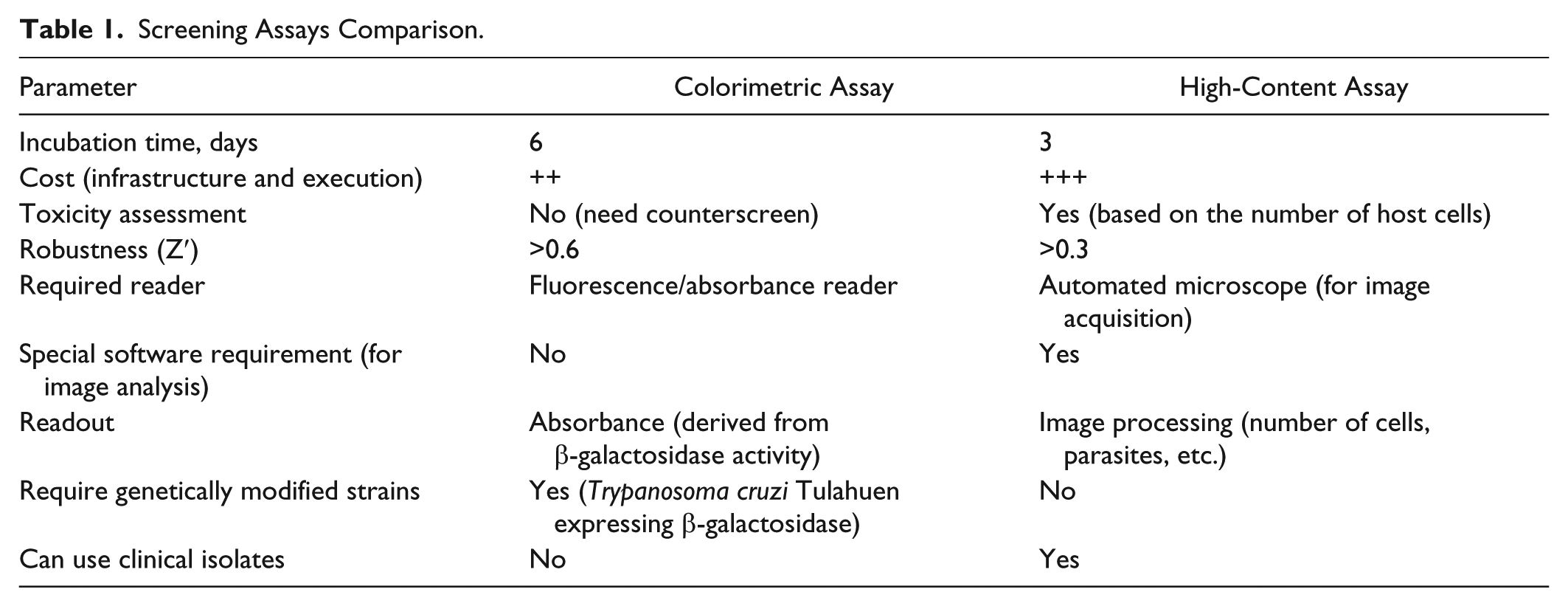
To compare the two screening assays, we screened 14,700 molecules from GNF’s ACL collection at a 6-µM concentration in the colorimetric reporter assay and at 10-µM concentration in the HCS assay. The reporter colorimetric assay had a Z′ > 0.6 and yielded 322 hits (2.2%) applying >50% normalized activity as the criteria for hit selection in the primary screening. The HCS screen had Z′ > 0.3 and identified 348 active compounds (2.4%) based on two standard deviations from the DMSO (untreated) control cutoff criteria for hit selection. A similar number of initial hits indicated that equivalent stringency was applied for the primary selection. Comparing both lists, of the 14,700 interrogated compounds, we identified 149 shared hits, approximately 1% of the total number of tested molecules (Venn diagram in Fig. 3A ). This can be considered a good overlap, given the assay differences that would contribute to a small list of common actives. For example, the image-based assay was tested with compounds at 10 µM as opposed to 6 µM in the colorimetric assay, favoring the selection of molecules with EC50 > 6 µM only in the HCS. The fact that the assays use different strains, different incubation times, and different readouts would also contribute to reduced overlap. Confirmation of hits was done in a dose-response format. Of 149 common hits, 48% reconfirmed activity in the colorimetric assay with EC50 lower than or equal to 2 µM (maximum tested concentration), and 55% of the hits reconfirmed in the HCS assay with EC50 lower than or equal to 10 µM (maximum tested concentration). Table 1 compares a number of parameters for each assay method.
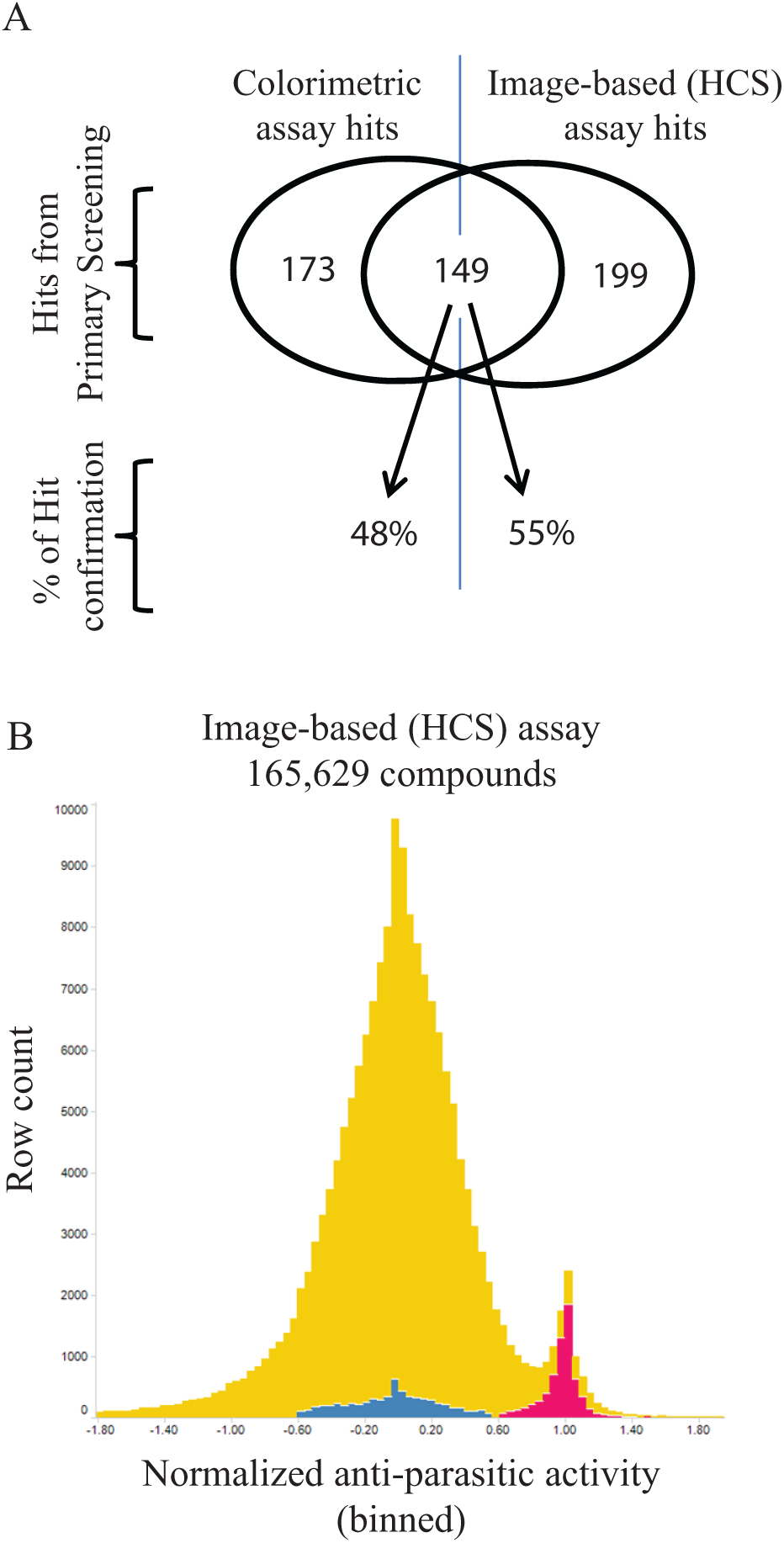
Screening of anti– Trypanosoma cruzi compounds. (
Screening Assays Comparison.
After comparing both assays, a large library of 165,629 compounds from the ACL was screened at a 10-µM concentration using the HCS assay. In total, 2385 (1.45%) compounds were identified as inhibiting T. cruzi growth, showing an inhibition of infection greater than two standard deviations from the DMSO control wells without overt toxicity to host cells (more than 50% reduction in host cells relative to the median number of cells per well). Figure 3B shows the compound-activity histogram distribution based on normalized antiparasitic activity. The negative (blue, DMSO only) and positive (pink, 10 µM benznidazole) controls are also shown.
Compound Bias of Colorimetric Reporter Assay versus HCS Assay
The hits from the ACL subset of 14,700 compounds screens were characterized in a dose-response format using both the colorimetric reporter and HCS assays, and inhibitors with sub-micromolar antiparasitic EC50s and minimal cellular toxicity (less than 50% reduction in host cell number at 10 µM) were selected for follow-up studies. One observation that emerged from this analysis was that some compounds had different potency in the colorimetric assay compared with the HCS assay. This discrepancy could be due to the differences in the parasite strains, host cells, or incubation time. Moreover, one important possibility is that alternative T. cruzi in vitro proliferation assays can exhibit strong biases toward different mechanisms of action/chemotypes, creating a bias in which types of compounds are then taken forward as drug leads.
Recent drug discovery work for antichagasic compounds has focused on inhibition of Cyp51 (the target of the “azole” inhibitors such as posaconazole). Cyp51 inhibition kills parasites by depleting ergosterol biosynthesis. In the context of the observed assay bias, we asked whether this particular mechanism of action also translates into different compound potencies in these two screening assays. The parasites carry a natural stock of ergosterol, and they must undergo several cell divisions after the onset of Cyp51 inhibition before growth inhibition and cell death occur. Because of the difference in colorimetric versus HCS assay duration (6 versus 3 days), the 6-day assay could be expected to report more potent EC50s for Cyp51 inhibitors than the 3-day assay. To test this hypothesis, we performed an ergosterol depletion assay on a subset of 74 confirmed hits in addition to posaconazole, a known T. cruzi Cyp51 inhibitor, as a reference drug. Posaconazole and 10 other compounds (13%) showed some degree of ergosterol depletion up to 20 µM, the maximum tested concentration. Posaconazole was estimated to be 16 times more potent in the colorimetric assay (EC50 = 0.7 nM 30 versus 11.5 nM in our HCS) and had a Cyp51 inhibition EC50 of 0.5 nM. Two other compounds had Cyp51 IC50 < 1 µM. Eight of 10 identified Cyp51 inhibitors were more potent in the colorimetric than in the HCS assay ( Fig. 4 ), five of which had more than a 10 times difference in potency. One identified Cyp51 inhibitor had similar potency in both assays, and one other was more potent in the HCS assay than in the colorimetric assay. As some compounds could inhibit more than one cellular target, it is possible that the latter compound, while being a Cyp51 inhibitor, actually kills T. cruzi by an alternative, more potent mechanism of action. The results support the hypothesis that compounds targeting the biosynthesis of ergosterol are preferentially detected by the colorimetric assay. This could occur as the consequence of extended assay duration or result from a greater sensitivity of the T. cruzi Tulahuen strain to this class of compounds compared with the CA-I/72 strain.
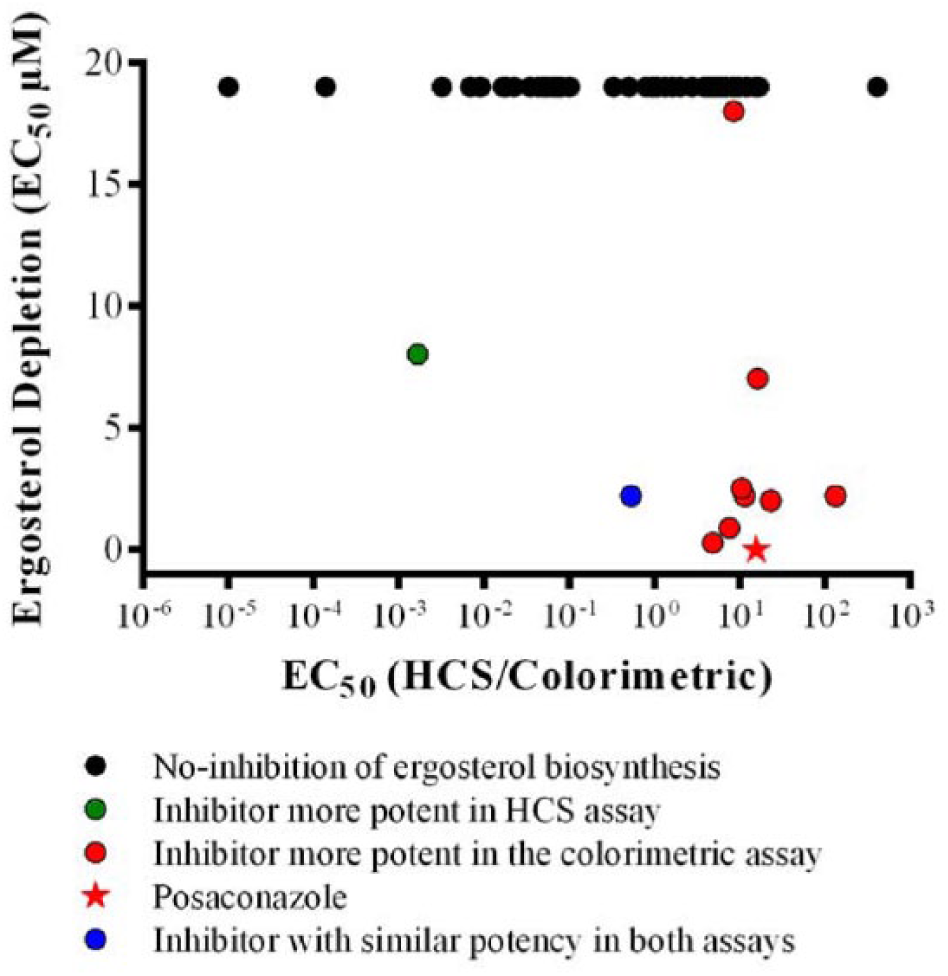
Ergosterol biosynthesis inhibitor potency in different assay methods. The x-axis represents in logarithmic scale the ratio of the EC50 calculated by the image-based assay (high-content screening [HCS]) by the EC50 calculated by the colorimetric assay. The y-axis represents the parasite ergosterol depletion EC50 in µM. The maximum tested concentration for the ergosterol depletion was 20 µM, so the compounds represented as black dots did not show ergosterol biosynthesis inhibition up to 20 µM. The green dot represents one ergosterol biosynthesis inhibitor more than 100 times more potent in the HCS assay. The blue dot represents one ergosterol biosynthesis inhibitor with similar potency in both assays, and the red dots represent ergosterol biosynthesis inhibitors more potent in the colorimetric assay. The red star represents posaconazole, a known T. cruzi Cyp51 inhibitor, as a reference drug.
A recent publication comparing T. cruzi strain susceptibility to a number of different compounds using an image-based assay reported 1.0 nM EC50 against the Tulahuen strain and 5.3 nM against the Dm28c strain 20 (from DTU I, same as CA-I/72), suggesting that using the same assay may reduce the difference in susceptibility of distinct strains, supporting that the variability in our case may be coming from the assay methods. Further investigation is needed to determine the cause of this assay bias toward a specific mechanism of action. Nonetheless, we believe that this is an important finding, since the colorimetric and the image-based HCS assays are the major methods used to screen anti–T. cruzi products, and the assay bias toward a particular mechanism of action should be acknowledged.
Hit-to-Lead Optimization of Xanthine Scaffold
The confirmed hits were further prioritized and grouped into six distinct, high-priority scaffolds with consideration given to novel anti-Chagas chemotypes, chemical tractability, lack of stereogenic centers, low molecular weight, and Lipinski rule-of-five compliance for oral bioavailability. We deprioritized chemotypes associated with the mechanism of action of known Chagas disease drugs, such as azoles and nitroheterocycles. In this article, we present characterization of one of our lead series derived from the xanthine scaffold.
An elaborated xanthine core was first exemplified by the screen hit GNF5689 (see
Table 2
). This inhibitor had a very good T. cruzi potency (EC50 = 250 nM in the colorimetric assay); however, the terminal methyl ester was rapidly hydrolyzed in mouse plasma (only 40.6% of the parent remained after 1 h), and the molecule was subject to oxidative metabolism (11% of the parent remained after a 1-h incubation with human microsomes). The structure-activity relationship (SAR) on the xanthine scaffold revealed that ester hydrolysis could be abrogated by employing an isopropyl substitution at the α position of the amino acid (GNF6732), with 100% of GNF6732 remaining intact after a 1-h incubation in mouse plasma. Potency was moderately improved (EC50 = 48 nM). However, the oxidative metabolism liability remained (6% of the parent remained after 1 h with human microsomes). GNF6732 had an oral bioavailability (F) of 12% in mouse, a low AUC of 1193 nM·h, and high clearance, consistent with the microsome data. Another solution to the ester hydrolysis problem was to replace the group with a terminal triazole ring (GNF7198). While the potency did improve for this analogue (EC50 = 160 nM), mouse plasma stability was still excellent (100% of the parent remaining after 1 h), and oxidative stability was improved in the presence of human microsomes (62% of GNF7198 remaining after 1 h). The in vitro ADME data accurately predicted in vivo exposure for this analogue, which showed a 10-fold improvement in oral AUC and an oral bioavailability of 44% (mouse) (
Properties of the Xanthine Analogues as Anti-Chagas Series.
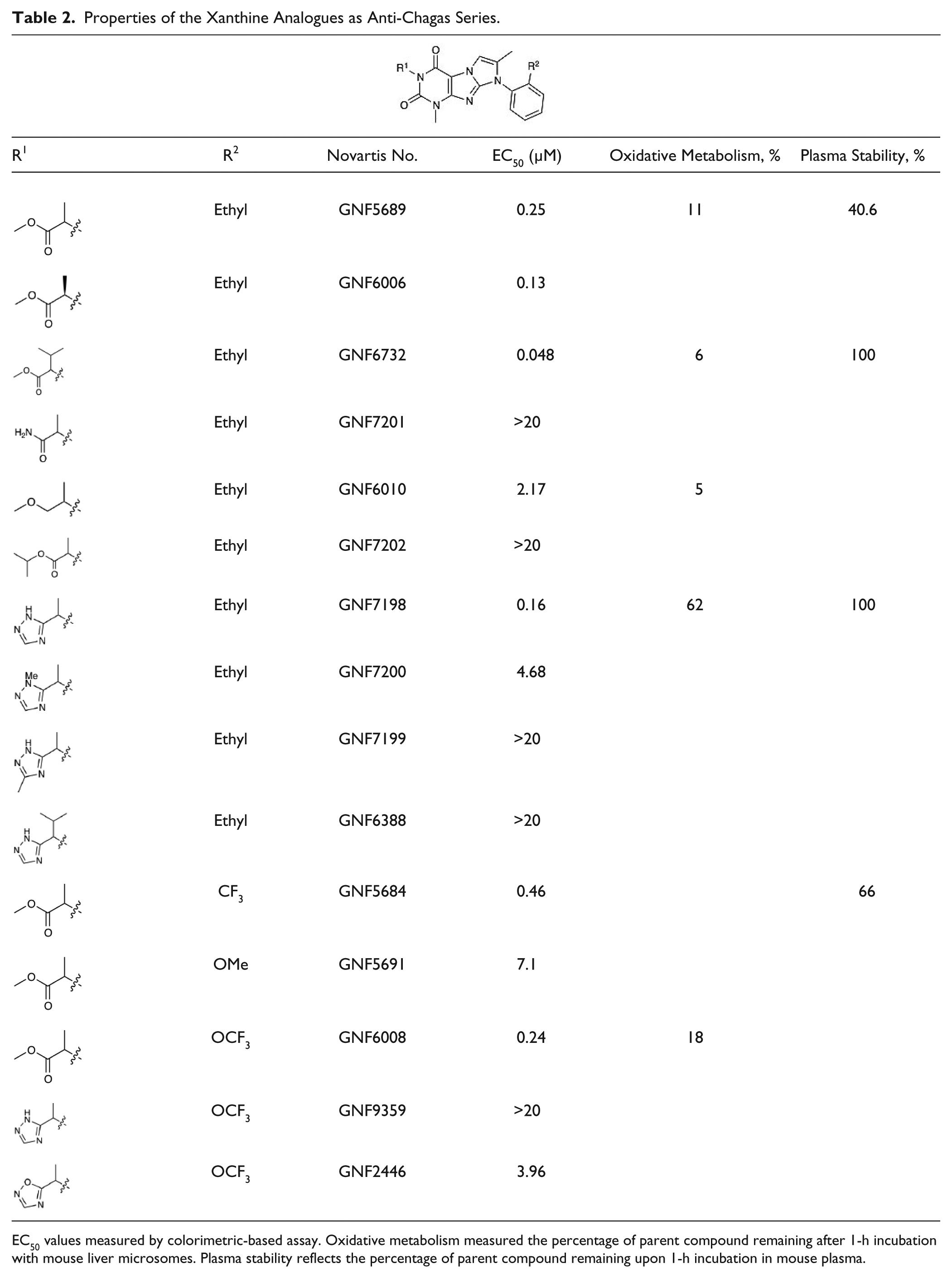
EC50 values measured by colorimetric-based assay. Oxidative metabolism measured the percentage of parent compound remaining after 1-h incubation with mouse liver microsomes. Plasma stability reflects the percentage of parent compound remaining upon 1-h incubation in mouse plasma.
In addition to the R1 modifications, we also explored the effects of substitutions at the R2 position ( Table 2 ) of GNF5689. The replacement of methyl with trifluoromethyl yielded an analogue (GNF5684) with a good predicted oral bioavailability (Caco-2 P(B – A)/P(A – B) = 0.69) and lipophilicity (cLogP = 3.09 at pH 6.8). As the methyl ester present in the original hit was retained, GNF5684 in vitro stability did not change substantially (mouse plasma stability: 66% of the parent remaining after 1 h; human microsome stability: 46% of the parent remaining). This in vitro profile of GNF5684 closely predicted mouse PK characterized by a high oral bioavailability (F = 44%) but also by a high clearance (Cl = 54 mL/min/kg) and short oral half-life (T1/2 = 1.9 h). Further in vitro characterization revealed that the analogue does not inhibit any of four major CYP450 isoforms (IC50 > 50 µM) or hERG channel conductance (automated patch clamp IC50 > 30 µM). Additional analogues with substitutions at the R2 position and their profiles are summarized in Table 2 .
In vivo efficacy of GNF7198 was evaluated in the mouse footpad model of acute Chagas disease. Briefly, mice were infected with the T. cruzi CL strain expressing the tdTomato reporter.
23
Seven days postinfection, the mice were dosed orally with 10 mg/kg, 30 mg/kg, and 100 mg/kg bid for 5 consecutive days. At day 9 postinfection, no significant reduction in florescent signal was observed. However, at day 11 postinfection, the observed T. cruzi signal reduction in the infected footpads was 83% at a 30-mg/kg dose and 65% at 10 mg/kg compared with the benznidazole control (normalized to 100%). Interestingly, the highest tested dose at 100 mg/kg bid did not have an antiparasitic effect. This observation has been reported for antibiotics and is known as a paradoxical effect or an Eagle effect.31,32 The complete profile of the GNF7198 lead series is presented in
In conclusion, the PPP between UCSF and the Genomics Institute of the Novartis Research Foundation resulted in optimization of an HCS assay and allowed us to screen more than 160,000 compounds for anti–T. cruzi activity. The combination of the academic expertise in Chagas models with the industrial scale and strength to generate robust preclinical data for lead selection is proving to be an effective formula. From this screen, with long-term support from the NIH, multiple novel antiparasitic chemotypes were identified, optimized, and characterized in terms of their ADME/PK profile.
Footnotes
Acknowledgements
We thank the GNF PK team members Barbara Saechao, Koyuki Takenaka, You-Qun He, Michael Shapiro, Todd Groessl, and the GNF in vitro ADME team members Thomas Hollenbeck, Patrick White, Kathy Senekeo-Effenberger, Michael Kwok, Ding Yuan, and John Isbell for their valuable contributions. We thank Dr. Frederick Buckner, University of Washington, for kindly donating T. cruzi Tulahuen expressing the β-galactosidase; Dr. Rick Tarleton, University of Georgia, for kindly donating T. cruzi CL expressing tdTomato; and Mr. Potter Wickware for proofreading of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research reported in this publication was supported by the National Institute of Allergy and Infectious Disease of the National Institutes of Health under grant number R01 AI090605. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.