
Abstract
Improved therapies for the treatment of Trypanosoma brucei, the etiological agent of the neglected tropical disease human African trypanosomiasis, are urgently needed. We targeted T. brucei methionyl-tRNA synthetase (MetRS), an aminoacyl-tRNA synthase (aaRS), which is considered an important drug target due to its role in protein synthesis, cell survival, and its significant differences in structure from its mammalian ortholog. Previous work using RNA interference of MetRS demonstrated growth inhibition of T. brucei, further validating it as an attractive target. We report the development and implementation of two orthogonal high-throughput screening assays to identify inhibitors of T. brucei MetRS. First, a chemiluminescence assay was implemented in a 1536-well plate format and used to monitor adenosine triphosphate depletion during the aminoacylation reaction. Hit confirmation then used a counterscreen in which adenosine monophosphate production was assessed using fluorescence polarization technology. In addition, a miniaturized cell viability assay was used to triage cytotoxic compounds. Finally, lower throughput assays involving whole parasite growth inhibition of both human and parasite MetRS were used to analyze compound selectivity and efficacy. The outcome of this high-throughput screening campaign has led to the discovery of 19 potent and selective T. brucei MetRS inhibitors.
Keywords
Introduction
Aminoacyl-tRNA synthases (aaRS) are essential in protein synthesis, catalyzing the synthesis of aminoacyl-tRNAs. The catalyzed reaction of aaRS usually proceeds via two steps: first the recognition of a specific amino acid and adenosine triphosphate (ATP) to form the aminoacyl-adenylate intermediate and, second, the recognition of associated tRNA and the transfer of the aminoacyl group to the terminal adenosine of the tRNA. Inhibition of either step results in disruption of protein biosynthesis, attenuating cell growth and compromising its viability. aaRSs are thus excellent drug targets for inhibition, delivering selective cytotoxicity versus pathogens. This has been demonstrated by the development of the drug mupirocin, which selectively inhibits isoleucyl-tRNA synthetase (IleRS) from archaea and eubacteria but does not inhibit eukaryotic IleRS. 1 Other aaRSs have been targeted in high-throughput screening (HTS) efforts, such as phenylalanyl tRNA synthetase (PheRS) and methionyl-tRNA synthetase (MetRS) from Staphylococcus aureus. 2 Herein we describe an HTS program targeting MetRS from the protozoan parasite Trypanosoma brucei, including an orthogonal counterscreen.
T. brucei is the causative agent of the neglected tropical disease human African trypanosomiasis (HAT), which threatens ~60 million people in sub-Saharan Africa. 3 When left untreated, HAT is fatal, and current treatment outcomes are unsatisfactory due to high toxicity, difficult administration regimes, and poor efficacy of existing drugs. 4 Hence, new drugs that are effective, nontoxic, and affordable are needed. Early studies using RNA interference (RNAi) demonstrated that MetRS from T. brucei is essential for parasite survival, making MetRS inhibition an attractive strategy for treating HAT.5–7 Potent aminoquinolone-based compounds, such as 2-((3-((3,5-dichlorobenzyl)amino)propyl)amino)quinolin-4(1H)-one (CID 18353708), have been identified but present poor bioavailability. 8 Moderately potent urea-based compounds and oxazolone-dipeptides have been reported.9–11 The goal of the HTS campaign presented here is to identify selective, potent, and drug-like small-molecule T. brucei MetRS inhibitors, with the ultimate potential to become novel therapeutics for HAT.
To minimize false leads, a key aspect of our effort was to use two orthogonal screens early in the program, using different assay technologies to identify novel inhibitors of the MetRS catalyzed reaction in vitro. This reaction is monitored by measuring the amount of residual ATP or adenosine monophosphate (AMP) from the MetRS reaction via luminescence and fluorescence polarization (FP), respectively. Excellent performance of both assays in the 1536-well plate format was achieved throughout the screen of the Molecular Libraries Small Molecule Repository (MLSMR) collection of 364,131 compounds. More than a dozen tractable compounds with sub-micromolar potency were identified using these assays.
Materials and Methods
Protein Expression and Purification
The full-length gene for T. brucei MetRS (Tb927.10.1500) was amplified from T. brucei brucei strain 427, expressed in Escherichia coli, and purified as previously described. 8 The N-terminal 6-His tag was retained. The human mitochondrial MetRS enzyme was expressed, as previously reported, with a plasmid provided by Dr. L. Spremulli. 12
Optimization and Miniaturization of MetRS Inhibition Assays
The assay for HTS required a robust, nonradiometric methodology. An ATP depletion assay based on a chemiluminescence was developed for the initial screen.13,14 Optimization with the Kinase-Glo reagent (Promega, Madison, WI) was initially done in a 96-well format using buffer conditions previously used for the radiometric aminoacylation assay
8
: 10 mM MgCl2, 25 mM HEPES-KOH (pH 7.9), 50 mM KCl, 2.5 mM dithiothreitol, 0.1 mg/mL bovine serum albumin, 0.2 mM spermine, and 10 U/mL pyrophosphatase. To achieve separation between the high controls (wells not containing cold L-methionine and inhibitors) and the low controls (wells not containing inhibitors), different variables (incubation time, substrate concentration, and enzyme concentration) were manipulated independently while all other conditions were held constant. Optimized substrate concentrations were as follows: L-methionine, 32 µM; bulk E. coli tRNA (Sigma-Aldrich, St. Louis, MO), 200 µg/mL; and ATP, 100 nM. The L-methionine is in the range of the reported Km of 54 ± 42 µM,
15
whereas the concentration of ATP is lower than the Km reported for the related MetRS enzyme of E. coli (20 µM).
12
It was necessary to use a low concentration of ATP to achieve optimal separation between the high and low controls. Once the substrate concentrations were established, enzyme reaction progress curves (
A stepwise protocol is depicted in
Ultra-High-Throughput Screening Campaign Summary and Results.
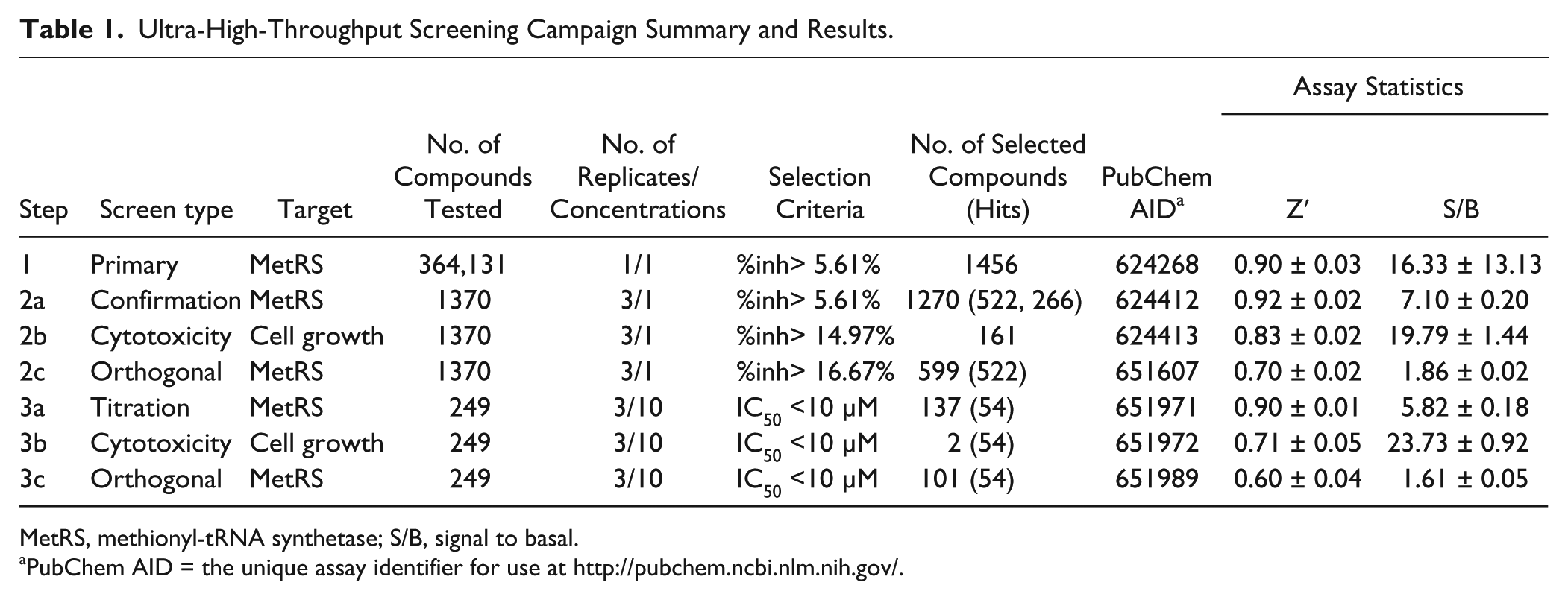
MetRS, methionyl-tRNA synthetase; S/B, signal to basal.
PubChem AID = the unique assay identifier for use at http://pubchem.ncbi.nlm.nih.gov/.
Cytotoxicity Counterscreen
Jurkat cells (clone E6.1, ATCC-TIB-152) were cultured in suspension with growth media containing RPMI-1640, 10% dialyzed fetal bovine serum, 0.1 mM nonessential amino acids (NEAA), 1 mM sodium pyruvate, 25 mM HEPES, 5 mM L-glutamine, and 1× Anti-Anti (Life Technologies, Carlsbad, CA) at 37 °C in 95% relative humidity and 5% CO2. Five hundred cells per well were added into 1536-well plates and incubated with compounds or DMSO alone for 18 h at 37 °C in 95% relative humidity and 5% CO2, at which time 5 µL of CellTiter-Glo (Promega) was added to all wells. Plates were centrifuged and incubated for 10 min at room temperature. Luminescence was measured on a ViewLux microplate reader (PerkinElmer). Doxorubicin was used a positive control. For more information, see Table 1 and PubChem AID 364.
Aminoacylation Assay (T. brucei and Human Mitochondrial MetRS)
To confirm the specificity of the hits from the HTS, a follow-up screen was performed using the aminoacylation assay that quantifies incorporation of [3H]L-methionine into tRNA. 8 Reactions were performed in 96-well filter plates with Durapore membranes (MSHVN4B10; Millipore, Billerica, MA) in volumes of 75 µL. The reaction buffer was 25 mM HEPES-KOH (pH 7.9), 10 mM MgCl2, 50 mM KCl, 0.2 mM spermine, 0.1 mg/mL bovine serum albumin, 2.5 mM dithiothreitol, 0.1 mM ATP, 240 nM [3H]L-methionine (83 Ci/mmol), 2% DMSO, and 0.1 U/mL pyrophosphatase (I1643; Sigma-Aldrich). Recombinant enzyme (10 nM) and test compounds (10 µM or 1 µM) were mixed with the buffer and preincubated for 15 min. To start the reaction, 400 µg/mL bulk E. coli tRNA (R4251; Sigma-Aldrich) was added and the plate was incubated without shaking at room temperature for 120 min. The reactions were stopped by the addition of 100 µL/well cold 10% trichloroacetic acid. The reaction components were separated from tRNA by filtration through a vacuum manifold and washed three times with 300 µL/well cold 10% trichloroacetic acid. The filter plates were dried, 25 µL/well scintillation fluid was added, and the counts on the plates were determined using a scintillation plate counter. Samples were run in triplicate, and the average activity of inhibitors was compared with that in control wells without inhibitors. Percent inhibition was calculated for the two concentrations (10 µM and 1 µM).
As a selectivity counterscreen, hit compounds were assayed against the human mitochondrial MetRS enzyme using the aminoacylation methodology as described above with a few differences. The assay was performed in a reaction buffer containing 50 mM Tris-HCl (pH 8.0), 6 mM MgCl2, 2.5 mM KCl, 0.2 mM spermine, 0.2 mg/mL bovine serum albumin, 2.5 mM dithiothreitol, 2.5 mM ATP, 240 nM [3H]L-methionine (83 Ci/mmol), 2% DMSO, and 0.1 U/mL pyrophosphatase. Recombinant enzyme (13 nM) and inhibitors (10 µM or 1 µM) were mixed with the buffer and preincubated for 15 min. To start the reaction, 200 µg/mL bulk E. coli tRNA was added. The plate was incubated without shaking at room temperature for 60 min and processed as described above.
T. brucei Growth Inhibition Assay
T. brucei (bloodstream form strain 427 from K. Stuart, Seattle Biomedical Research Institute, Seattle, WA) was cultured in HMI-9 medium 17 containing 10% fetal bovine serum, penicillin, and streptomycin at 37 °C with 5% CO2. Drug sensitivity of the T. brucei strain was determined in 96-well microtiter plates in triplicate with an initial inoculum of 1 × 104 trypomastigotes per well. Compound stock solutions were prepared in DMSO at 20 mM and/or 10 mM and added in serial dilutions for a final volume of 200 µL/well. Parasite growth was quantified at 48 h by the addition of AlamarBlue (Alamar Biosciences, Sacramento, CA). 18 Pentamidine isethionate (Aventis, Dagenham, UK) was included in each assay as a positive control. Compounds were tested in triplicate using a 4-point serial 3-fold dilution system starting at a test concentration of 10 µM. Inhibition at various concentrations was determined and the EC50 value was calculated by the online service CDD (www.collaborativedrug.com) using an equation to describe the sigmoidal dose-response curve.
Screening Data Acquisition, Normalization, Representation, and Analysis
Raw luminescence data or FP values were uploaded into the SRIMSC institutional HTS database (Symyx Technologies, Santa Clara, CA). Raw luminescence data were imported directly while FP values were calculated using the following equation prior to upload:
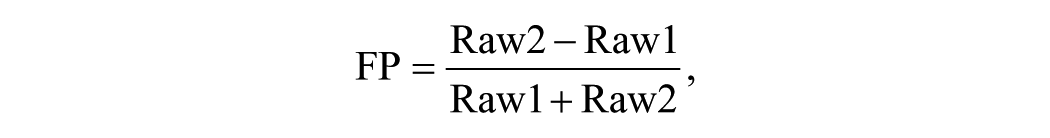
where Raw1 is defined as polarized fluorescence in the P channel and Raw2 is defined as polarized fluorescence in the S channel.
In either case, response data were normalized using the following equation:
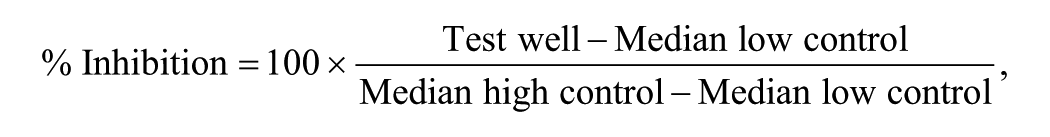
where high control represents wells from the same plate containing 1.5 µM control compound CID 18353708 or, in the case of the cytotoxicity assay, 8 µM doxorubicin, and low control represents wells treated with DMSO only.
Data were normalized on a per-plate basis, and each assay plate underwent a quality control check; a Z′ value greater than 0.5 was required for acceptance of data. 19 CRC data were analyzed by plotting the average percent inhibition of the triplicate results against the compound concentration. A four-parameter equation describing a sigmoidal concentration-response curve was then fitted with adjustable baseline using Assay Explorer software (Symyx). Similarly, for the graphs shown in this article, CRCs and pIC50 values were generated by Prism (GraphPad Software, San Diego, CA). All further results from the screens can be found at the National Institute of Health’s (NIH’s) PubChem website (http://pubchem.ncbi.nlm.nih.gov/) using the AID listed in Table 1 .
Cheminformatics
Shared scaffolds of active compound families from the confirmation screen and CRC experiments were identified using a Maximum Common Substructure hierarchical clustering (ChemAxon LibraryMCS 5.10.2; ChemAxon, Cambridge, MA). The physical properties (i.e., molecular mass, topological polar surface area, chiral atoms, H-bond acceptors/donors, ring count, and rotatable bonds) of the compounds tested in dose-response format were calculated (ChemAxon Instant JChem 6.2.2).
Chemicals
The MLSMR library was provided by BioFocus DPI (South San Francisco, CA) through the NIH’s Roadmap Molecular Libraries Initiative. Details regarding compound selection for this library can be found online at http://mli.nih.gov/mli/compound-repository/mlsmr-compounds/. Briefly, this library is a highly diversified collection of small molecules (more than 50% in the molecular weight range of 350–410 g/mol) and comprises both synthetic and natural products, from either commercial or academic sources, that can be grouped into the four following categories: (1) specialty sets of known bioactive compounds such as drugs and toxins (0.65%), (2) focused libraries aimed at specific target classes (2.85%), (3) noncommercial sources (7.4%), and (4) diversity sets covering a large area of the chemical space (89.1%).
The positive control compound CID18353708 was synthesized as described elsewhere. 8
Results
Implementation of the Orthogonal HTS Assays
A luminescence-based assay was designed and run as a primary screen to identify inhibitors of T. brucei MetRS by measuring the ATP depletion during the aminoacylation reaction. Residual ATP was measured through the luciferase-catalyzed reaction of the mono-oxygenation of luciferin substrate (Kinase-Glo) in which the luminescence signal is inversely proportional to MetRS activity ( Fig. 1 ).
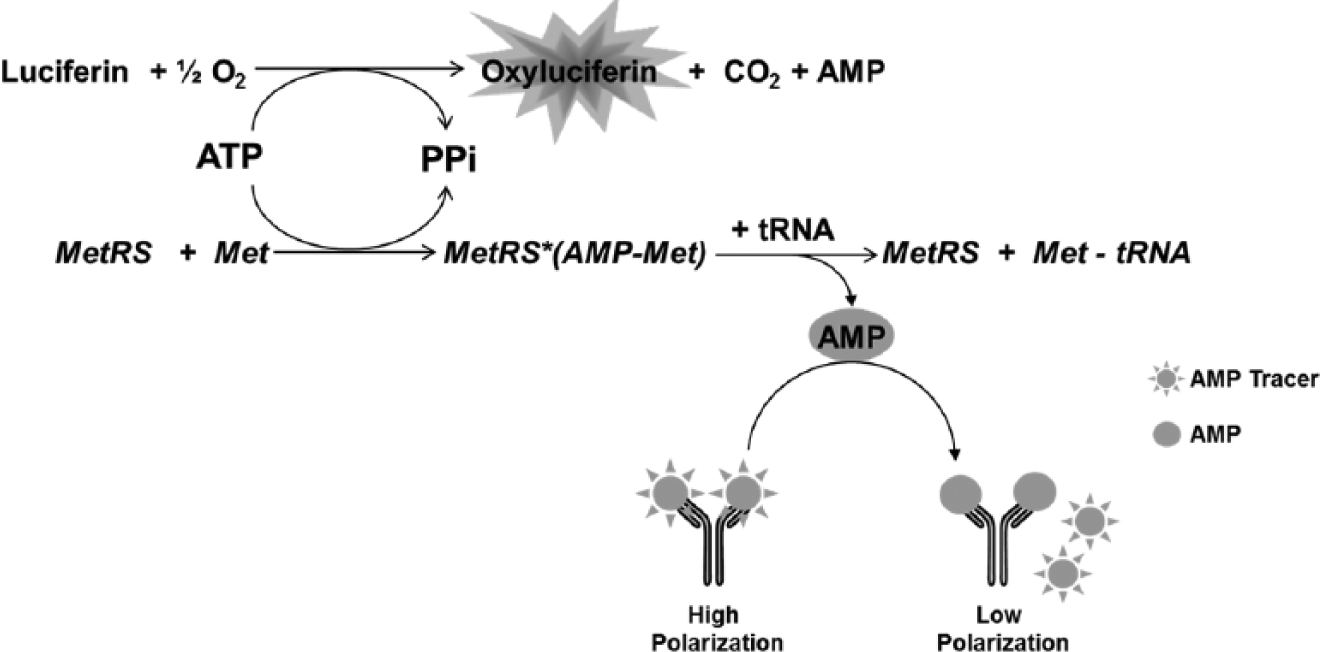
Trypanosoma brucei methionyl-tRNA synthetase (MetRS)–catalyzed reaction. The primary screen assay focuses on the luminescence signal generated from the luciferase-catalyzed reaction using residual adenosine triphosphate (ATP). The orthogonal counterscreen focuses on the fluorescence polarization signal generated from the displacement of a labeled adenosine monophosphate (AMP) molecule from a selective antibody when AMP is generated from the T. brucei MetRS-catalyzed reaction.
To reduce the cost and increase the throughput of the HTS campaign, the assay was miniaturized and implemented in a 1536-well plate format. Optimization of the assay was performed by assessing the effect of critical variables that affect the aminoacylation reaction (i.e., MetRS concentration, ATP concentration, and DMSO tolerance; see the supplemental material). Three sets of controls (n = 24 per set) were placed on every assay plate: the high control (1.5 µM CID 18353708 final), the low control containing DMSO (0.9% final), and a 50% inhibition control containing CID 18353708 at its IC50 concentration (40 nM). These controls were used to (1) ensure T. brucei MetRS activity, (2) normalize the data, and (3) monitor the data quality by measuring Z′ and signal to basal (S/B). Two additional controls were placed on every plate: a positive control containing ATP and tRNA but no L-methionine and also a background control without ATP to ensure proper compound and reagent dispensing. Good separation of the controls was observed as shown in the plot of the raw luminescence values measured during the primary screen (
Fig. 2
). All primary HTS data were normalized to the inhibition of compound CID 18353708 versus the DMSO-only wells and were used to produce a scatterplot to aid in visualization of the activity across the HTS campaign (
Fig. 3
). Notably, the positive control compound has been formerly shown to produce sub-micromolar inhibition of T. brucei MetRS.
18
Under the final conditions listed in
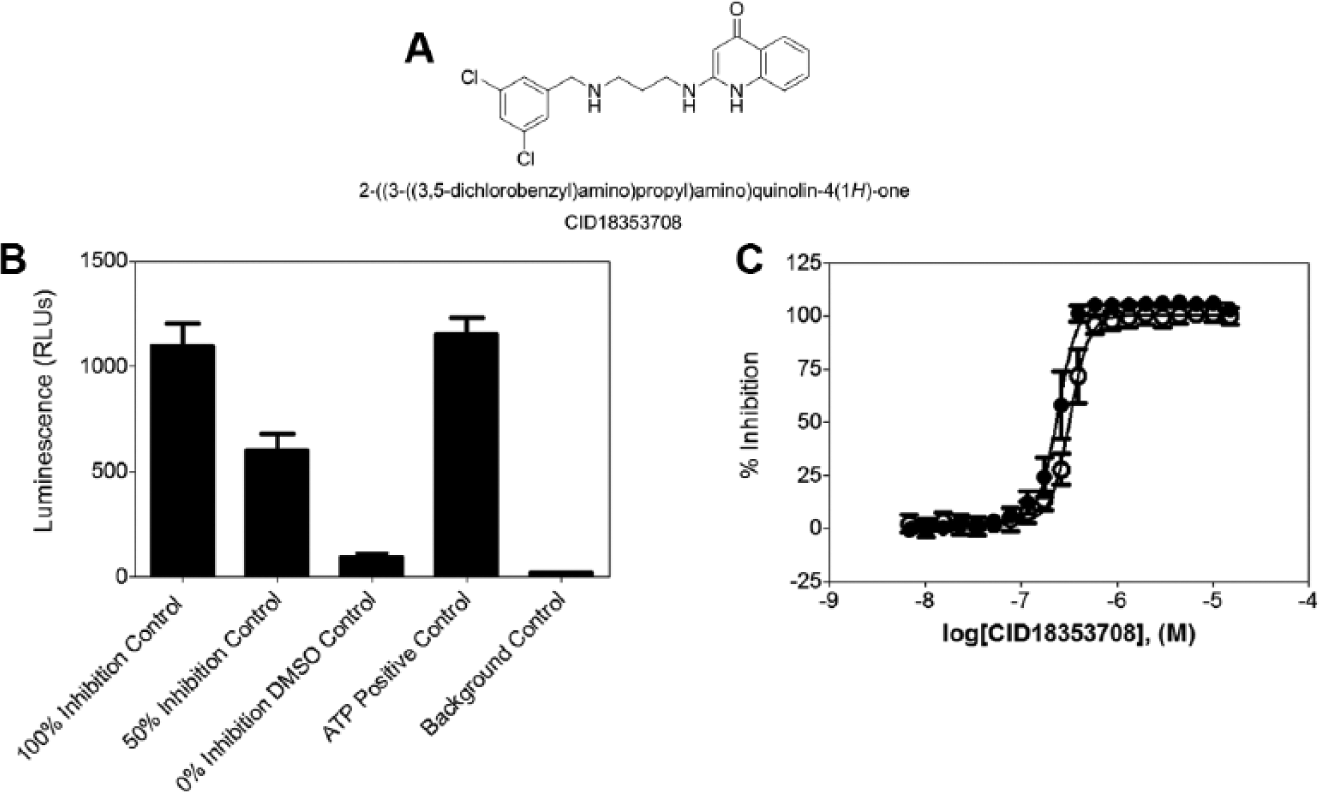
Assay controls. (
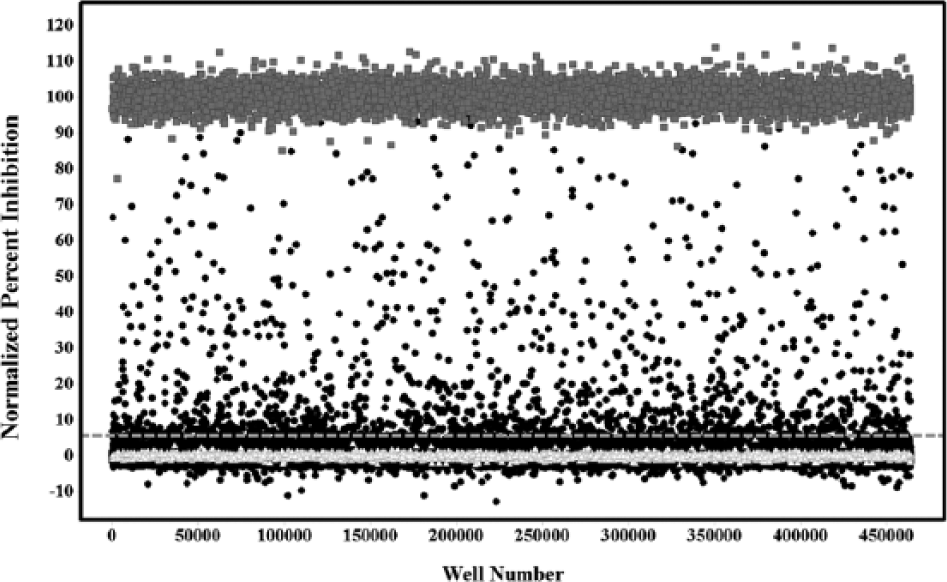
Primary screen scatter plot. (△) Represent low control wells containing DMSO only, (●) represent data wells containing compounds, and (■) represent high control wells containing compound CID 18353708. The dashed line represents the hit cutoff. Compounds with inhibition above this line were considered primary actives or hits.
The orthogonal FP counterscreen was implemented to help prioritize hit selection by removing potential artifacts from the luminescent detection system (quenchers, luciferase inhibitors, etc.). This assay measures AMP production in the T. brucei MetRS-catalyzed aminoacylation reaction as a function that is proportional to MetRS activity. Optimal assay conditions were in accord with the manufacturer’s recommendations (BellBrook Labs), and all subsequent experiments were performed using the conditions listed in
T. brucei MetRS Inhibition Screening Campaign Results
The T. brucei MetRS inhibition screening campaign is summarized in Table 1 . A total of 364,131 distinct samples from the MLSMR were tested during the primary screen at a nominal concentration of 12 µM. While the Z′ data indicate robustness at each step throughout the HTS campaign, we did observe a high degree of variation within the S/B ratio for the MetRS luminescence assay. This variability between batches was due to basal signal levels approaching zero in terms of raw data relative light units. Hence, even a very small shift in the raw data resulted in a fairly significant shift in the normalized S/B. From the primary screen 1456 compounds (i.e., 0.4% of the total library) were identified as actives, exhibiting a percent inhibition greater than the nominal hit cutoff of 5.61% (average background + 3 SD). Of these, 1370 were available from the MLSMR for testing at the confirmation and counterscreen stages.
To confirm inhibitory activity, eliminate luminescent artifacts, and eliminate broadly cytotoxic compounds, the 1370 samples were retested in triplicate in three different assays: (1) the luminescence MetRS ATP depletion assay, (2) the fluorescence polarization AMP production assay, and (3) the luminescence-based Jurkat cell cytotoxicity assay. The 161 compounds found to be active in the cytotoxicity assay were eliminated from further follow-up. Noncytotoxic compounds with activity in both the ATP depletion and AMP formation assays were prioritized for CRC studies (522 compounds). To facilitate compound selection for titration studies, the 522 compounds were classified into 86 clusters based on their most common substructure. The compounds for CRC studies were selected based on two main criteria: (1) the most potent compounds from each cluster were selected to ensure chemical diversity, and (2) all compounds that exhibited a 1:1 ± 0.6 correlation between the IC50s of the two target-based assays were selected to proceed. Under these criteria, 266 compounds were selected for IC50 titration, of which 249 compounds were available from the MLSMR.
The CRCs for the 249 compounds were determined via testing in a triplicate 10-point 3-fold serial dilution format against the same three assays. Individual CRCs offered better insight into the selectivity of the compounds and their inhibition profiles between the different assays, facilitating the triage of cytotoxic compounds such as CID 16312830 and luminescent artifacts such as CID 2810697 (
Fig. 4
and
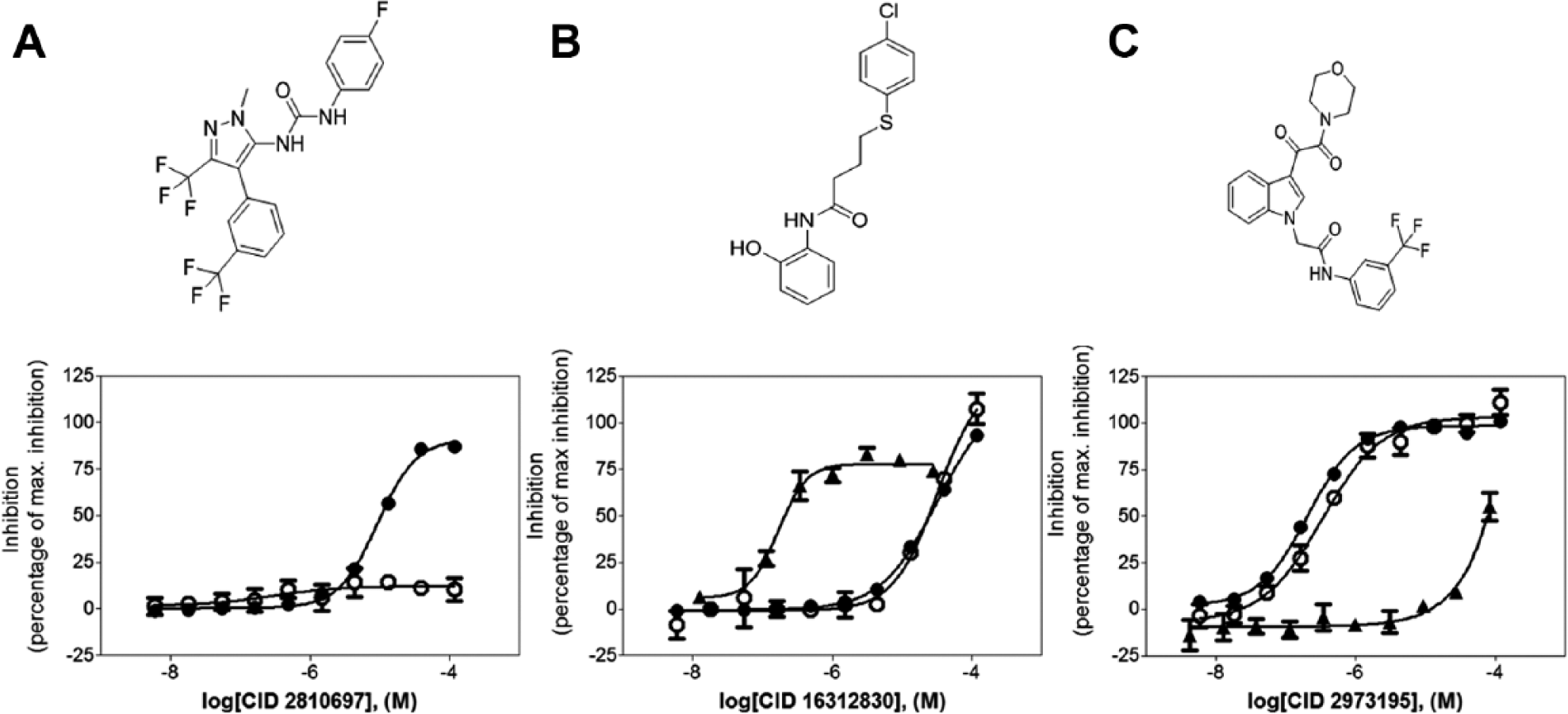
Compound dose-response curves. (●) Luminescence curve, (○) fluorescence polarization assay curve, and (▲) cytotoxicity assay curve. (
Low-Throughput Secondary Assays
The 54 confirmed active compounds were tested for activity in an aminoacylation assay versus T. brucei cells grown in culture. The data are shown in
To assess parasite versus human selectivity, the 54 confirmed active compounds were assayed against the human mitochondrial MetRS using the aminoacylation methodology. Forty-four compounds (81%) showed weak or no inhibition of the human mitochondrial MetRS (i.e., <20% at 10 µM). Five compounds had >30% inhibition against the human enzyme, indicating that the screen was successful in identifying numerous active compounds that are specific for the trypanosome over the mammalian MetRS. Only one compound (CID 46902334) was moderately potent toward the human mitochondrial MetRS (80.3% inhibition at 10 µM, 14.3% inhibition at 1 µM). As noted earlier, one of the selection criteria for compound progression was low cytotoxicity: a CC50 against Jurkat cells of >10 µM.
Finally, the 54 compounds were tested for growth inhibition activity (EC50) of T. brucei cultures. Twelve compounds (22.2%) showed some degree of inhibition at 10 µM. The most potent compounds had apparent EC50s in the 2-µM range. These compounds are screening hits that can penetrate cells well, an important criterion in drug discovery efforts.
Discussion
Orthogonal screens were successfully developed and implemented to identify potent small-molecule inhibitors of T. brucei MetRS, using ATP and AMP levels as reporters of MetRS activity via luminescence and fluorescence polarization detection methods, respectively. Both assay technologies demonstrated robust performance on the Kalypsys GNF µHTS platform. As noted, batch-to-batch variability was observed for the overall S/B ratio of the primary screen. This finding was of little concern given that (1) the data were normalized on a plate-per-plate basis, (2) the relative shift in the raw data was minor yet resulted in substantial shifts in S/B, and (3) the pharmacological control behaved well, giving the expected IC50. We maintained Z′ values >0.8 and an average throughput of approximately 15,000 compounds per hour throughout the HTS campaign (
Use of an orthogonal FP-based counterscreen to eliminate detection format-specific hits (such as CID 2810697) eliminated nuisance compounds early in the process and allowed a high confirmation rate in subsequent secondary assays. In addition, cytotoxic compounds (i.e., CID 16312830) were eliminated from advancement based on results in the Jurkat assay. This battery of complementary assays proved to be an effective way to quickly identify inhibitors of aaRS. Initial SAR analysis for CID 2973195, the indole oxalamide shown in Figure 4C , demonstrated excellent “out-of-the-box” potency in the primary and FP assays and relatively no cytotoxicity; pIC50 values in respective order are 6.77, 6.53, and <4.08 (IC50 values of 171 nM, 292 nM, and >83 µM, respectively).
Virtually all compounds selected for low-throughput screening (52/54) were confirmed to be active in the T. brucei aminoacylation assay. This methodology measures the whole MetRS enzyme reaction by quantifying the incorporation of [3H]L-methionine into tRNA. These confirmatory results were reassuring and showed that the HTS assay cascade used was effective.
Since human mitochondrial MetRS is a potential off-target for these hits, we tested the same set of compounds against this human mitochondrial enzyme. It should be noted that human cells also contain a cytoplasmic MetRS, but the homology to the T. brucei MetRS in the active site is relatively low (46% identical residues), whereas the homology of the human mitochondrial MetRS to the active site of T. brucei MetRS is higher (79% identical residues). The human selectivity screen showed that 81% of compounds had ≤20% inhibition of the human mitochondrial MetRS, meaning that if developed as drugs, relatively few compounds are likely to cause host toxicity, inhibiting this human enzyme in patients with HAT. This selectivity in the hit set was likely enhanced by earlier implementation of the Jurkat cytotoxicity screen. It can be concluded that the differences between the human mitochondrial and trypanosomal MetRS (21% amino acid sequence difference in the ATP-methionine binding sites) are sufficient to make selective inhibition by small molecules possible. Structural studies of the compounds bound to the T. brucei MetRS may aid understanding and guide further enhancements in potency and selectivity of the hit compounds.
Finally, 12 of the 54 HTS hit compounds (22%) inhibited growth of T. brucei grown in culture. It is assumed that in each case, the growth inhibition observed is due to inhibition of the MetRS target, although further work will be necessary to confirm this hypothesis. We have shown in previous work that aminoquinolone MetRS inhibitors in fact block protein synthesis in cultured T. brucei cells. 10 It is likely that many of 42 compounds inactive versus cultured T. brucei have poor membrane permeability and thus do not reach the target. Also, the weaker MetRS inhibitors may not sufficiently bind the target to affect growth at the concentrations tested, even if they enter the cells.
The screening approach used during this campaign exhibited great robustness and the ability to rapidly identify and triage potent, noncytotoxic compounds. In combination with secondary assays, several promising selective compounds (with pIC50 values as high as 7.36 or IC50 as low as 44 nM) were identified and will be pursued in future studies for further development as novel potent molecular probes, perhaps ultimately leading to a drug to treat HAT. This orthogonal screening approach, using an automated platform, has proven to be an excellent tactic to screen large compound libraries that can certainly be extended to other aaRSs.
Footnotes
Acknowledgements
We thank Pierre Baillargeon and Lina DeLuca (Lead Identification, Scripps Florida) for compound management and our MSGPP coworkers for contributions to the early stages of this project.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health’s Roadmap Initiative through grants U54MH084512 (LPR, CE, FM, SB, JL, TB, LS, TS, PH), PO1AI067921 (Medical Structural Genomics of Pathogenic Protozoa [MSGPP]), and RO1AI084004 to WGJH and RO1 AI097177 to FB and EF.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.