
Abstract
As a label-free technology, mass spectrometry (MS) enables assays to be generated that monitor the conversion of substrates with native sequences to products without the requirement for substrate modifications or indirect detection methods. Although traditional liquid chromatography (LC)–MS methods are relatively slow for a high-throughput screening (HTS) paradigm, with cycle times typically ≥60 s per sample, the Agilent RapidFire High-Throughput Mass Spectrometry (HTMS) System, with a cycle time of 5–7 s per sample, enables rapid analysis of compound numbers compatible with HTS. By monitoring changes in mass directly, HTMS assays can be used as a triaging tool by eliminating large numbers of false positives resulting from fluorescent compound interference or from compounds interacting with hydrophobic fluorescent dyes appended to substrates. Herein, HTMS assays were developed for multiple protease programs, including cysteine, serine, and aspartyl proteases, and applied as a confirmatory assay. The confirmation rate for each protease assay averaged <30%, independent of the primary assay technology used (i.e., luminescent, fluorescent, and time-resolved fluorescent technologies). Importantly, >99% of compounds designed to inhibit the enzymes were confirmed by the corresponding HTMS assay. Hence, HTMS is an effective tool for removing detection-based false positives from ultrahigh-throughput screening, resulting in hit lists enriched in true actives for downstream dose response titrations and hit-to-lead efforts.
Introduction
Proteases are a class of enzymes that function to catalyze the degradation of proteins and peptides by the hydrolysis of amide bonds, and they are estimated to encompass at least 500–600 members, or about 2% of the human genome. 1 Within the protease enzyme family, several different catalytic mechanisms have evolved to cleave peptides, including serine proteases, cysteine proteases, and threonine or amino-terminal proteases that possess an amino acid side chain that participates in covalent catalysis. 2 Other classes, such as aspartyl proteases and metalloproteinases, carry out peptide bond cleavage noncovalently through generation of an activated water molecule that functions as a nucleophile. 2 Proteases play a key role in many signaling pathways and proteolytic cascades with misregulation implicated in a wide range of diseases, including cardiovascular disorders, neurodegeneration, infectious diseases, and cancer.1,2 Multiple examples of successful marketed drugs exist that target proteases. Recent examples include telaprevir and boceprevir, which target the NS3 serine protease of the hepatitis C virus; 3 the antihypertensive aliskiren, which inhibits the aspartyl protease renin; 4 and the antidiabetic class of dipeptidyl peptidase 4 inhibitors, exemplified by sitagliptin. 5
When a protease of therapeutic interest is discovered, efforts to identify small-molecule inhibitors of the enzyme’s activity tend to focus on converting known substrates into stable peptidomimetic inhibitors, chemically modifying known inhibitors, or performing screens of chemical libraries to identify novel small-molecule hit classes with the desired inhibitory, pharmacokinetic and pharmacodynamic, and selectivity profiles.2,6 Many technologies that read out enzyme activity by monitoring changes in light are amenable to interrogating large chemical libraries of >1 million compounds in 1536- or 3456-well plate formats. Representative technologies include fluorescent intensity (FLINT), fluorescence resonance energy transfer (FRET), time-resolved FRET (TR-FRET), and luminescent readouts. These approaches have successfully led to the identification of lead classes for multiple proteases; however, the identification of validated hit classes is hindered by a high number of false positives. Assay-dependent false positives can be due to many causes, including aggregation and fluorescent compound interference. 7 Because commonly used assay formats for proteases require the modification of a peptide recognition sequence with a bulky aromatic fluorescent dye, a quencher, a luciferase substrate, or a europium or terbium chelate, the potential of compounds to interact with the substrate itself must also be considered. Recently, a study with caspase 6 identified a highly selective lead series that inhibited the enzyme via an uncompetitive mechanism. Further investigation discovered that the presence and character of the fluorophore appended to the P1’ position of the peptide substrate in the enzymatic assays directly lead to additional compound–substrate interactions that modulate the binding affinity. 8 Studies have also demonstrated that activation of the NAD+-dependent protein deacetylase SIRT1 by resveratrol is dependent on the presence of the fluorophore on the Fluor-de-Lys peptide substrate (Enzo Life Sciences, Farmingdale, NY).9–11
To reduce wasted time and monetary expenditures, effective triaging strategies must be implemented to remove detection-based false positives from high-throughput screening (HTS) hit lists by introduction of an orthogonal confirmation assay or assays. Mass spectrometry offers an ideal functional readout to remove detection-based false positives. Mass spectrometry–based assays are quantitative and label-free, allowing the use of substrates and products with native sequences without the requirement for modifying substrates with bulky fluorophores or the application of indirect detection methods. 12 Without the requirement for fluorophores, there is a reduction in false positives due to compound interference or interactions with the fluorescent dyes. Some common challenges with mass spectrometry screening are the requirement for sample desalting and well-based instead of plate-based reading, which results in traditional liquid chromatography methods that typically require a cycle time ≥60 s. For a typical ultrahigh-throughput screening (uHTS) confirmation screen wherein ≥30,000 compounds are screened at least in duplicate, about 50 days of continuous run time on one instrument would be required. With a cycle time averaging 5 to 10 s per sample, or about 1 h per 384-well plate, however, the Agilent RapidFire High-Throughput Mass Spectrometry (HTMS) system (Agilent, Wakefield, MA) could complete the same confirmation assay in about 8 days of continuous run time on one instrument. Therefore, HTMS has enabled the rapid analysis of compounds at throughputs compatible with those of HTS.12–21 RapidFire functions by combining automated microscale solid-phase extraction in place of high-performance liquid chromatography (HPLC) for sample desalting with a triple quadrupole mass spectrometer for detection and quantitation of analytes. 12
Recently, several HTS campaigns of protease targets have been run, including thrombin, caspase 6, β-secretase (BACE1), and HIV protease. Thrombin, a serine protease responsible for converting soluble fibrinogen to fibrin, plays a central role in the coagulation cascade for which inhibition is an established mechanism for preventing thromboembolism. 22 Caspase 6, a cysteine protease, has been identified as a critical component of apoptosis in neurons, especially in Alzheimer’s disease (AD) and Huntington’s disease (HD);23,24 therefore, inhibitors of caspase 6 are a potential treatment for neurodegeneration. 25 BACE1 is an aspartyl protease responsible for initiating a critical step in the production of Aβ40 and Aβ42 peptides, which are a major constituent of amyloid plaques in AD; therefore, inhibitors of BACE1 may be a potential mechanism for treating the disease. 26 Finally, HIV protease, an aspartyl protease, comprises a critical component of the HIV replication cycle by processing polypeptides transcribed from the gag and pol genes late in the viral cycle. 27 Inhibition of this enzyme leads to production of immature noninfectious viral progeny. Herein, we describe the development and application of RapidFire HTMS assays as orthogonal confirmation screens for the 4 proteases described above. Independent of the detection technology applied as the primary assay to screen a library of >1 million compounds, the confirmation rate for each protease assay by HTMS averaged <30%; however, all compounds designed to inhibit the enzyme and previously validated hit classes were confirmed. The results demonstrate that HTMS is an effective tool for removing potential detection-based false positives in uHTS screens, thus providing hit lists enriched in true actives for downstream dose response titrations and hit-to-lead efforts.
Materials and Methods
Reagents and Materials
HEPES buffer, HPLC-grade water, and bovine serum albumin (BSA) were obtained from Fisher Scientific (Pittsburgh, PA). Sodium acetate pH 5.5 was obtained from USB Corporation (Santa Clara, CA). HPLC-grade acetonitrile, DMSO, formic acid, Triton X-100, glycerol, Brij-35, sodium acetate, pH 4.5, trifluoroacetic acid, dithiothreitol, and sodium chloride were purchased from Sigma-Aldrich (St. Louis, MO). Data analysis was performed using the Graphpad Prism (GraphPad Software, La Jolla, CA) and Tibco Spotfire (Boston, MA) software packages. Protocols for the uHTS assays are reported in the Supplemental Material.
Caspase 6 HTMS Assay
Recombinant human caspase 6 containing both the 18 and 11 kDa subunits (Enzo Life Sciences) was diluted in caspase 6 buffer (10 mM HEPES, pH 7.2, 2 mM dithiothreitol, 0.005% BSA, 0.001% Brij-35), and 15 uL was dispensed to columns 1–22 of a 384-well REMP polypropylene plate (Brooks, Poway, CA) for a final caspase 6 concentration of 0.5 U/uL. As a background control, 15 uL of caspase 6 buffer was added to columns 23 and 24. Compounds from a 2 mM DMSO stock were added via a 100 nl pin tool to all wells for a final compound concentration of 10 uM and 1% DMSO. Plates were incubated for 20 min at room temperature. Post incubation, 5 uL of substrate peptide with a sequence of QVEIDG (CPC Scientific, Sunnyvale, CA) was added to each well of the plate for a final substrate concentration of 72 uM. Plates were incubated for 200 min at 25 °C, followed by quenching with 40 uL water containing 2% vol/vol formic acid (FA). Plates were sealed and centrifuged (1500×g for 5 min) prior to storage at −80 °C. Prior to analysis, plates were thawed at room temperature, then centrifuged at 1500×g for 5 min. Plates were loaded onto the Agilent RapidFire RF300 HTMS system, wherein samples were sipped for 600 ms, and the sip sensor detection signals the diversion of sample from the loop onto a C4 solid phase extraction (SPE) cartridge, where salts are washed off for 3000 ms with a mobile phase of water with 0.1% vol/vol FA and 0.01% vol/vol trifluoroacetic acid (TFA) at a flow rate of 1.5 ml/min. The column path is switched, and the sample is eluted off the cartridge and flowed into the mass spectrometer for 3500 ms with a mobile phase of 80% vol/vol acetonitrile, 0.1% vol/vol FA, and 0.01% vol/vol TFA at a flow rate of 1.5 ml/min. The aspirator tip was washed separately with water and acetonitrile between sample aspirations (each for 600 ms) to minimize carryover contamination. Mass spectrometry was carried out using a Sciex API4000 triple quadrupole mass spectrometer (Applied Biosystems, Concord, ON, Canada) equipped with electrospray ionization (ESI) operated in the positive-ion mode. The substrate QVEIDG and product QVEID were detected using multiple reaction monitoring (MRM) with Q1/Q3 transitions at m/z 660.4 to 357.3 and m/z 603.2 to 200.2, respectively, with a dwell time of 100 ms for each transition. The mass spectrometer used an ESI voltage of 5000 V and a source temperature of 650 °C. Extracted ion chromatograms for each transition were integrated and processed using the RapidFire Integrator software. The data for each well were normalized by monitoring product conversion with the ratio of AUCproduct / (AUCproduct + AUCsubstrate).
For the determination of the Michaelis constant (Km) of the caspase 6 substrate, the initial rates of the reaction with 0.5 U/ml caspase 6 were measured, then fit to the Michaelis–Menten equation with GraphPad Prism.
BACE1 HTMS Assay
To a 384-well REMP polypropylene plate (Brooks), 0.5 uL of 2 mM compound or control in 75% DMSO and 25% water was pre-plated via an ATS100 acoustic dispenser (EDC Biosystems, Fremont, CA) for a final compound concentration of 40 uM. Recombinant soluble human BACE1, composed of amino acids 1–454, was expressed and purified from Escherichia coli. 28 The N-terminal pro-domain was autocatalytically cleaved to yield soluble mature BACE1 (autoBACE1) composed of amino acids 41–454. AutoBACE1 was diluted in BACE1 buffer (20 mM NaOAc, pH 4.5, 5% glycerol, 0.01% Brij-35), and 15 uL was dispensed to columns 1–22 of a 384-well REMP polypropylene plate for a final BACE1 concentration of 4 nM. As a background control, 15 uL of BACE1 buffer was added to columns 23 and 24. Plates were centrifuged (1500×g for 1 min) and incubated at ambient temperature for 30 min. Post incubation, 10 uL of the substrate peptide with a sequence of KTEEISEVNLDAEFRHDK (Genscript, Piscataway, NJ) was added to each well of the plate for a final substrate concentration of 10 uM and 1.7% DMSO. Plates were incubated for 210 min at 25 °C followed by quenching with 25 uL water with 2% formic acid containing 600 nM of the internal standard with the sequence KTEEISEVN[L-U7]-OH (CPC Scientific). Plates were sealed and centrifuged (1500×g for 5 min) prior to storage at −80 °C. Prior to analysis, plates were thawed at room temperature, then centrifuged at 1500×g for 5 min. Plates were loaded onto the Agilent Rapidfire RF300 HTMS system, wherein samples were sipped for 600 ms, and the sip sensor detection signals the diversion of sample from the loop onto a C4 SPE cartridge, where salts are washed off for 2500 ms with a mobile phase of water with 0.1% vol/vol FA and 0.01% vol/vol TFA at a flow rate of 1.5 ml/min. The flow path is switched, and the sample is eluted off the cartridge and flowed into the mass spectrometer for 3000 ms with a mobile phase of 80% vol/vol acetonitrile, 0.1% vol/vol FA, and 0.01% vol/vol TFA at a flow rate of 1.5 ml/min. The aspirator tip was washed separately with water and acetonitrile between sample aspirations (each for 600 ms) to minimize carryover contamination. Mass spectrometry was carried out using a Sciex API4000 triple quadrupole mass spectrometer (Applied Biosystems) equipped with ESI operated in the positive-ion mode. The reaction product KTEEISEVNL and internal standard KTEEISEVN[L-U7]-OH were detected using MRM with Q1/Q3 transitions at m/z 581.8 to 246.6 and m/z 584.9 to 253.1, respectively, with a dwell time of 100 ms for each transition. The mass spectrometer used an ESI voltage of 5000 V and a source temperature of 650 °C. Extracted ion chromatograms for each transition were integrated and processed using the RapidFire Integrator software. The data for each well were normalized by monitoring product conversion with the ratio of AUCproduct / (AUCinternal standard).
HIV-1 Protease HTMS Assay
To a 384-well REMP polypropylene plate (Brooks), 15 uL of full length HIV-1 protease (Proteros, Planegg, Germany) in HIV protease assay buffer (50 mM NaOAc, pH 5.5, 100 mM NaCl, 0.0015% Triton X-100, 0.01% BSA) was added to columns 1–22. Buffer was added to columns 23 and 24. Then, 0.1 uL of compound in 75% DMSO and 25% water was transferred via a Janus pintool (PerkinElmer, Waltham, MA) to the wells and incubated at room temperature for 15 min. Reactions were initiated with the addition of 5 uL of the peptide substrate KVSLNFPIL (CPC Scientific) diluted in assay buffer, followed by 60 min incubation at room temperature. The final assay conditions are 1.6 nM HIV-1 protease, 60 uM peptide substrate, and 10 uM compound in assay buffer. The enzyme reactions were quenched with 20 uL of 2% formic acid containing 2 uM internal standard [P-U6]IL (CPC Scientific). Plates were sealed and centrifuged (1500×g for 5 min) prior to storage at −80 °C. Prior to analysis, plates were thawed at room temperature, then centrifuged at 1500×g for 5 min. Plates were loaded onto the Agilent RapidFire RF300 HTMS system, wherein samples were sipped for 600 ms, and the sip sensor detection signals the diversion of sample from the loop onto a C4 SPE cartridge, where salts are washed off for 2500 ms with a mobile phase of water with 0.1% vol/vol FA and 0.01% vol/vol TFA at a flow rate of 1.5 ml/min. The column path is switched, and the sample is eluted off the cartridge and flowed into the mass spectrometer for 3000 ms with a mobile phase of 80% vol/vol acetonitrile, 0.1% FA vol/vol, and 0.01% vol/vol TFA at a flow rate of 1.5 ml/min. The aspirator tip was washed separately with water and acetonitrile between sample aspirations (each for 600 ms) to minimize carryover contamination. Mass spectrometry was carried out using a Sciex API4000 triple quadrupole mass spectrometer (Applied Biosystems) equipped with ESI operated in the positive-ion mode. The reaction product PIL and internal standard [P-U6]IL were detected using MRM with Q1/Q3 transitions at m/z 342 to 183 and m/z 348 to 189.3, respectively, with a dwell time of 100 ms for each transition. The mass spectrometer used an ESI voltage of 5000 V and a source temperature of 650 °C. Extracted ion chromatograms for each transition were integrated and processed using the RapidFire Integrator software. The data for each well were normalized by monitoring product conversion with the ratio of AUCproduct / (AUCinternal standard).
Thrombin HTMS Assay
Recombinant human α-thrombin (Factor IIa; Enzyme Research Labs, South Bend, IN) was diluted in thrombin buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 0.001% Brij-35, 0.05% BSA), and 15 uL was dispensed to columns 1–22 of a 384-well REMP polypropylene plate (Brooks) for a final thrombin concentration of 5 nM. As a background control, 15 uL of thrombin buffer was added to columns 23 and 24. Plates were centrifuged (1500×g for 1 min), and compounds were added via a 100 nl pin tool for a final compound concentration of 10 uM, then incubated at ambient temperature for 30 min. Nafamostat (Cayman Chemical Co., Ann Arbor, MI), a thrombin inhibitor, 29 was added at its IC50 concentration of 150 nM final to column 1, and DMSO was added to column 2 as a 0% inhibition control. Post incubation, 5 uL of the substrate peptide with a sequence of DFLAEGGGVRGPRV (CPC Scientific) was added to each well of the plate for a final substrate concentration of 29 uM, which is equivalent to 0.5 Km. This amino acid sequence corresponds to the fibrinogen Aα chain cleavage site, which thrombin cleaves between the arginine and glycine residues. 30 Plates were incubated for 30 min at 25 °C, followed by quenching with 20 uL water with 2% vol/vol FA. Plates were sealed and centrifuged (1500×g for 5 min) prior to storage at −80 °C. Prior to analysis, plates were thawed at room temperature, then centrifuged at 1500×g for 5 min. Plates were loaded onto the Agilent Rapidfire RF300 HTMS system, wherein samples were sipped for 600 ms, and the sip sensor detection signals the diversion of sample from the loop onto a C4 SPE cartridge, where salts are washed off for 2500 ms with a mobile phase of water with 0.1% vol/vol FA and 0.01% vol/vol TFA at a flow rate of 1.5 ml/min. The column path is switched, and the sample is eluted off the cartridge and flowed into the mass spectrometer for 3000 ms with a mobile phase of 80% vol/vol acetonitrile, 0.1% vol/vol FA, and 0.01% vol/vol TFA at a flow rate of 1.5 ml/min. The aspirator tip was washed separately with water and acetonitrile between sample aspirations (each for 600 ms) to minimize carryover contamination. Mass spectrometry was carried out using a Sciex API4000 triple quadrupole mass spectrometer (Applied Biosystems) equipped with electrospray ionization operated in the positive-ion mode. The substrate DFLAEGGGVRGPRV and reaction product GPRV were detected using MRM with Q1/Q3 transitions at m/z 715.7 to 235.2 and m/z 428.2 to 329.2, respectively, with a dwell time of 100 ms for each transition. The mass spectrometer used an ESI voltage of 5000 V and a source temperature of 650 °C. Extracted ion chromatograms for each transition were integrated and processed using the RapidFire Integrator software. The data for each well were normalized by monitoring product conversion with the ratio of AUCproduct / (AUCsubstrate + AUCproduct).
Results and Discussion
Protease uHTS Primary Screens
Screening programs were initiated for identifying chemical matter for hit-to-lead efforts for 4 protease targets for different therapeutic indications. The Merck compound collection, which is >1 million compounds, was screened against each of these targets in either 1536-well plate or 3456-well plate format assays using a number of different detection strategies, all dependent on monitoring either the increase or decrease of light. For BACE1, an aspartyl protease, activity was monitored in a 3456-well plate format assay using a quenched TR-FRET (qTR-FRET) format wherein a peptide based on the Swedish mutant sequence of amyloid precursor protein (APP, which is associated with early-onset Alzheimer’s disease) was appended with a europium–terpy chelate and a QSY7 quencher. 31 Cleavage was detected as an increase in the emission of light at 618 nm after excitation at 340 nm via time-resolved fluorescence with a delay time of 70 us. Primary screens for the aspartyl HIV protease and the serine protease thrombin used the quenched FRET peptide substrates SensoLyte 520 HIV Protease substrate (Anaspec, Fremont, CA) and Sensolyte 520 Thrombin substrate (Anaspec), respectively. The substrates append peptide recognition sequences with a 5-carboxyfluorescein (5-FAM) fluor and a QXL520 quencher. Enzymatic cleavage is monitored by the increase in fluorescence of 5-FAM (excitation, 480 nM; emission, 540 nM). In contrast, the activity of caspase 6 was monitored by luminescence in a 3456-well plate format using a caspase 6-glo substrate purchased from Promega (Madison, WI), wherein enzyme activity is monitored by enzymatic release of aminoluciferin, which is hydrolyzed by luciferase, thereby generating luminescence. Based on overall statistics, all assays had mean signal-to-background ratios between 11- and 74-fold and mean Z-prime values between 0.74 and 0.88 ( Fig. 1A ). Z-prime is a measure of assay quality that takes into account both the assay window and the standard deviation. 32 Scores >0.5 are typically deemed to be good screening assays that have a large enough window to overcome assay variability to detect hits. The uHTS assays performed consistently throughout the duration of the screen with examples of a few outlier plates based on Z-prime and S/B (signal-to-background ratio; Fig. 1B–E ). Generally, inconsistent plates were rescheduled unless an obvious outlier in the control region was negatively affecting the statistical analysis. The thrombin primary screen did demonstrate a greater variation from plate to plate during the course of the screen, as indicated by the larger deviation in both S/B and Z-prime values ( Fig. 1E ). To rescue hits that may be obscured by systematic plate effects, primary hit selection was based on the union of 4 scoring approaches: % activity, % activity adjusted based on the B-score algorithm, B-score, and Z-score. 33 For BACE1, HIV protease, and thrombin, the union of the compounds that scored at the top 1% for each statistical value was repicked and progressed into the confirmation screens ( Fig. 1A ). For caspase 6, due to a relatively high hit rate, the primary hits were selected based on a cutoff of 48% inhibition, which correlated to a 2% hit rate.
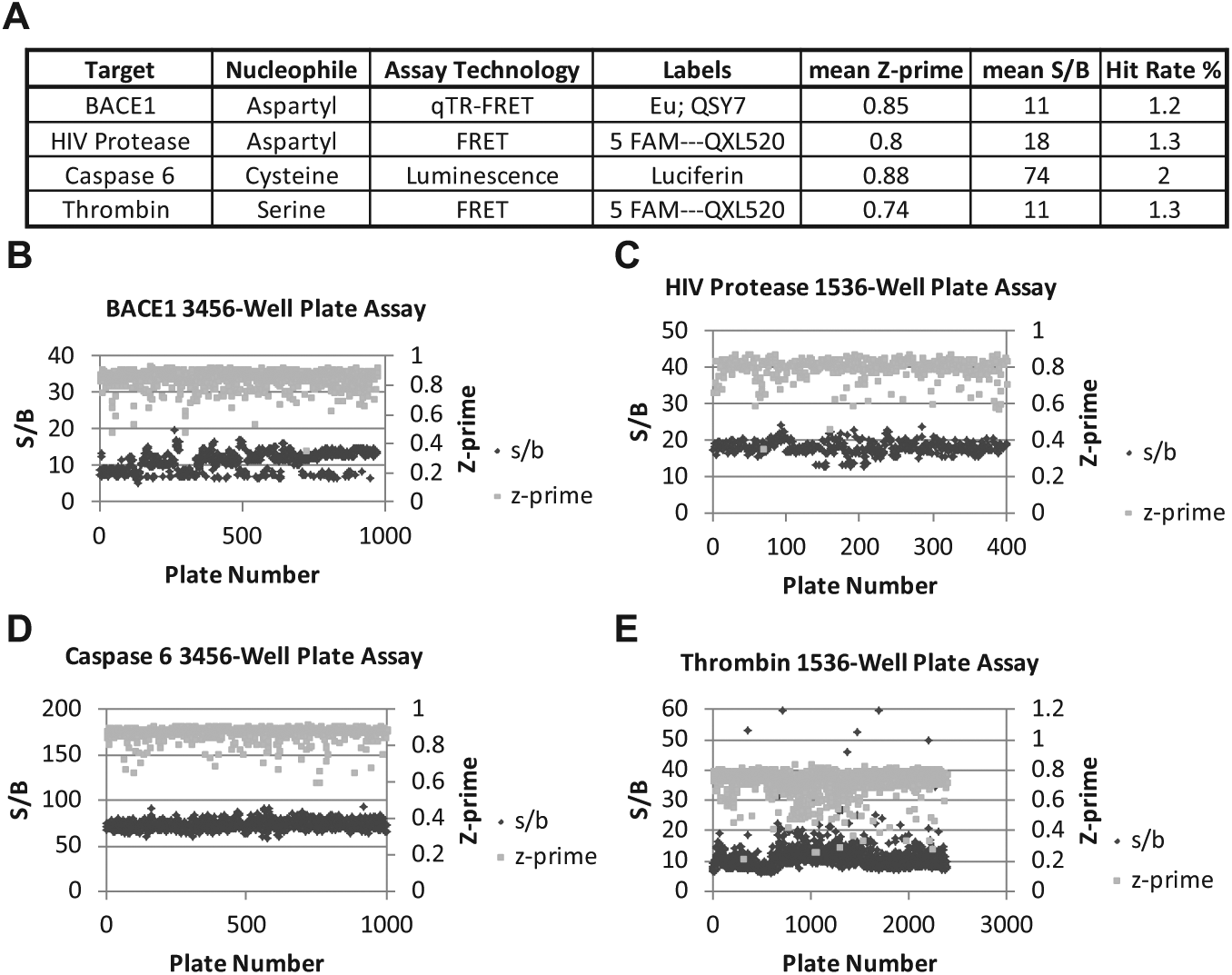
Protease primary ultrahigh-throughput screening (uHTS) assay results. (
Development of RapidFire HTMS Protease Assays
For each protease program, an MS-based assay was developed for the RapidFire HTMS platform. The RapidFire platform couples the capability to monitor a wide array of peptidic or small-molecule–metabolite analytes with the throughput to analyze the 20,000–80,000 samples in a 384-well plate format required to support confirmation screens of uHTS programs. RapidFire uses small SPE cartridges to retain the analytes of interest and remove any salts that would interfere with MS-based detection. Coupled with the resolution of a triple quadrupole mass spectrometer to distinguish analytes from other assay components, RapidFire can process samples at a speed of 5–7 s/sample, or about 45 min to 1 h per 384-well plate.
For the HTMS assays, the first step in assay development involves selection of an unlabeled peptide substrate. For caspase 6, the substrate sequence QVEIDG was selected based on the standard recognition sequence used for caspase 6 along with the tolerability of a G amino acid on the C-terminal side of the cleavage site.
34
For BACE1, the peptide sequence matches that of the qTR-FRET substrate used in the primary assay with the absence of the Europium chelate and QSY7 labels (
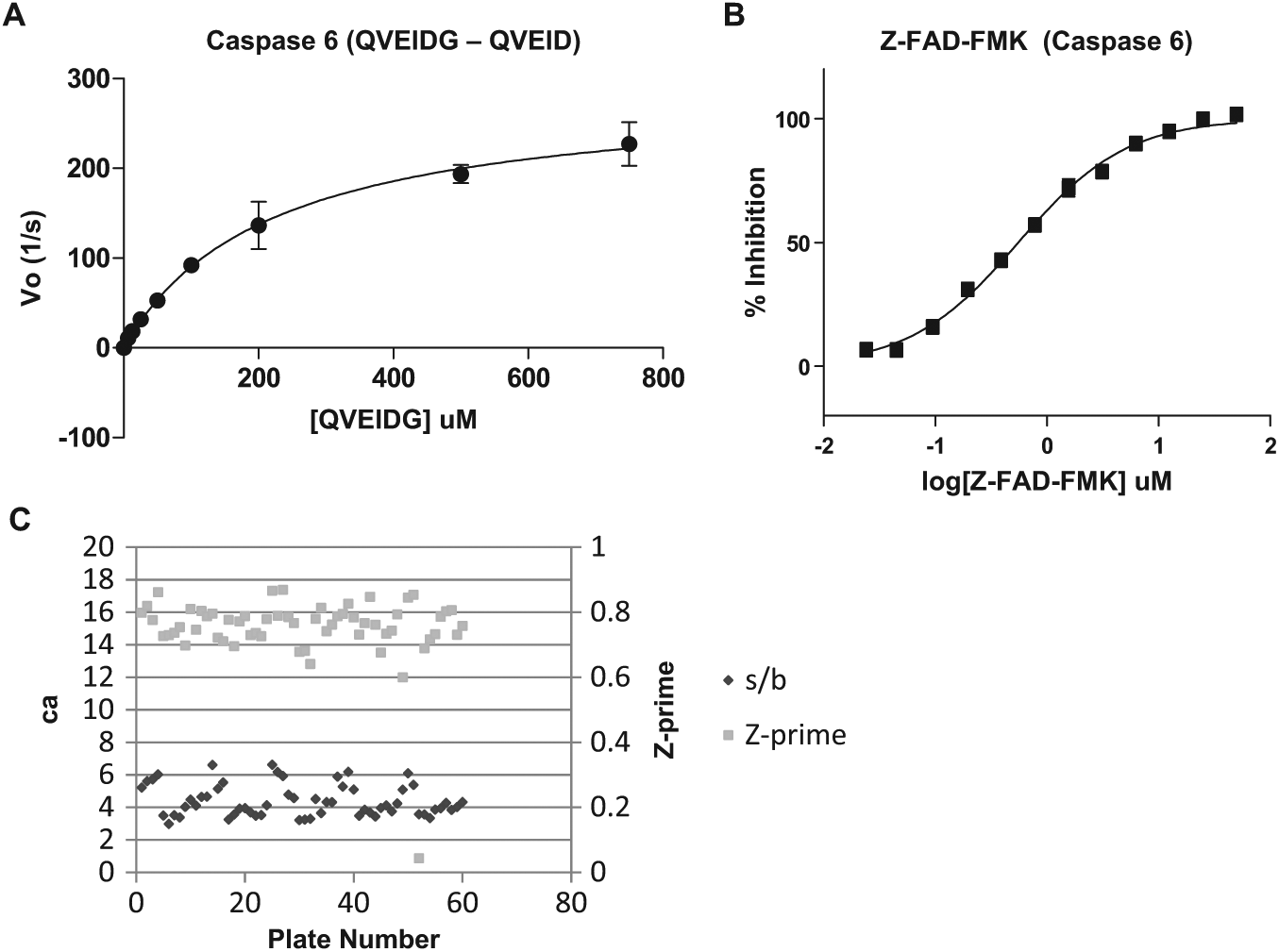
Caspase 6 384-well plate high-throughput mass spectrometry (HTMS) confirmation screen assay development. (
Hit Confirmation Assays
At the hit confirmation stage of screening, compounds selected as hits from the primary screen are selected and plated into 1536- or 3456-well plates, then screened in triplicate at a single concentration. For the protease targets, strategies were selected for hit confirmation that paired replication of activity in the primary assay with methods to remove detection-based artifacts via positive selection assays using orthogonal detection formats or negative selection assays using the same detection strategy with an unrelated enzyme. For BACE1 and thrombin, the primary assay was coupled with an HTMS assay for hit confirmation (
Table 1
). For HIV protease, the HTMS assay was the only assay applied to the confirmation of hits; however, a subset of 2560 primary hits was screened in the primary FRET assay to estimate a confirmation rate for that assay format. Alternatively, for caspase 6, due to the high hit rate in the luminescent Caspase-Glo 6 primary assay format, hits were first confirmed in the Caspase 6-Glo assay as well as in a positive selection orthogonal TR-FRET format. In addition, a negative selection assay using a DPP-IV-Glo luminescent format was applied to further eliminate detection-based false positives. The hits from this set were progressed to dose titration assays in parallel with analysis in the caspase 6 HTMS assay. Based on full plate statistics, all assays performed well with average Z-prime values >0.7 and S/B ratios >4 (
Fig. 2C
Confirmation Assay Results.
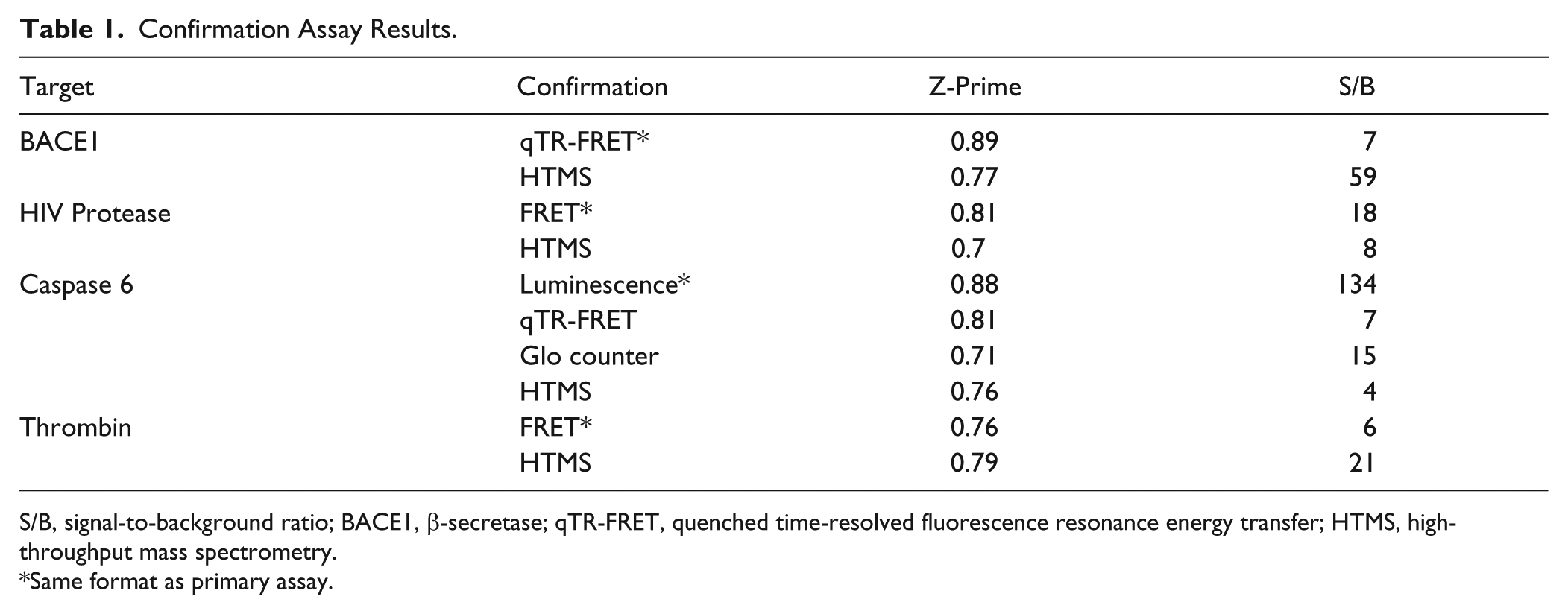
S/B, signal-to-background ratio; BACE1, β-secretase; qTR-FRET, quenched time-resolved fluorescence resonance energy transfer; HTMS, high-throughput mass spectrometry.
Same format as primary assay.
For the light-based confirmation assays, a compound was deemed a confirmed hit if the median percent inhibition of the 3 replicates had a value greater than or equal to the percent activity cutoff applied to the primary screen ( Fig. 3A , Table 2 ). For BACE1, 30,784 compounds or 90.4% of primary assay hits were confirmed with >45% inhibition in the qTR-FRET assay. For HIV protease, based on the results of the subset of compounds, 16,716 or 80% of compounds would have been confirmed with >30% inhibition in the 1536-well plate Sensolyte 520 FRET assay used in the primary. Compounds with >40% inhibition in the thrombin FRET assay were confirmed as hits, which correlated to 26,440 compounds or 69.6% of the primary hits. Finally, for caspase 6, hits were selected based on demonstrating >30% inhibition in the Caspase-Glo 6 luminescence assay, >25% inhibition in an orthogonal qTR-FRET assay, and < 30% inhibition in a DPP-4 Glo luminescent counterscreen. Using this strategy, 6255 compounds or 10.7% of the primary hits were confirmed, indicating that the application of orthogonal assay formats is highly effective at removing assay-dependent artifacts.
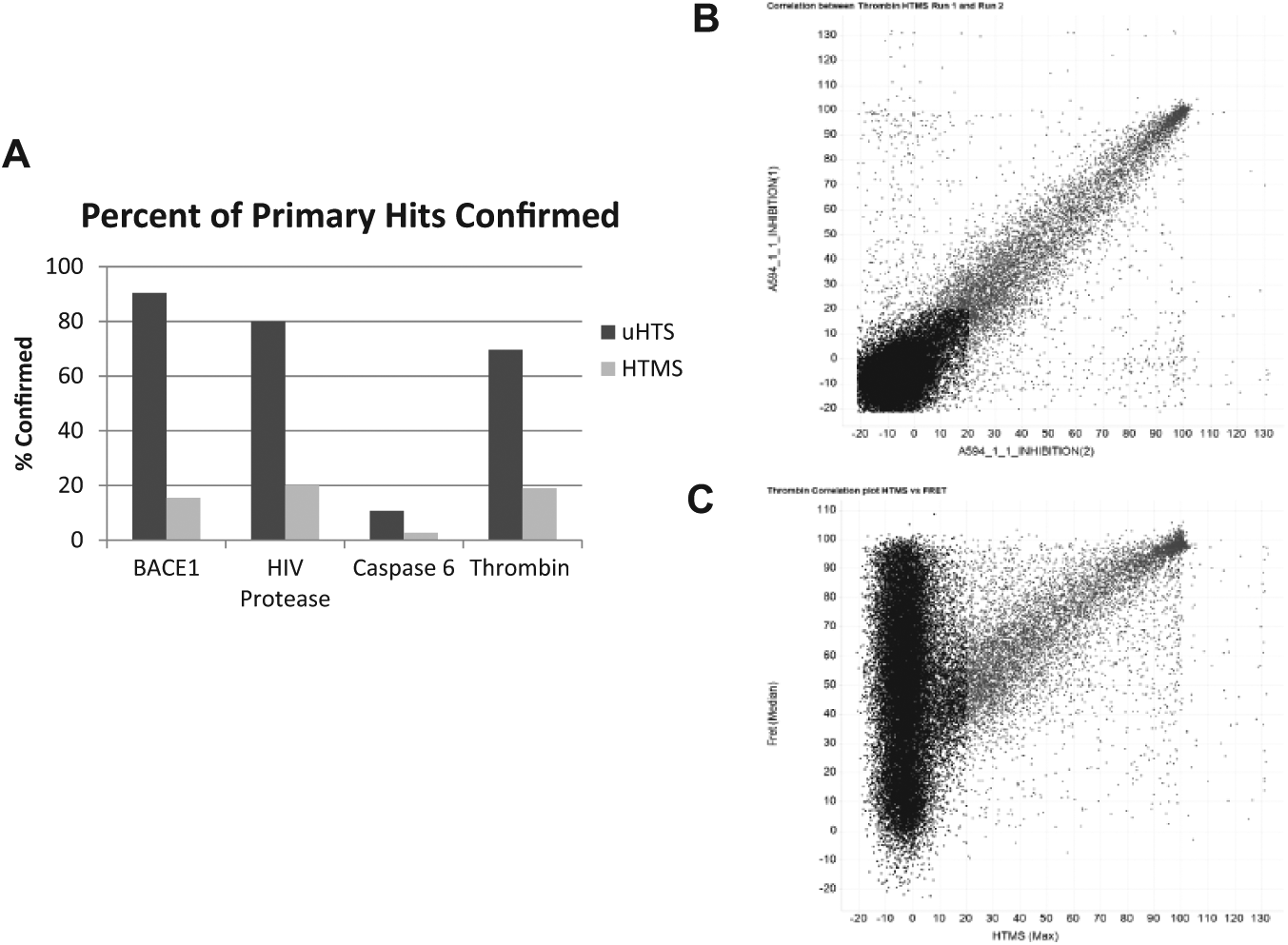
High-throughput mass spectrometry (HTMS) confirmation assay results. (
Assay Hit Rates.
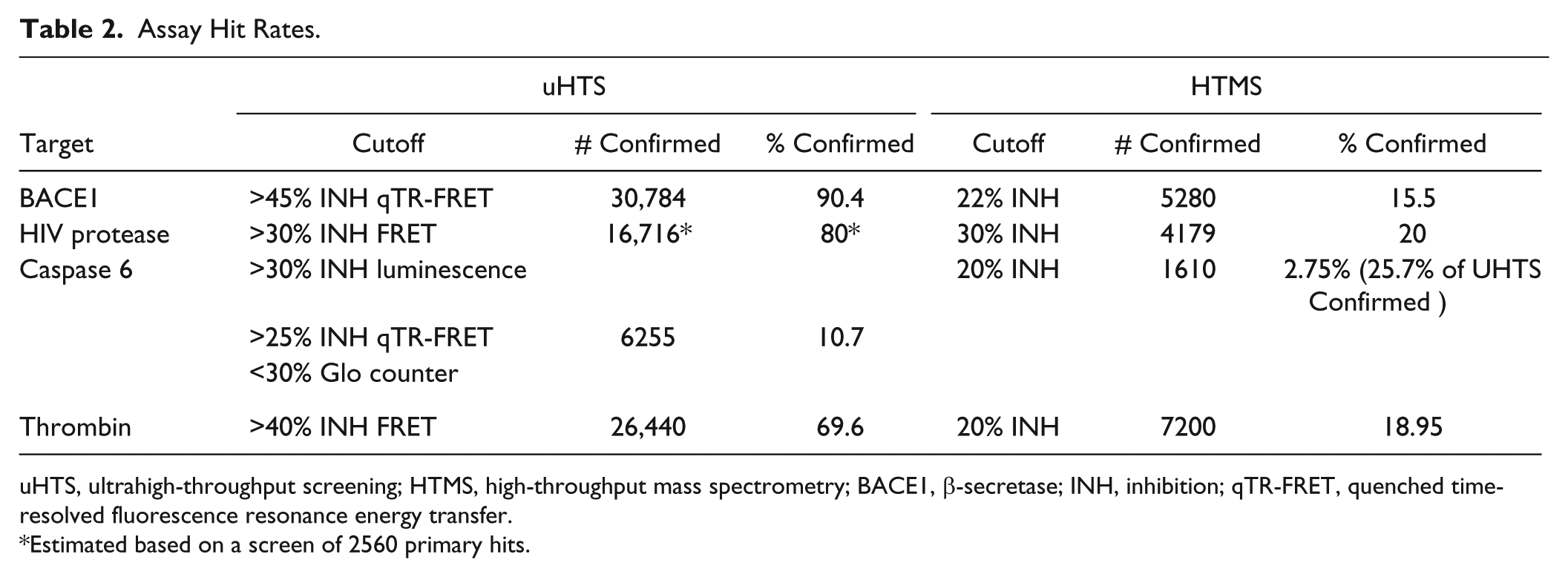
uHTS, ultrahigh-throughput screening; HTMS, high-throughput mass spectrometry; BACE1, β-secretase; INH, inhibition; qTR-FRET, quenched time-resolved fluorescence resonance energy transfer.
Estimated based on a screen of 2560 primary hits.
For the HTMS assays, due to a relatively lower throughput compared to assays with light-based readouts, each compound is screened in duplicate. Replicates of the HTMS assays tend to be highly correlative, as indicated by the scatterplot of the thrombin HTMS assay ( Fig. 3B ), wherein the correlation of the percent inhibition of replicate 1 depicted on the x-axis and replicate 2 on the y-axis has an R2 value of 0.8. There is, however, a scattering of compounds that demonstrate a variable percent inhibition between the 2 replicates. To avoid missing true hits in which 1 of the 2 replicates is low due to a systematic error or a missed injection on the RapidFire, hits were selected based on either or both replicates being greater than the mean+3-sigma cutoff for the assay. For BACE1, based on a 22% inhibition cutoff, 4280 compounds, or 15.5% of hits, were confirmed in the HTMS assay ( Fig. 3A , Table 2 ). About 20% of the primary hits, or 4179 compounds, were confirmed in the HIV protease HTMS assay that applied a 30% inhibition cutoff. With a 20% inhibition cutoff, 7200 compounds or 19% of the primary hits in the thrombin FRET assay were confirmed in the thrombin HTMS assay. Finally, the 6255 hits confirmed in the 3 light-based confirmation assays for caspase 6 were further screened in the HTMS assay. Based on a 20% inhibition cutoff, 1610 compounds were confirmed correlating to 25.7% of the uHTS confirmed hits. A subset of compounds selected from the set of primary hits in the Caspase-Glo 6 assay that were not confirmed by the light-based assays was screened in the HTMS assay. None of the compounds analyzed demonstrated any inhibition of caspase 6, further indicating that this set of compounds was correctly eliminated as false positives.
Notably, the protease HTMS assays confirmed only between 2% and 20% of the primary hits independent of the detection technology used in the primary screen, whereas, depending on the triage strategy applied, between 10% and 90% of hits were confirmed in the light-based confirmation assays. The correlation plot between the thrombin HTMS assay and the thrombin FRET assay clearly indicates that the subset of compounds identified as hits in both formats (light gray in Fig. 3C ) displays correlation based on the percent inhibition values determined in both assays. The scatterplot also depicts a high number of compounds that demonstrate inhibition in the thrombin FRET assay but show no inhibition in the HTMS format.
HTMS Confirmation Assays Are Detecting True Positives
On average, only 1 out of 5 compounds was confirmed as a hit in the assays applying MS as a detection strategy. The relatively low confirmation rate raises the concern that true positives are not being captured by the HTMS assay. One approach to determine if the assays are capturing true hits is to observe whether compounds annotated as being synthesized for a specific program in the database are captured as hits in the HTMS assay. Using this criterion, 14 out of 15 compounds annotated as BACE1 program compounds were detected as inhibitors in the HTMS confirmation assay ( Fig. 4A ). Looking further into the data reveals that the one potential false negative in the HTMS assay was annotated as a 2.5 uM inhibitor in a TR-FRET assay with a maximum of 150% inhibition. For HIV protease ( Fig. 4B ) and caspase 6 ( Fig. 4C ), 304 out of 304 and 26 out of 26 program compounds were identified as hits by the HTMS assays, respectively. For thrombin, 98 out of 101 compounds were identified as hits in the thrombin HTMS confirmation assay ( Fig. 4D ). The 3 compounds that were synthesized for the thrombin program but were putative false negatives in the HTMS assay reported no inhibition in a thrombin assay in the corporate database, indicating that these compounds are true negatives. These data indicate that HTMS-based assays are effective at triaging false positives while capturing true-positive inhibitors of proteases.
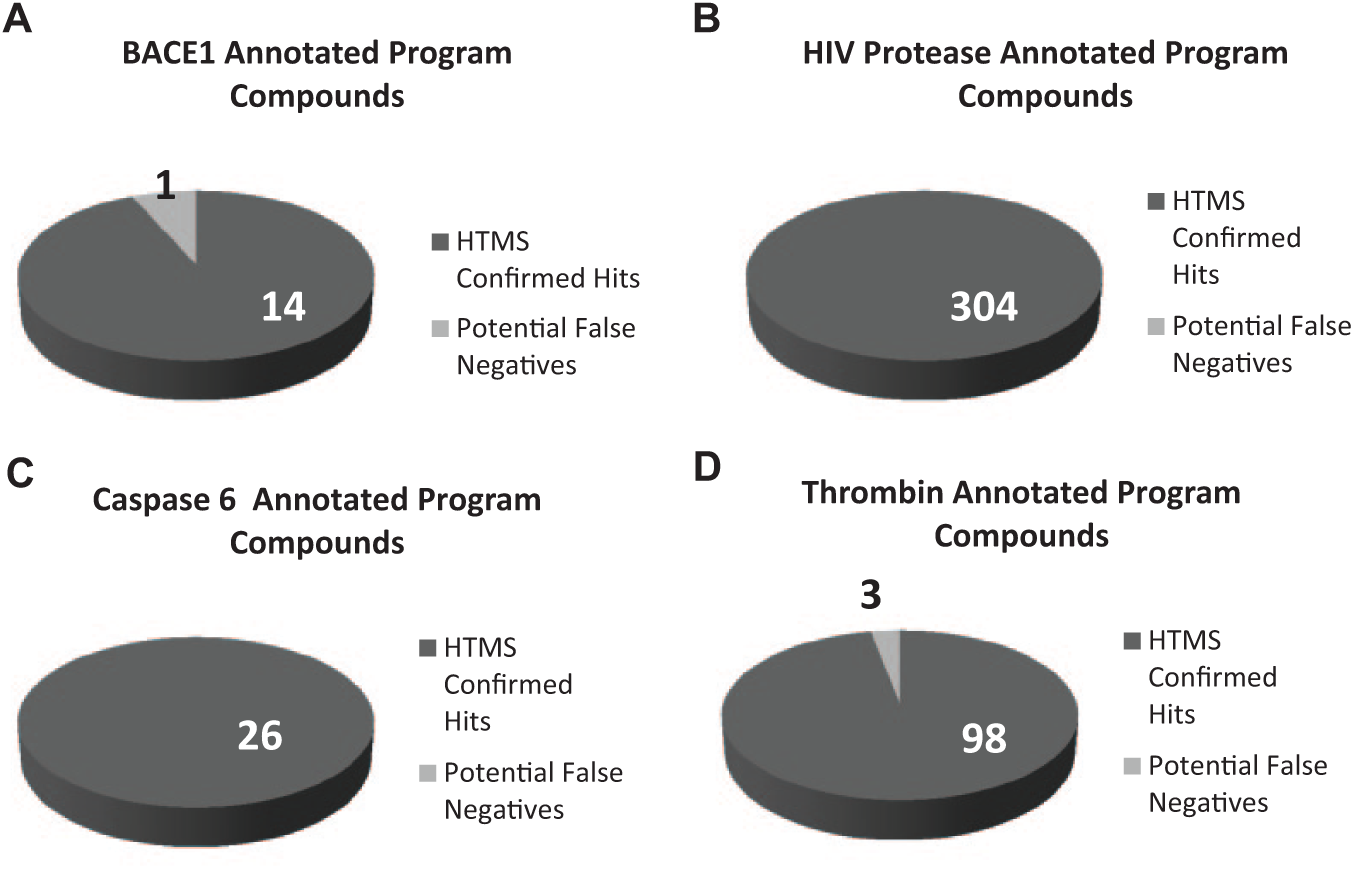
Program compounds are confirmed as hits in high-throughput mass spectrometry (HTMS) assays. Determination of whether program compounds generated for the specific target were confirmed as hits in the HTMS assay for (
Based on the data reported here, confirmation assays for protease programs applying RapidFire HTMS as a detection strategy are effective at removing potential false positives from uHTS hit lists. Independent of the class of protease (cysteine, serine, or aspartyl) or the assay technology applied for the primary screen (TR-FRET, FRET, or luminescence), HTMS assays effectively remove up to 80% of the hits as false positives. Comparing the different primary approaches reveals that the luminescent assay (the Caspase-Glo 6 assay) has the highest rate of potential assay artifacts, with only about 3% of compounds confirmed after an extensive triage. Based on this detection methodology, false positives could be due to luminescent compounds or luciferase inhibitors. Considering that the FRET assay monitors fluorophores that exhibit prompt fluorescence, and the TR-FRET assay monitors the time-resolved fluorescence of a lanthanide cryptate, it is surprising that the confirmation rates between the FRET and TR-FRET assays are comparable. By monitoring time-resolved fluorescence, it is anticipated that a reduction in fluorescence-based interference would be observed because most compounds are expected to exhibit prompt fluorescence.
Because the protease uHTS screens demonstrate a high number of compounds not confirmed by MS-based assays across multiple detection strategies, mechanisms other than light-based interference may be large contributors to the false-positive totals. Work by the Schoichet lab has highlighted the prevalence of aggregation-based artifacts in screening data; 35 however, compound aggregation that interferes with the enzyme activity is expected to occur in both MS-based and light-based assay formats. By including biophysical or aggregation-based counterscreens, compounds that bind to the enzyme in a super-stoichiometric manner may be eliminated. Another potential difference between light-based and MS-based assays to consider is the structural differences between the substrates. A common feature of the substrates for MS assays uses native sequences without labels, whereas all of the substrates used for the light-based assays require a peptide substrate appended with a fluorescent tag, quencher, or aminoluciferin. These tags are relatively hydrophobic conjugated ring systems that may result in interactions with members of the small-molecule libraries. One consideration is that appending a fluorophore onto a peptide substrate may make it more susceptible to substrate–compound interactions, such as substrate absorption into small-molecule aggregates. There are several instances in the literature wherein inhibitory activity observed for compounds is dependent on the presence of a fluorescent tag appended to the substrate. One example is substrate-specific activation of Sirt1 by resveratrol and a series of compounds generated by Sirtris.9-11 Also recently, a study of caspase 6 by researchers at Genentech identified a series of N-furoyl-phenylalanine compounds that were found to bind in a fluor-dependent manner to a complex of the enzyme and the substrate. 8 This series was prioritized due to a novel uncompetitive mechanism of inhibition of caspase 6 and optimized from a 10 uM hit to a 0.011 uM lead. This series, however, demonstrated a 100-fold reduction in potency when the rhodamine label on the substrate was replaced with amino-methylcoumarin. These studies highlight the importance of screening protease hits in label-free assays such as HTMS to remove substrate-related artifacts. Future efforts will focus on parsing out the cause of the compounds not confirmed by MS in the different assay technologies and on expanding this screening approach to other protein classes. It is also of interest to interrogate smaller compound collections directly with a mass spectrometry–based assay through an iterative focused approach to determine if hit classes are missed in the light-based uHTS assays.
Based on the results presented herein, the combination of a light-based primary screen to interrogate a full screening file of >1 million compounds and the incorporation of an MS-based confirmation assay is an effective way to generate a concise hit list enriched in true positives. The reduction in the numbers of compounds progressed to dose titration assays saves resources and avoids the requirement for a computational selection process to select hits for dose titration. The extra time required, which averages an additional 2–3 weeks, to run the HTMS assay on 30,000+ compounds is minimal when compared to the amount of time and resources saved on focusing downstream hit-to-lead efforts on true positives instead of assay artifacts.
Footnotes
Acknowledgements
We would like to acknowledge Ed Hudak and Alex Wolicki for compound management support; Anthony Kreamer, Dr. Andy Liaw, and Tim Hare for informatics support; and Lou Locco, Alex May, Carissa Quinn, and Todd Smith for robotics support. Also, we would like to thank Dr. William LaMarr, Dr. Patty Sun, Dr. Kari Schlicht, and Dr. Maxine Jonas of Agilent Biotechnologies for their advice and technical support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.