
Abstract
Recently, dual-specificity phosphatase 16 (DUSP16) emerged as a promising therapeutic target protein for the development of anti-atherosclerosis and anticancer medicines. The present study was undertaken to identify the novel inhibitors of DUSP16 based on the structure-based virtual screening. We have been able to find seven novel inhibitors of DUSP16 through the drug design protocol involving homology modeling of the target protein, docking simulations between DUSP16 and its putative inhibitors with the modified scoring function, and in vitro enzyme assay. These inhibitors revealed good potency, with IC50 values ranging from 1 to 22 µM, and they were also screened computationally for having desirable physicochemical properties as drug candidates. Therefore, they deserve consideration for further development by structure-activity relationship studies to optimize the inhibitory activity against DUSP16. Structural features relevant to the stabilization of the newly identified inhibitors in the active site of DUSP16 are addressed in detail.
Introduction
The mitogen-activated protein kinase (MAPK) pathway controls the cellular functions associated with the growth, differentiation, and death of cells. 1 In this signaling cascade, MAPKs are activated by the phosphorylation in the active loop residues. MAPK phosphatases (MKPs) dephosphorylate MAPKs and thereby turn off their activities. 2 Of various MKPs, dual-specificity phosphatase 16 (DUSP16; also known as MKP-7) dephosphorylates JUN kinases (JNKs) specifically and thereby regulates T-cell differentiation, 3 which can be responsible for the pathogenesis of Burkitt’s lymphoma 4 and cerebral ischemia. 5 Recently, DUSP16 was also found to induce the expression of VCAM-1 through the suppression of JNK signaling. 6 Because VCAM-1 plays a pivotal role in the development of atherosclerosis and tumor progression, 7 DUSP16 can serve as a potential target for the development of anti-atherosclerosis and anticancer medicines. The merit of DUSP16 as a target for drug discovery was further supported by the experimental finding that the knockout of DUSP16 gene by small interfering RNA inhibited the expression of VCAM-1. 6
Although the three-dimensional (3D) structure of DUSP16 is not yet known, X-ray crystal structures of its highly homologous proteins such as DUSP9 were reported in complex with a substrate analogue. 8 To accommodate the small phosphorylated substrate group, DUSP9 has a planar and shallow active site in which the negatively charged substrate group can be stabilized by the hydrogen bonds with the backbone amide groups and the side-chain guanidinium ion of the Arg residue located near the catalytic cysteine residue (Cys290). Structural information about the active site and the interactions with a small-molecule ligand is very useful for designing new potent inhibitors that can develop into drug candidates. Indeed, many potent and selective small-molecule phosphatase inhibitors have been identified based on such a structure-based molecular design approach.9,10 Nonetheless, the discovery of DUSP16 inhibitors has lagged behind the biological and pharmaceutical studies. No small-molecule DUSP16 inhibitor has been reported so far in the literature. This may be attributed in a large to the lack of 3D structure of DUSP16, which makes it difficult to apply the rational design approaches for finding the novel inhibitors.
In this study, we try to identify the potent DUSP16 inhibitors through the structure-based drug design protocol, which involves homology modeling of the 3D structure of DUSP16, virtual screening of a chemical library with docking simulations, and an in vitro enzyme assay. Virtual screening using molecular docking has not always been useful because of the inaccuracy in the scoring function that estimates the binding affinity between the target protein and a putative inhibitor. 11 Therefore, prior to carrying out the virtual screening, we improve the existing scoring function by the implementation of an accurate solvation model to reflect the effect of desolvation cost for a ligand on the binding affinity to the target protein. This modification seems to have an effect of increasing the hit rate in the enzyme assay because the neglect of the solvation term often leads to the overestimation of the binding affinity of a ligand with many polar atoms. 12 We find in this study that the docking simulations with the improved scoring function can be a useful computational tool for elucidating the activities of the identified inhibitors, as well as for enriching the chemical library with molecules that are likely to have desired biological activities.
Materials and Methods
Because of the lack of structural information on DUSP16, we performed homology modeling using the X-ray crystal structure of DUSP9 (PDB entry: 2HXP) [8] as the template to build the 3D structure of DUSP16 necessary for the structure-based virtual screening. The sequence alignment between the phosphatase domains of DUSP9 and DUSP16 was derived with version 2.1 of the ClustalW program, 13 using the BLOSUM matrices for scoring the alignments. Opening and extension gap penalties were changed systematically, and the derived alignments were inspected for the violation of structural integrity in the structurally conserved regions. More specifically, we adopted only the alignments in which the sequences of active-site residues of DUSP9 (CLAGVSR) and DUSP16 (CLAGISR) were matched because all DUSPs share the catalytic core in a form of CXXGXXR. Using the best-scored sequence alignment, the 3D structure of the catalytic domain of DUSP16 was constructed using the MODELLER program (version 9.12). 14 In this homology modeling, we applied the optimization method involving conjugate gradients and molecular dynamic simulations to minimize the violations of the spatial restraints. With respect to the structure of the gap regions, the coordinates were built from a randomized distorted structure that resides approximately between the two anchoring regions as implemented in the MODELLER program. To refine the calculated structures, loop modeling was also carried out based on the enumeration algorithm. 15 WPD (residues 209–216) and Q (residues 279–285) loops were included in this structural refinement. As the final structural model for DUSP16, we selected the one with the lowest value of MODELLER objective function. Finally, we calculated the conformational energy of the predicted structure of DUSP16 with the ProSa 2003 program 16 for the purpose of evaluation.
The atomic coordinates obtained from the homology modeling were used as the receptor model in the virtual screening of DUSP16 inhibitors. Special attention was paid to the determination of the protonation states of the ionizable residues of the homology-modeled structure. The side chains of Asp and Glu residues were assumed to be neutral if either of the carboxylate oxygens was directed toward a hydrogen-bond accepting group such as the backbone aminocarbonyl oxygen within the distance of 3.5 Å, which is a generally accepted distance limit for the hydrogen bond of moderate strength. 17 Similarly, the side chains of lysine residues were assumed to be protonated unless the NZ atom stayed in proximity to a hydrogen-bond donating group. The same procedure was also applied for determining the protonation states of His residues. As a consequence of the inspection for protonation states, Asp199, Lys234, and Asp264 were found to reside in the neutral form in the final structural model of DUSP16. After the assignment of the protonation states, we carried out 200 cycles of energy minimization for DUSP16 with the AMBER program of version 12 to remove the bad steric contacts. In this step, it should be kept in mind that the energy minimization should be terminated once all bad van der Waals contacts were removed because the full minimization in the gas phase can make the protein structure converge to a physically unacceptable one because of the overestimation of intramolecular electrostatic interactions.
The docking library for DUSP16 comprising about 260,000 compounds was constructed from the chemical database distributed by Interbioscreen (http://www.ibscreen.com) containing approximately 477,000 synthetic and natural compounds. Prior to performing the docking simulations for virtual screening, they were filtrated according to Lipinski’s “Rule of Five” to select only the compounds with the physicochemical properties of potential drug candidates 18 and without reactive functional group(s). To remove the structural redundancies in the chemical library, the structurally similar compounds with Tanimoto coefficient larger than 0.8 were clustered into a single representative molecule. As a consequence of the filtering process, a docking library containing ~260,000 compounds was constructed. These compounds were then processed with the CORINA program to obtain their 3D atomic coordinates, followed by the assignment of atomic charges with the Gasteiger-Marsilli method. 19 We used version 3.5 of the AutoDock program 20 in the virtual screening of DUSP16 inhibitors. AMBER force field parameters were used for calculating the van der Waals interactions and the internal energy of a ligand as implemented in the original AutoDock program. Docking simulations with AutoDock were then carried out in the active site of DUSP16 to score and rank the compounds in the docking library according to their calculated binding affinities.
In the docking simulations for virtual screening, we used the modified AutoDock scoring function to which the solvation free energy term for organic molecules was added to estimate the desolvation cost for protein-ligand association. This modified scoring function can be written in the following form.
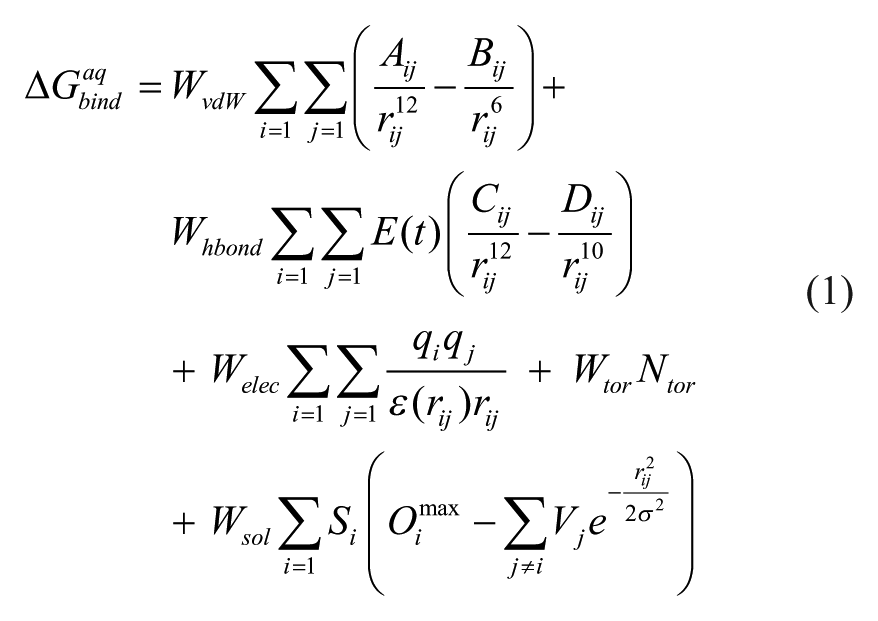
Here, WvdW, Whbond, Welec, Wtor, and Wsol are the weighting factors of van der Waals, hydrogen bond, electrostatic interactions, torsional term, and solvation free energy of a putative inhibitor, respectively. rij represents the interatomic distance, and Aij, Bij, Cij, and Dij are associated with the depths of the potential energy well and the equilibrium separations between the two atoms. The hydrogen bond term has an additional weighting factor, E(t), to represent the angle-dependent directionality. With respect to the distance-dependent dielectric constant (ϵ(rij)), a sigmoidal function proposed by Mehler et al. 21 was used for the calculation of the interatomic electrostatic interactions between DUSP16 and the putative inhibitors. In the entropic term, Ntor denotes the number of rotatable bonds in the ligand. Although the original AutoDock scoring function included a simple solvation energy term, we replaced it with the improved one shown in the last term of eq 1. In this new solvation free energy term, Si and Vi are the solvation parameter and the fragmental volume of atom i, 22 respectively, whereas Oimax stands for the maximum atomic occupancy that means the volume of atom i exposed to bulk solvent in the absence of the other solute atoms. The negative and positive values of Si parameter indicate the stabilization and destabilization of the solute atom i, respectively, due to the combined effects of intermolecular interactions with water molecules and intramolecular interactions with the rest of the solute atoms. In the calculation of the molecular solvation free energy term in eq 1, we used the atomic parameters developed by Choi et al. because they proved to be successful in predicting the solvation free energies of a variety of organic molecules. 23 This modification of the scoring function seems to increase the accuracy in virtual screening of DUSP16 inhibitors because the underestimation of ligand solvation often leads to the overestimation of the binding affinity of an inhibitor with many polar atoms. Indeed, the superiority of this modified scoring function to the previous one was well appreciated in recent studies for virtual screening of kinase and phosphatase inhibitors.24,25
The catalytic domain of DUSP16 was overexpressed in Escherichia coli and purified. For this purpose, E. coli cells containing the DUSP16 expression vector were induced with 0.2 mM IPTG at 18 °C overnight. Cell pellets were lysed in a buffer containing 50 mM Tris-HCl (pH 7.5), 500 mM NaCl, 1% phenylmethylsulfonyl fluoride, 4 mM 2-mercaptoethanol, and 5% glycerol. The His-tagged DUSP16 was purified by Ni-NTA agarose column and dialyzed against 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, 2 mM DTT, and 5% glycerol.
A total of 148 compounds selected from the virtual screening were evaluated for in vitro inhibitory activity against the purified DUSP16. These phosphatase enzyme assays were carried out by using 6,8-difluoro-4-methyl-umbelliferyl phosphate (DiFMUP) as a fluorogenic substrate, which has a Km value of approximately 20 µM for protein tyrosine phosphatases (PTPs). 26 To determine the conditions for the phosphatase assay, we performed the preliminary assay experiments with varying concentrations of DUSP16 and DiFMUP. Then, we selected a combination of the concentrations (3 µM DUSP16 and 10 µM DiFMUP) that generated an ideal progression curve for fluorescence measurements. Under these assay conditions, the fluorescence intensity showed a linear increase in the early time region and reached the maximum after 10 to 20 min. In case of the reaction buffer, we used the conditions under which most DUSPs were found to exhibit an optimal activity. 27 Actually, the usefulness of the assay conditions described above was confirmed by elucidating the potency of various known phosphatase inhibitors such as NSC 95397 and Cpd 5. 28
The purified DUSP16, DiFMUP, and a candidate inhibitor were incubated in the reaction mixture containing 20 mM Tris-HCl (pH 8.0), 0.01% Triton X-100, and 5 mM DTT for 20 min. The resulting fluorescence was measured by using the PerkinElmer 2030 instrument with excitation and emission wavelengths of 355 and 460 nm, respectively. Initial velocities of reactions were estimated for various inhibitor concentrations (0–50 µM). The inhibitory activities of the putative DUSP16 inhibitors were measured in duplicate at the concentrations of 0.0, 0.625, 1.25, 2.5, 5, 10, 20, and 50 µM to obtain the dose-response curve fits. The IC50 value of each inhibitor was then determined from direct regression analysis using the four-parameter sigmoidal curve as implemented in the SigmaPlot program.
Results and Discussion
The peptide sequence of human DUSP16 comprising 665 amino acids was retrieved from UniProtKB protein knowledgebase (http://www.uniprot.org, accession number: Q9BY84). Although the 3D structure of DUSP16 has not been known, we found from a database search at NCBI BLAST (http://blast.ncbi.nlm.nih.gov) that DUSP9 could serve as a structural homolog for DUSP16. As shown in the supplementary data (
The structure of the phosphatase domain of DUSP16 generated from the homology modeling was then evaluated with the ProSa 2003 program 16 by investigating whether the interactions of each amino acid residue with the rest of the protein structure could be maintained favorably. This program has been widely used for estimating the reasonableness of a predicted protein structure because the calculated knowledge-based mean fields could serve as a yardstick to judge the quality of protein folds. Figure 1 compares the ProSa energy profiles of the homology-modeled structure of DUSP16 and the X-ray crystal structure of DUSP9. It is seen that the ProSa energy remains negative for all amino acid residues of the template and the target, indicating that both protein structures would be physically acceptable. We also note that the homology-modeled DUSP16 structure appears to be relatively unstable in the N-terminal region when compared with the X-ray structure of DUSP9, and vice versa in the C-terminal region. The overall similarity of the conformational stability of the target to that of the template indicates that the 3D structure of DUSP16 can be predicted with accuracy from homology modeling.
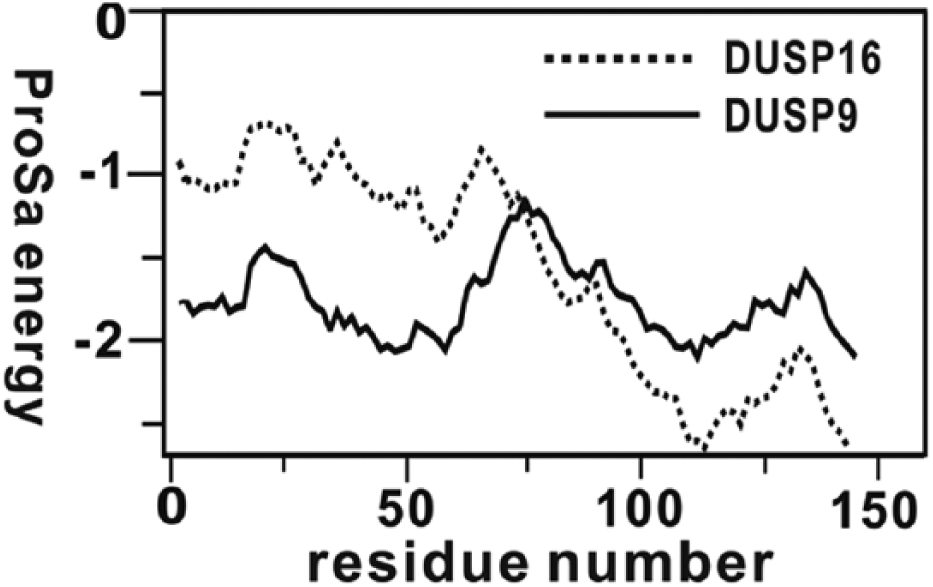
Comparative view of the ProSa energy profiles for the X-ray crystal structure of DUSP9 and the homology-modeled DUSP16 structure. For convenience, the amino acids are renumbered from 1 instead of retaining the original numbers.
Although it is known that the accuracy of virtual screening can be enhanced significantly by taking into account the effect of protein structure flexibility,
30
we could not consider such a dynamic property in the virtual screening of DUSP16 inhibitors because the receptor had to be treated as a rigid body in the current version of the AutoDock program. Of the 260,000 compounds screened with docking simulations in the active site of the homology-modeled DUSP16 structure, 150 top-scored compounds were selected as the virtual hits. A total of 148 of them were commercially available from the compound supplier and tested for inhibitory activity against DUSP16 by in vitro enzyme assay. As a result, we found seven compounds that inhibited the catalytic activity of DUSP16 by more than 50% at the concentration of 20 µM, which were selected to measure the IC50 values. The molecular structures and the inhibitory activities of the newly found inhibitors are shown in
Figure 2
and
Table 1
, respectively. The dose-response behaviors of the inhibitors measured to obtain their IC50 values are presented in
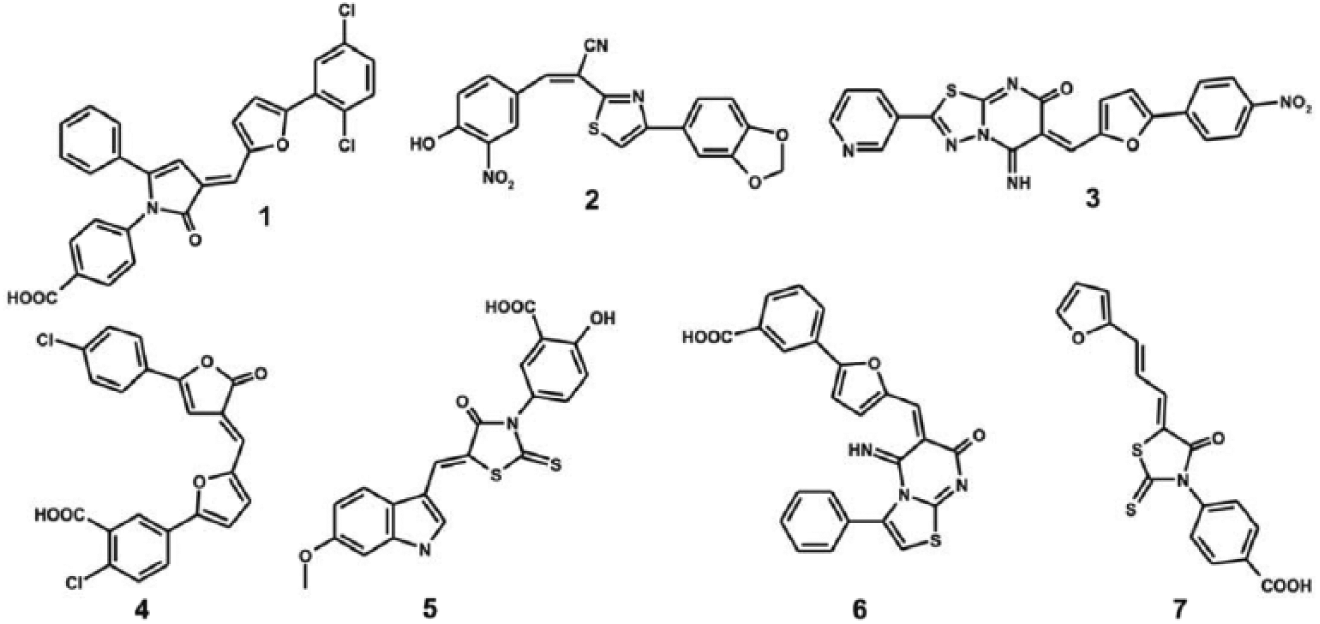
Chemical structures of seven DUSP16 inhibitors identified with virtual screening.
IC50 values (in µM) of
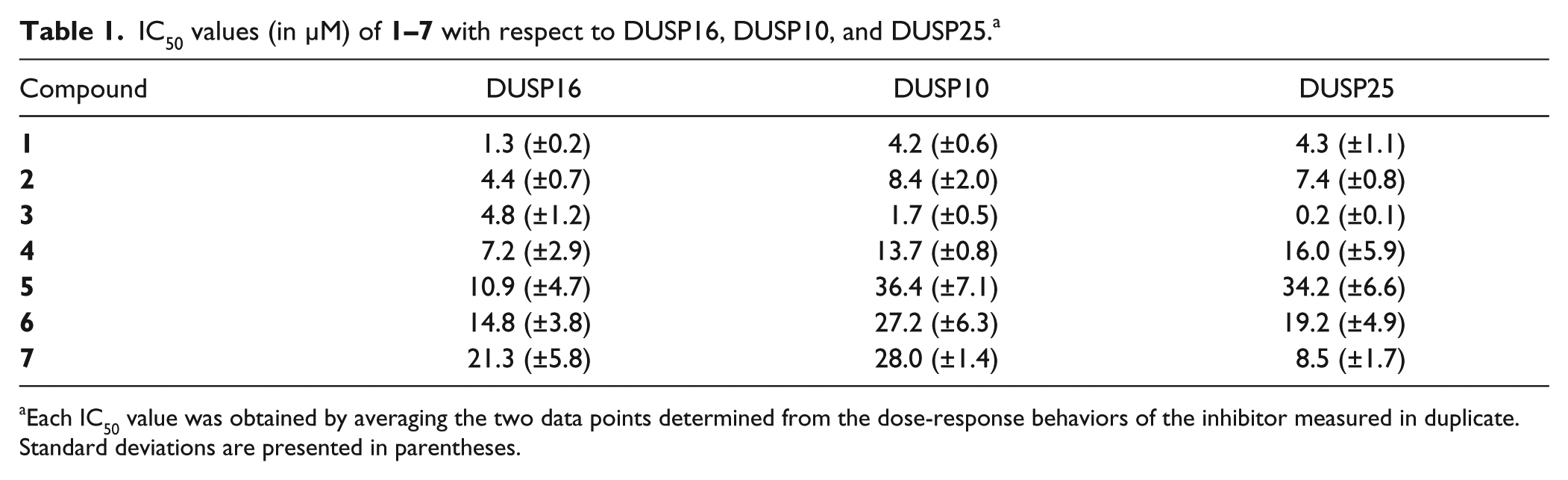
Each IC50 value was obtained by averaging the two data points determined from the dose-response behaviors of the inhibitor measured in duplicate. Standard deviations are presented in parentheses.
Because
Because the selectivity has been one of the most important issues in the development of phosphatase inhibitors, we determined the inhibitory activities of
Related to the imperfections in the simple desolvation model implemented in the original AutoDock program, we found that the rankings of
To gain structural insight into the inhibition mechanisms for the identified DUSP16 inhibitors, we examined their binding modes in the active site in a comparative fashion.
Figure 3
displays the lowest-energy conformations of
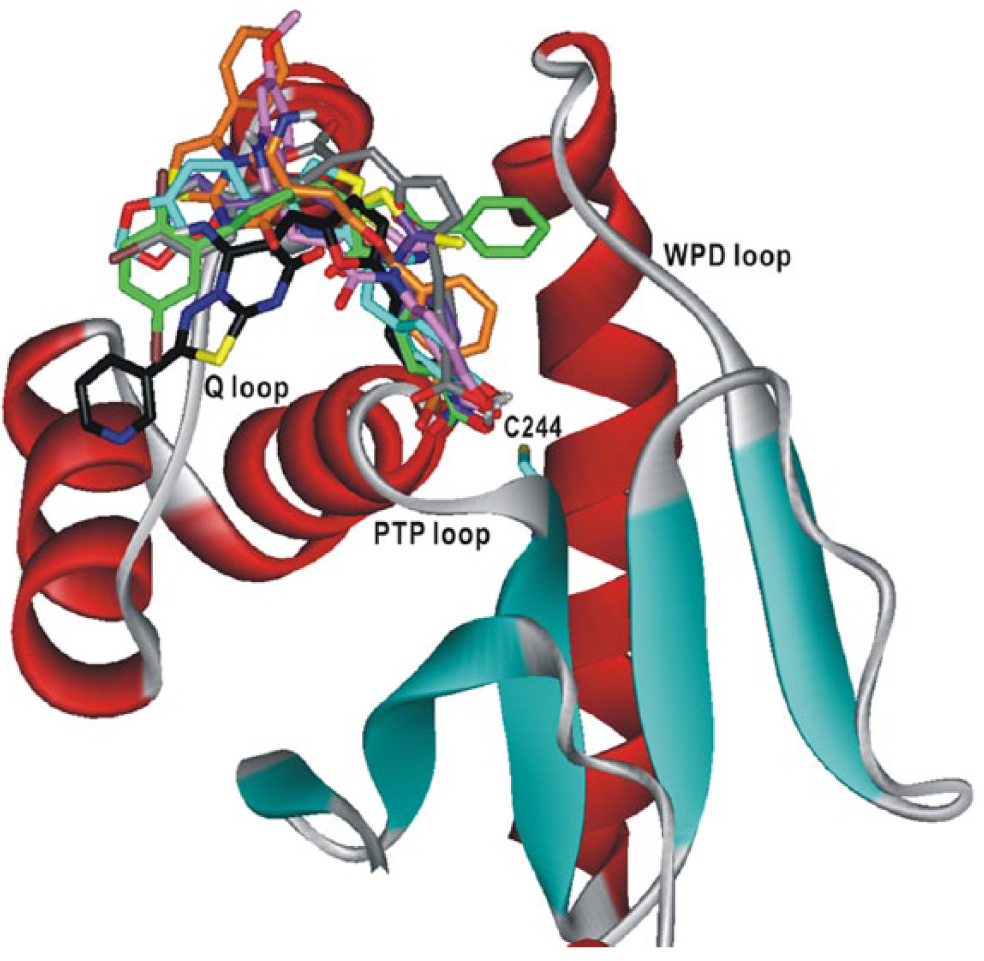
Comparative view of the binding modes of
The calculated binding mode of
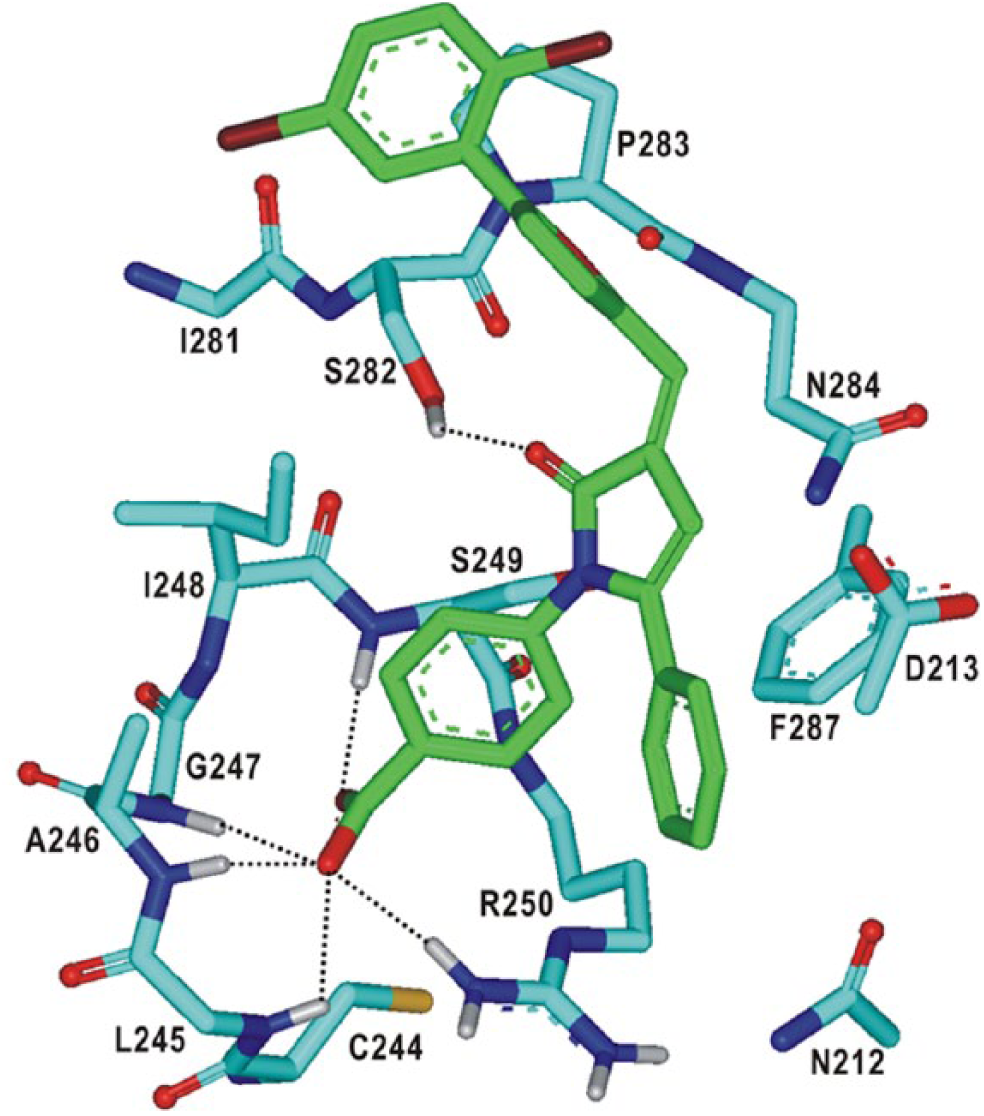
Calculated binding mode of
Figure 5
shows the lowest-energy binding mode of
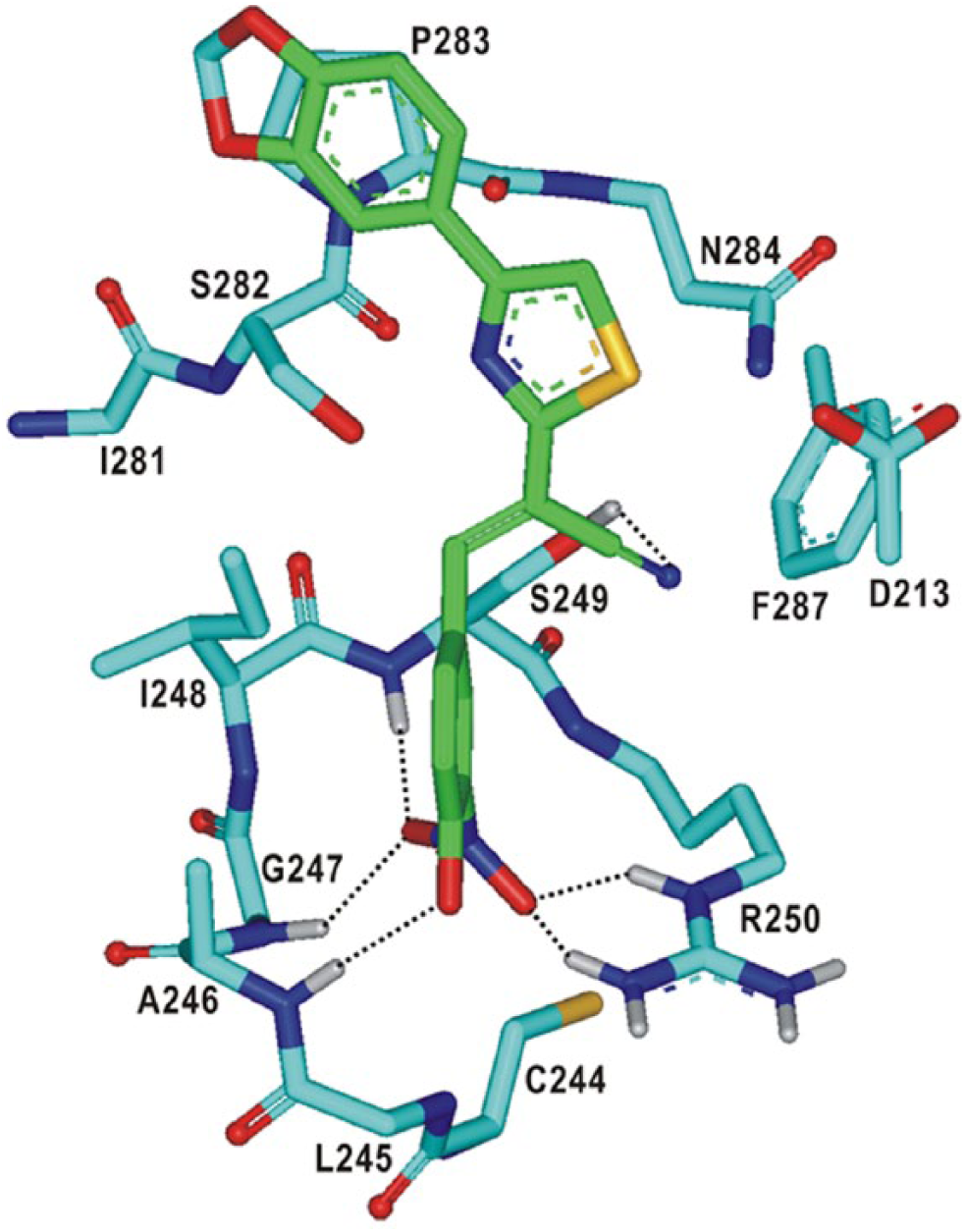
Calculated binding mode of
As can be inferred from the overlaid docking poses in
Figure 3
,
In conclusion, we have identified seven novel inhibitors of DUSP16 by means of a computer-aided drug design protocol involving the homology modeling and the structure-based virtual screening. To increase the accuracy in the calculated binding affinity between DUSP16 and the putative inhibitors, the scoring function was improved in such a way as to reflect the effect of ligand solvation on protein-ligand docking. The newly found inhibitors revealed a good potency, with IC50 values ranging from 1.3 to 21.3 µM. Because these inhibitors were also found to have significant inhibitory activity for other DUSPs, future SAR studies to develop the lead compounds for a new anticancer medicine should be carried out in such a way as to improve the selectivity in the inhibition of DUSP16 as well as to optimize the inhibitor potency. Detailed binding mode analyses with docking simulations indicated that the inhibitors could be stabilized through the multiple hydrogen bonds and the van der Waals contacts established simultaneously in the active site of DUSP16.
Footnotes
Acknowledgements
The authors thank the Olson group at Scripps Research Institute for kindly providing the AutoDock program.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2011-0022858), and a Hanyang University internal grant.