
Abstract
VH1-like phosphatase Z (VHZ) has proved to be a promising target for the development of therapeutics for the treatment of human cancers. Here, we report the first example for a successful application of structure-based virtual screening to identify the novel small-molecule inhibitors of VHZ. These inhibitors revealed high potencies with the associated IC50 values ranging from 3 to 20 µM and were also screened for having desirable physicochemical properties as a drug candidate. Therefore, they deserve consideration for further development by structure-activity relationship studies to optimize inhibitory and anticancer activities. Structural features relevant to the stabilization of the newly identified inhibitors in the active site of VHZ are discussed in detail.
Introduction
Protein tyrosine phosphatases (PTPs) are often involved in mitogenic signal transduction for cell growth and differentiation. VH1-like phosphatase Z (VHZ), which is also called dual-specificity protein phosphatase 23 (DUSP23), catalyzes the dephosphorylation reactions of phosphotyrosyl and phosphoseryl/threonyl residues in specific protein substrates. 1 VHZ is one of the smallest phosphatases with 150 amino acids and contains the minimal set of secondary structure elements conserved in all known phosphatases. 2 Recently, it was demonstrated that the activation and the knockdown of VHZ would, respectively, have an effect of enhancing and retarding the progression of the cell cycle associated with carcinogenesis from the G1 to S phase. 3 Furthermore, VHZ was shown to be overexpressed in multiple human primary cancers, including breast cancer. 3 These series of experimental observations indicated that VHZ should be a promising target for the development of therapeutics for human cancers.
VHZ can dephosphorylate p44-ERK1 (MAPK3) 4 and enhance the activation of c-Jun N-terminal kinase (JNK) and p38 (MAPK14) stimulated by several osmotic stress, and it functions as an enhancer of osmotic stress. 5 X-ray crystal structures of human VHZ were reported in complex with the substrate analogue, D-malate. 6 The presence of structural information about the nature of the interactions between the active-site residues of VHZ and the small-molecule ligand can make it a plausible task to design the potent inhibitors that may develop into an anticancer medicine. Indeed, the usefulness of such structural information has been well appreciated in designing the potent and selective small-molecule inhibitors of various PTPs. 7 Nonetheless, the discovery of VHZ inhibitors has lagged behind the biochemical and structural studies. To the best of our knowledge, no small-molecule VHZ inhibitor has been reported so far in the literature or in patents.
In the present study, we aim to identify the potent VHZ inhibitors by means of a structure-based drug design protocol involving virtual screening with docking simulations and an in vitro enzyme assay. The characteristic feature that discriminates our virtual screening approach from the others lies in the implementation of an accurate solvation model in calculating the binding free energy between VHZ and its putative inhibitors, which would have an effect of increasing the hit rate in the enzyme assay. 8 We find in this study that the docking simulations with the improved binding free energy function can be a useful computational tool for elucidating the activities of the identified inhibitors, as well as for enriching the chemical library with molecules that are likely to have desired biological activities.
Materials and Methods
The atomic coordinates in the X-ray crystal structure of human VHZ 6 (PDB entry: 2IMG) were selected as the receptor model in the virtual screening with docking simulations. The docking library for VHZ, comprising about 240 000 compounds, was constructed from the latest version of the chemical database distributed by InterBioScreen (Moscow, Russia) containing approximately 477 000 synthetic and natural compounds. Prior to the virtual screening with docking simulations, they were filtrated on the basis of Lipinski’s “Rule of Five” to adopt only the compounds with the physicochemical properties of potential drug candidates 9 and without reactive functional group(s). All of the compounds included in the docking library were then processed with the CORINA program (Molecular Networks, Erlangen, Germany) to generate their 3D atomic coordinates, which was followed by the assignment of Gasteiger-Marsilli atomic charges. We used the AutoDock program (AutoDock, La Jolla, CA) 10 in the virtual screening of VHZ inhibitors because the outperformance of its scoring function over those of the others had been shown in several target proteins. 11 Docking simulations with AutoDock were carried out in the active site of VHZ to score and rank the compounds in the docking library according to their calculated binding affinities.
In the actual docking simulation of the compounds in the docking library, we used the empirical AutoDock scoring function improved by the implementation of a new solvation model for a compound. The modified scoring function has the following form:
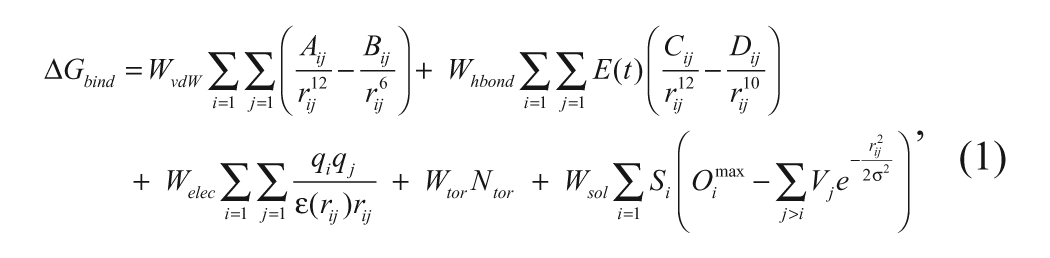
where WvdW, Whbond, Welec, Wtor, and Wsol are the weighting factors of van der Waals, hydrogen bond, electrostatic interactions, torsional term, and desolvation energy of a putative inhibitor, respectively. rij represents the interatomic distance, and Aij, Bij, Cij, and Dij are related to the depths of the potential energy well and the equilibrium separations between the two atoms. The hydrogen bond term has an additional weighting factor, E(t), representing the angle-dependent directionality. In the entropic term, Ntor is the number of rotatable bonds in the ligand. In the desolvation term, Si and Vi are the solvation parameter and the fragmental volume of atom i, 12 respectively, whereas Omax i stands for the maximum atomic occupancy. In the calculation of the molecular solvation free energy term in equation (1), we used the atomic parameters developed by Kang et al. 13 This modification of the solvation free energy term is expected to increase the accuracy in virtual screening because the underestimation of ligand solvation often leads to the overestimation of the binding affinity of a ligand with many polar atoms. 8 Indeed, the superiority of this modified scoring function to the previous one was well appreciated in recent studies for virtual screening of kinase and phosphatase inhibitors.14,15
The gene for residues 1 to 150 of VHZ was cloned into Escherichia coli expression vector pET28a. Cells containing the vector were induced with 0.2 mM IPTG and grown further at 18 °C for 16 h. Cell pellets were resuspended in the lysis buffer containing 50 mM Tris-HCl pH (7.5), 0.5 M NaCl, 1% PMSF, and 5% glycerol. The resuspension was then lysed by sonication on ice. The His-tagged VHZ protein was purified by an Ni-NTA agarose column and dialyzed against 20 mM Tris-HCl (pH 8.0), 5 mM EDTA, 50 mM NaCl, and 5% glycerol. In total, 142 compounds selected from the virtual screening were evaluated for their in vitro inhibitory activity against the purified VHZ. These enzyme assays were performed by monitoring the extent of hydrolysis of 6,8-difluoro-4-methyl-umbelliferyl phosphate (DiFMUP) with the spectrofluorometric assay. The purified VHZ (100 nM), DiFMUP (10 µM), and a candidate inhibitor were incubated in the reaction mixture containing 20 mM Tris-HCl (pH 8.0), 0.01% Trition X-100, and 5 mM dithiothreitol (DTT) for 20 min. The resulting fluorescence was measured at 460 nm by using the PerkinElmer 2030 instrument (PerkinElmer, Waltham, MA). To determine the IC50 values of the inhibitors, their inhibitory activities were measured in duplicate at the concentrations of 0.0, 0.1, 0.5, 1.0, 2.0, 5.0, 10, 20, and 50 µM to obtain the dose-response curve fits. IC50 values of the inhibitors were then determined from direct regression analysis using the four-parameter sigmoidal curve as implemented in the SigmaPlot program (Systat Software, Chicago, IL).
Results and Discussion
Of the 240 000 compounds screened with docking simulations, 150 compounds were selected as virtual hits. In total, 142 were available from the compound supplier and were tested for inhibitory activity against VHZ by an in vitro enzyme assay. As a result, we identified seven compounds that inhibited the catalytic activity of VHZ by more than 50% at the concentration of 20 µM, which were selected to determine the IC50 values. The chemical structures and the inhibitory activities of the newly identified inhibitors are shown in
Figure 1
and
Table 1
, respectively. The dose-response behaviors of the inhibitors measured to obtain their IC50 values are presented in the supplementary data. We note that compounds
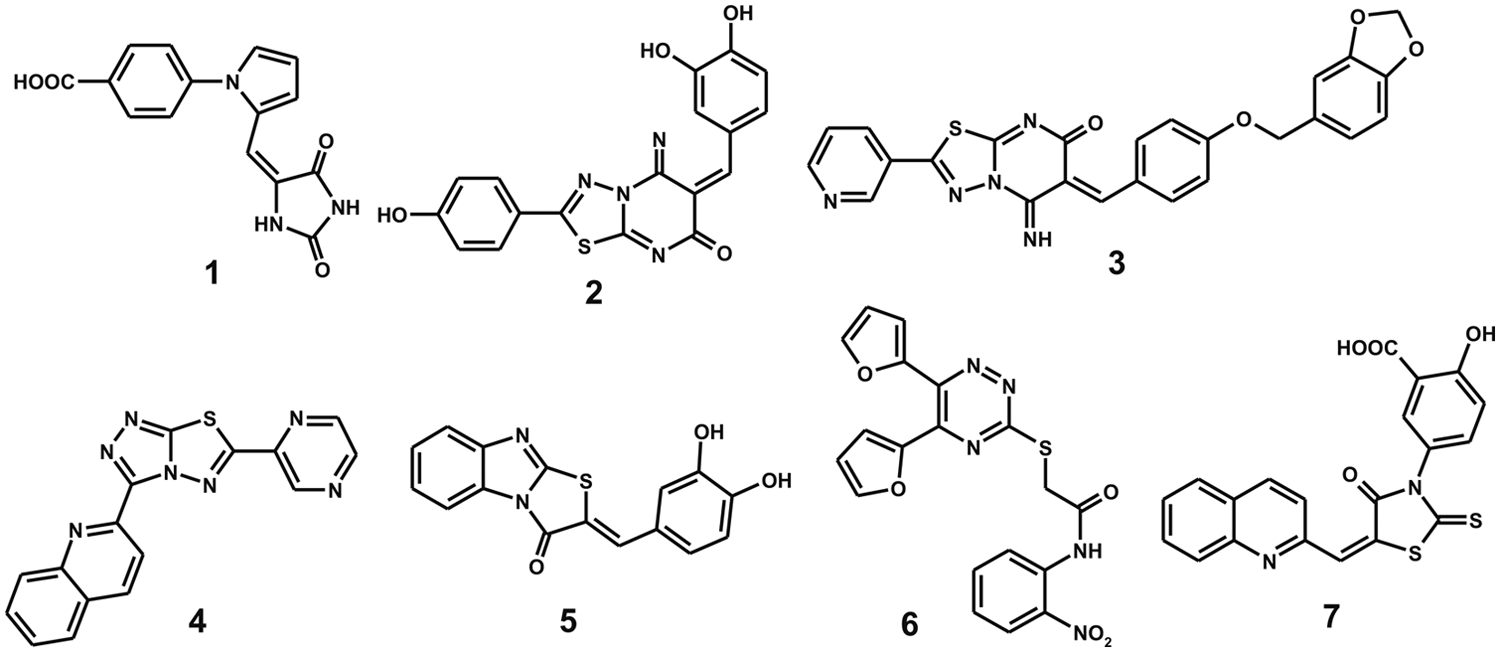
Chemical structures of the newly identified VHZ phosphatase inhibitors.
IC50 Values (in µM) against Various Dual-Specificity Protein Phosphatases and the Fluorescence Interference (FI) in Enzyme Assay for
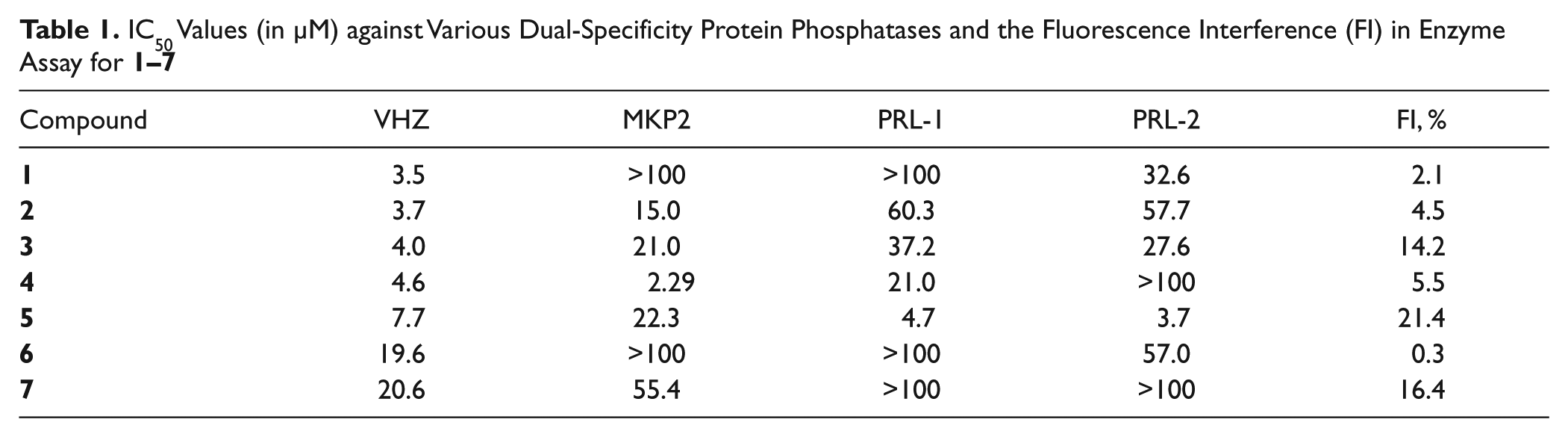
To confirm the inhibitory activity of a phosphatase inhibitor, one must disprove the possibility that the inhibitor would fluorescently interfere with the assay signal. This validation study started with the incubation of an enzymatic reaction mixture for 20 min without the treatment of an inhibitor to obtain the maximum fluorescence signal. At this point, the enzymatic reaction was terminated by the treatment of vanadate, which was followed by the addition of each inhibitor in the reaction mixture to determine its effect of fluorescence interference. The extent of the decrease in the fluorescence signal was then measured at the inhibitor concentration of 10 µM after being incubated for 5 min. As can be seen in the last column of Table 1, the fluorescence signals appear to be reduced by only 0.3% to 21.4% due to the interference effects from the inhibitors. These small interference effects indicate that the major contribution to the decrease in fluorescence signal in enzyme assays should come from the inhibition of enzymatic activity rather than from the role of the fluorescence quencher played by the inhibitors.
Because selectivity has been one of the most important issues in the development of phosphatase inhibitors, we determined the inhibitory activities of
To obtain structural insight into the inhibitory mechanisms of the identified VHZ inhibitors, we investigated their binding modes in the active site in a comparative fashion. The active site of VHZ comprises a distinctive HCXXGXXRS(T) signature motif, which is known as the PTP loop and is characteristic of cysteine-dependent PTPs. The PTP loop includes the conserved catalytic Cys95 that plays the role of nucleophile to attack the phosphorous atom in the catalytic dephosphorylation of pTyr/Thr/Ser substrates. The lowest-energy conformations of
We next turn to the identification of the detailed interactions responsible for the stabilization of the newly found inhibitors in the active site. The calculated binding mode of
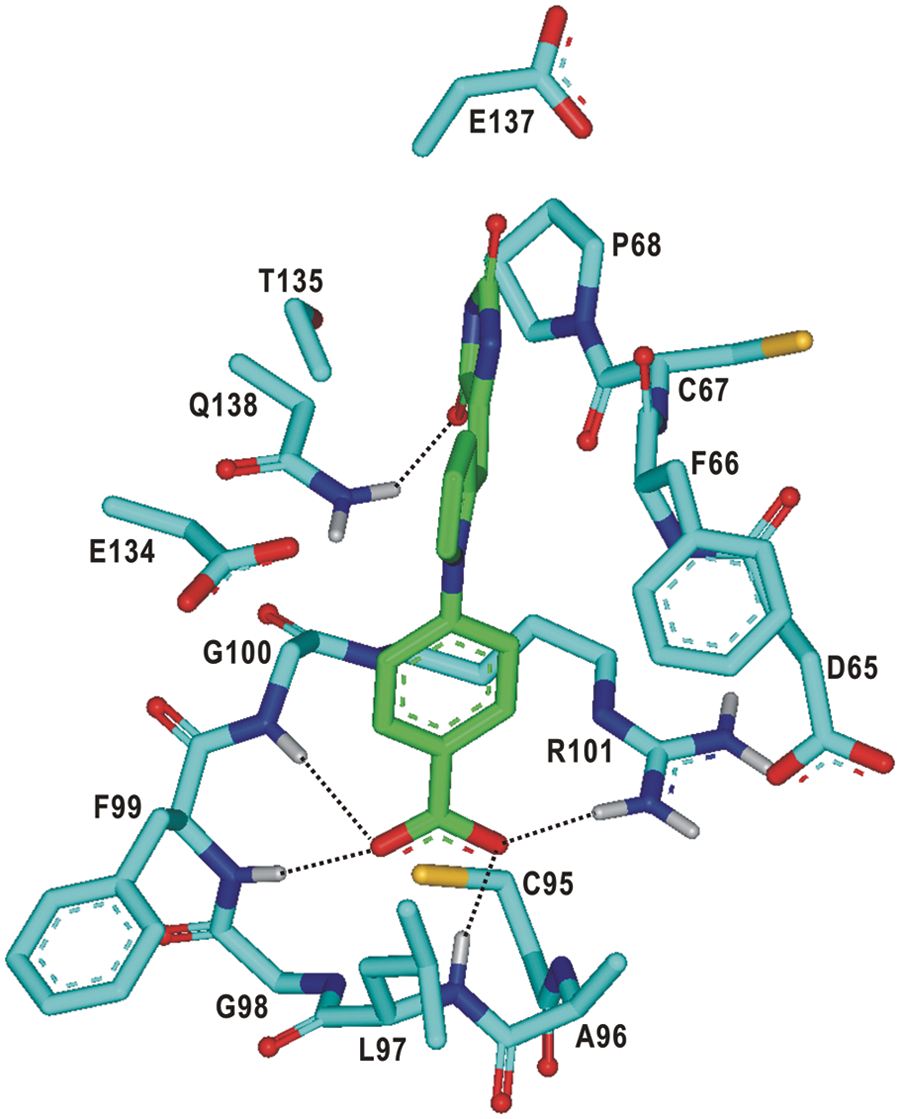
Calculated binding mode of
Figure 3
shows the lowest-energy binding mode of
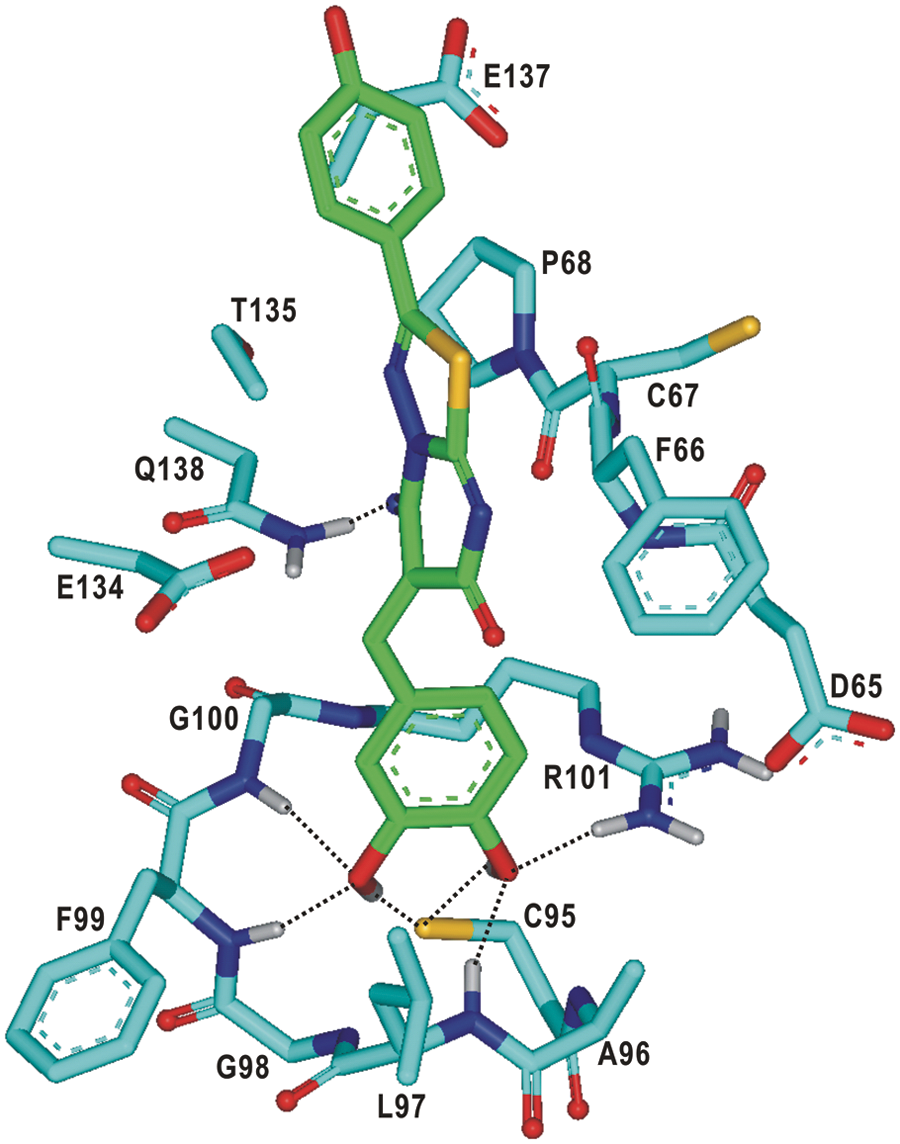
Calculated binding mode of
In conclusion, we have identified seven novel inhibitors of VHZ by applying a computer-aided drug design protocol involving the structure-based virtual screening with docking simulations under consideration of the effects of ligand solvation in the scoring function. These inhibitors have desirable physicochemical properties as a drug candidate and reveal a high potency with IC50 values ranging from 3 to 20 µM. Therefore, each of the newly discovered inhibitors deserves consideration for further development by SAR studies to optimize inhibitory and anticancer activities. Detailed binding mode analyses with docking simulations show that the inhibitors can be stabilized in the active site of VHZ by the simultaneous establishment of multiple hydrogen bonds with the active-site residues and van der Waals contacts with the hydrophobic side chains of the residues in the WPD loop.
Footnotes
Acknowledgements
We thank Prof. Olson at Scripps Research Institute for providing the AutoDock program.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2011-0022858), and a Hanyang University internal grant.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.