
Abstract
The transfer RNA (tRNA)–dependent pathway for lipid aminoacylation is a two-step pathway composed of (1) a tRNA aminoacylation step catalyzed by an aminoacyl-tRNA synthetase, forming a specific aa-tRNA, and (2) a tRNA-dependent transfer step in which the amino acid acylating the tRNA is transferred to an acceptor lipid. The latter step is catalyzed by a transferase located within the cytoplasmic membrane of certain bacteria. Lipid aminoacylation modifies the biochemical properties of the membrane and enhances resistance of some pathogens to various classes of antimicrobial agents and components of the innate immune response. Lipid aminoacylation has also been linked to increased virulence of various pathogenic bacteria. Inhibition of this mechanism would render pathogens more susceptible to existing drugs or to natural defenses of a host organism. Because lipid aminoacylation is widespread in many bacterial genera and absent from eukaryotes, and because the tRNA aminoacylation step of this pathway is also used in protein biosynthesis (a process essential for bacterial life), this pathway represents an attractive target for drug design. We have reconstituted the lipid aminoacylation pathway in vitro and optimized it for high-throughput screening of libraries of compounds to simultaneously identify inhibitors targeting each step of the pathway in a single assay.
Keywords
Introduction
Aminoacyl-phosphatidylglycerol synthases (aaPGSs) are integral membrane proteins responsible for the transfer of amino acids (aa) from aminoacyl–transfer RNAs (aa-tRNAs) to the head groups of phosphatidylglycerol (PG) to form aminoacyl-phosphatidylglycerols (aa-PGs) within the bacterial membrane. Some aaPGSs are specific for a single aa-tRNA (e.g., Lys- or Ala-tRNA), while others exhibit relaxed substrate specificity and can use up to three different aa-tRNAs as aa donors (i.e., Lys-, Ala-, and Arg-tRNA).1–3 A novel lipid (different from PG) was recently shown to be included in the repertoire of substrates that are modified by these enzymes. An aaPGS homolog, characterized in Corynebacterium glutamicum, was shown to be an alanyl-diacylglycerol synthase (AlaDAGS), which alanylates diacylglycerol (DAG) instead of PG. 4
Bacteria exhibiting aa-PGs in the membrane display enhanced resistance to environmental stressors (e.g., low pH, osmolytes), as well as to a variety of antimicrobial molecules. Specifically, aa-PGs increase bacterial resistance to several glycopeptides, 5 β-lactams, lipopeptides, and various cationic antimicrobial peptides (CAMPs), such as those secreted by the innate immune systems of certain multicellular organisms as a defense against pathogenic invasion (see Roy 6 and Ernst and Peschel 7 for review). The newly characterized AlaDAGS was likewise found to increase the fitness of C. glutamicum when challenged against several CAMPs and lactic acid. 4 Lastly, Lys-PG in the membrane was shown to increase the virulence of various bacterial pathogens (e.g., Staphylococcus aureus, 8 Listeria monocytogenes, 9 Bacillus anthracis, 10 and Mycobacterium tuberculosis 11 ) in experiments using cell lines as well as animal models. To date, over 600 aaPGS homologs have been identified, encompassing nearly 150 bacterial genera. 12 The broad distribution of aaPGSs in bacterial species, along with their absence in eukaryotes, makes these enzymes appealing drug targets for which inhibitors could render pathogens more susceptible to existing antibiotics or to natural defenses of a host organism’s innate immune response (i.e., CAMPs).
The first step of the lipid aminoacylation pathway is carried out by specific aminoacyl-tRNA synthetases (aaRSs), which produce the aa-tRNAs acting as substrates for aaPGSs. aaRSs are essential for cellular life as they also provide the aa-tRNAs necessary for protein synthesis. Several natural inhibitors targeting these enzymes have been identified (e.g., mupirocin 13 and microcin C 14 ). Therefore, aaRSs represent longstanding, validated drug targets for antibiotic development. Indeed, certain microbial and parasitic aaRSs, lacking homology to the corresponding human aaRSs, have been used as molecular targets for the recent development of anti-infective drugs (for review, see Pham et al. 15 and Vondenhoff and Van Aerschot 16 ). In contrast to aaRSs, there are currently no known inhibitors of the aaPGSs responsible for the second step of lipid aminoacylation.
In the present study, we developed a nonradioactive assay to monitor the activity of the two-step pathway for lipid aminoacylation. The high sensitivity and reliability, as well as low cost and low hazard qualities of the assay, provide a strong foundation for its applicability as a high-throughput screening (HTS) method for simultaneously identifying inhibitors of both enzymes (i.e., aaRS and aaPGS) constituting the lipid aminoacylation pathway.17,18
Materials and Methods
General Procedures
The plasmid encoding tRNAAla (UGC) and the strain expressing the alanyl-tRNA synthetase (AlaRS) from Escherichia coli bearing a N-terminal 6xHis tag were both a gift from K. Musier-Forsyth (The Ohio State University). 19 The strain expressing the AlaDAGS483–832 (Cg1103 483–832) from C. glutamicum deprived of its membrane domain and conditions for expression and purification of histidine-tagged proteins were described previously. 4
In Vitro Transcription of tRNAAla
In vitro transcription of the tRNAAla (UGC) from E. coli was conducted as described earlier. 20 Transcripts were extracted with phenol/chloroform, precipitated with ethanol, and purified on 12% (v/v) acrylamide gel containing 8 M urea. Transcripts were recovered from the gel by electroelution in a dialysis bag (molecular weight cutoff 5 kDa) subjected to 100 V for 2 h. The concentration of active tRNA was determined using the aminoacylation assay with [14C]-Ala (see below).
Determination of the Concentration of Active tRNA Transcript by Aminoacylation with Radiolabeled Ala
Aminoacylation was performed in 100 mM Hepes-NaOH (pH 7.2), 30 mM KCl, 2 mM adenosine triphosphate (ATP), 10 mM MgCl2, and 50 µM L-[14C]Ala (200 cpm/pmol). E. coli AlaRS (0.1 µM) was used with various amounts of tRNAAla transcript from E. coli. After 10 min of incubation at 37 °C, aliquots (15 µL) were periodically removed and spotted on 3MM filter discs (Whatman International Ltd, Maidstone, UK), washed 3 times with 10% trichloroacetic acid, and dried. The amount of active tRNA corresponds to the amount of L-[14C]Ala-tRNAAla retained on each filter disc at the plateau of the reaction and was determined by liquid scintillation counting.
Preparation of Diacylglycerol/Triton X-100 Mixed Micelles and Monoacylglycerol/Diacylglycerol Emulsions
To obtain DAG/Triton X-100 mixed micelles, dried lipids were resuspended in water with varying amounts of Triton X-100 via gentle sonication. Various ratios of DAG/Triton X-100 were assayed and the total concentration was held constant at 1 mM. To obtain monoacylglycerol (MAG)/DAG emulsions (8:1), dried lipids were resuspended by vortexing and gentle sonication to a final concentration of 1.6 mM in buffer containing 50 mM Hepes-NaOH (pH 7.2), 30 mM KCl, 10 mM MgCl2, and 1 mM dithiothreitol (DTT). DAG (1-palmitoyl-2-oleoyl-sn-glycerol) and MAG (1-(9Z-octadecenoyl)-rac-glycerol) were purchased from Avanti Polar Lipids, Inc., Alabaster, AL, USA.
Malachite Green Assay for Monitoring In Vitro Lipid Aminoacylation
Lipid aminoacylation reactions contained 50 mM Hepes-NaOH (pH 7.2), 30 mM KCl, 0.4 mM ATP, 10 mM MgCl2, 1 mM DTT, 2 U/mL PPase (Hoffmann-La Roche Ltd, Basel, Switzerland), 0.3 mM L-Ala, 10 µM tRNAAla, AlaRS from E. coli and AlaDAGS483–832 from C. glutamicum (as indicated), and either 1 mM DAG mixed micelles (Triton X-100 + DAG; Avanti Polar Lipids) or 1.6 mM MAG/DAG emulsion. After various times of incubation at room temperature, 15-µL aliquots were removed and the reaction was stopped by addition of 15 µL RNaseA (0.1 mg/mL) in a 96-well plate. The malachite green reagent was prepared daily as described in Biswas et al. 21 by combining stock solutions of 1.75 mM malachite green oxalate, 2.32% (w/v) polyvinyl alcohol, 292 mM ammonium molybdate (in 6M HCl), and water in a 2:1:1:2 volumetric ratio. Then, 150 µL of the malachite green reagent was added to the samples, and after 30 min of incubation at room temperature, the absorbance of each well was measured at 630 nm (Synergy H1 Hybrid Reader; BioTek Instruments, Winooski, VT). Phosphate was quantified using a NaK(PO4)2 standard curve. The amount of contaminating phosphate present at the beginning of the reaction (independently of the tRNA aminoacylation reaction) was determined in a mixture deprived of tRNA. This value was subtracted from amounts determined with complete reaction mixtures.
Results
In Vitro Quantification of Lipid Aminoacylation Activity Using the Malachite Green Assay
The current method for quantifying the activity of the lipid aminoacylation pathway uses radioactivity 6 and was recently used to identify the membrane lipid DAG as a new component for lipid aminoacylation. 4 However, this method is impractical for HTS due to the hazardous nature and high cost of radiolabeled amino acids. Moreover, this method requires several labor-intensive extraction steps to isolate the radiolabeled aminoacylated lipids prior to their quantification, which makes this assay incompatible with an HTS system.
A spectrophotometric assay for measuring aaRS activity was recently reported. 22 In this assay, pyrophosphatase (PPase) within the assay converts the inorganic pyrophosphate (PPi) released during the tRNA aminoacylation step into inorganic phosphate (Pi). The Pi is then quantitatively detected at 620 nm using a malachite green reagent. We developed a similar assay to monitor the two-step tRNA-dependent pathway for lipid aminoacylation by exploiting the high amount of PPi generated upon recycling of the tRNA as the amino acids are transferred during the lipid aminoacylation step ( Fig. 1A ). In this scheme, recycling of the tRNA by AlaDAGS results in increased accumulation of PPi relative to a system composed of AlaRS alone. Measuring the Pi levels with the malachite green reagent permits monitoring of the activity of the complete pathway when the tRNA recycling step remains the rate-limiting step of the overall system. To quantify the Pi accumulation resulting from this reaction, a standard curve was generated using inorganic phosphate ( Fig. 1B ). The two-step pathway for lipid aminoacylation was reconstituted in vitro using the purified AlaRS and tRNAAla transcript from E. coli and the soluble form of the AlaDAGS (deprived of its membrane domain encompassing residues 483–832) from C. glutamicum ( Fig. 1C ). 4 Since we were unable to obtain a lipid emulsion using DAG alone, we assessed the initial sensitivity of the assay using MAG/DAG vesicles. As predicted, in the absence of AlaDAGS, alanylation of 10 µM tRNAAla yielded approximately 20 µM Pi, which corresponds to twice the concentration of tRNAAla present in the medium. Addition of AlaDAGS and MAG/DAG vesicles to the system allowed for recycling of the tRNA, and the concentration of Pi increased approximately 7-fold. In the absence of AlaRS, AlaDAGS could not promote formation of Pi. This was considered background and subtracted from the other samples ( Fig. 1C ). While the pathway was successfully reconstituted in vitro, the MAG/DAG emulsion was not stable and tended to precipitate, leading us to further optimize the addition of DAG in the assay.
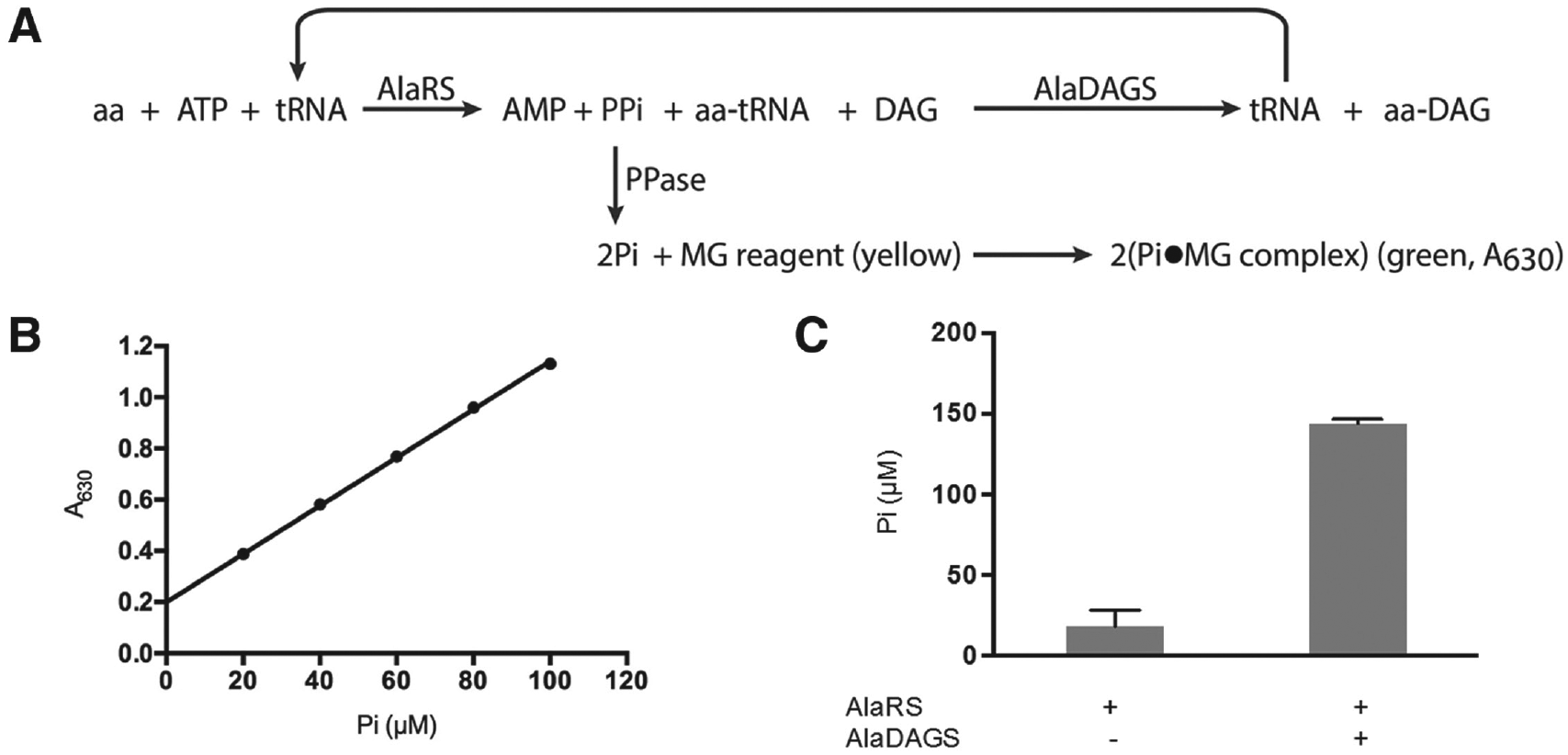
Spectrophotometric detection of the transfer RNA (tRNA)–dependent pathway for lipid aminoacylation. (
Addition of DAG in the Form of Mixed Micelles with Triton X-100
Following sonication, the MAG/DAG emulsions often precipitated rendering the samples unreadable after addition of the malachite green reagent (data not shown). In addition, the high cost of purified MAG prompted us to optimize the addition of DAG in the assay. A method to generate DAG/Triton X-100 mixed micelles (previously developed for DAG kinase studies) was used, and various ratios of the two components were tested in the assay for tRNA-dependent lipid modification ( Fig. 2 ). 23 The sum of DAG and Triton X-100 (DAG + Triton X-100) in each mixture was held constant at a total concentration of 1 mM. DAG and Triton X-100 added in a 1:9 ratio yielded the highest velocity of Pi accumulation, suggesting that DAG provided at a lower ratio is more accessible to AlaDAGS than DAG provided at a higher ratio. It is worth noting that the amount of PPase (2 U/mL) used to generate the data presented in Figure 3 was non–rate limiting. This amount of enzyme ensured a large kinetic excess of PPase, and twice this amount (4 U/mL) in the assay did not increase the kinetic of accumulation of Pi (data not shown). DAG/Triton X-100 mixed micelles (1:9) were further used to optimize the concentrations of enzymes (i.e., AlaRS and AlaDAGS) to be used in the assay.
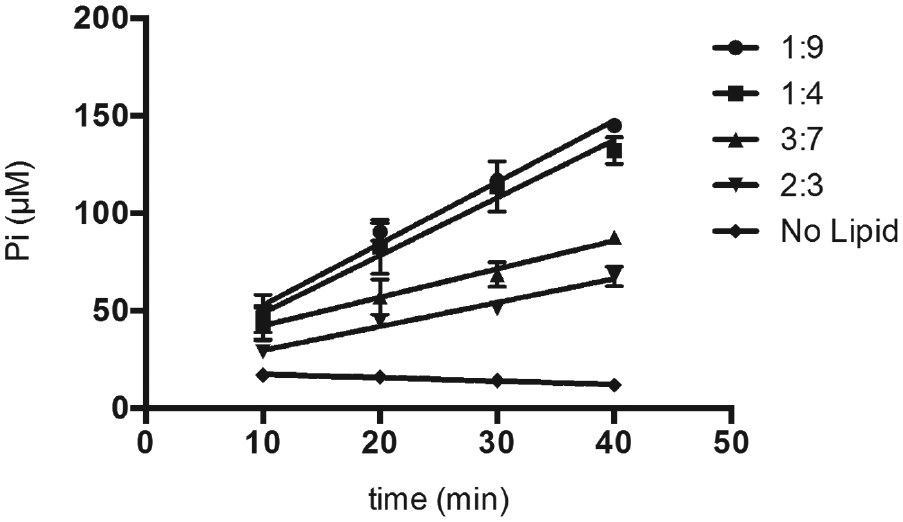
Kinetics of diacylglycerol (DAG) aminoacylation using DAG/Triton X-100 mixed micelles as substrates. The total concentration of combined lipids and detergent was held constant at 1 mM with the ratios (indicated at right) representing the relative amounts of DAG and Triton X-100. The concentrations of alanyl-tRNA synthetase (AlaRS) and alanyl-diacylglycerol synthase (AlaDAGS) were 0.05 µM and 2 µM, respectively. Inorganic phosphate (Pi) accumulation was determined kinetically by removal of 15-µL aliquots of the reaction mixture over time as described in Materials and Methods.
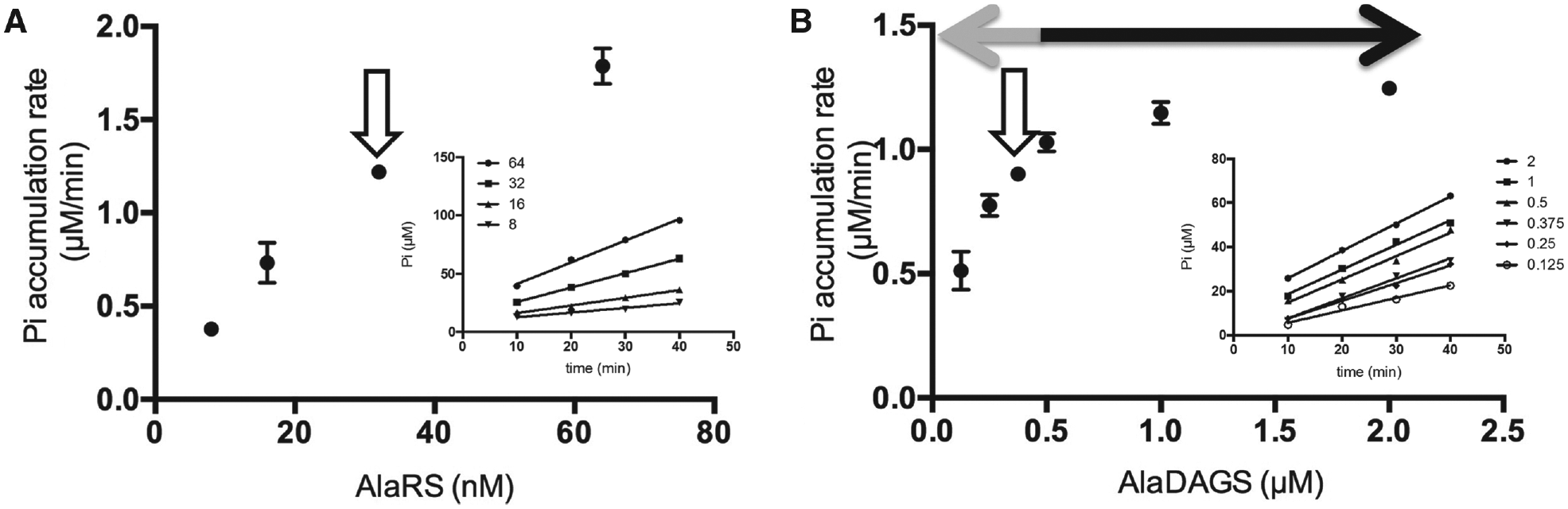
Optimization of the amounts of enzymes for the high-throughput screening (HTS) assay. (
Optimization of AlaRS and AlaDAGS Concentrations for an HTS Assay
The concentration of each substrate targeted in an HTS assay is an important parameter to consider, especially when identifying competitive inhibitors of enzyme activity. For instance, substrate levels that are too high (i.e., >>10 × KM) may prevent binding of weak inhibitors, while levels that are too low can decrease the measurable readout of the assay. Most inhibitors of aaRSs discovered to date compete with small substrates of the tRNA aminoacylation reaction such as aa and ATP. 16 The KM values of the E. coli AlaRS for its substrates have been determined (KM for L-Ala is 0.3 mM; KM for ATP is 0.14 mM), 24 and the concentrations of substrates used in the assay described here (i.e., 0.3 mM L-Ala and 0.3 mM ATP) are within close proximity (1- to 2-fold) of the reported values. The KM of AlaDAGS for Ala-tRNAAla is not known but is likely to be comparable to that of similar enzymes (i.e., AlaPGS and LysPGS from Clostridium perfringens 2 ), for which the values were determined to be in the micromolar range. The concentration of Ala-tRNAAla used in this assay is therefore likely to be within an acceptable range (1- to 2-fold) of the KM value of AlaDAGS.
To detect inhibition of either step of the lipid aminoacylation pathway, it is essential to optimize the concentration of AlaRS and AlaDAGS used in the assay. To this end, tRNA recycling must be maintained as the rate-limiting step of the overall reaction. In these conditions, an inhibitor targeting the tRNA aminoacylation step will directly decrease the rate of accumulation of Pi. Alternatively, an inhibitor targeting the lipid aminoacylation step will indirectly affect the rate of Pi accumulation by decreasing the rate of tRNA recycling. In addition, to minimize the amount of inhibitors to be tested, the concentrations of both enzymes in the reaction need to be minimized but sufficient to allow for sensitive and accurate detection of Pi. Lastly, because end-point determination of Pi accumulation will be used in the HTS assay, it is essential that the end-point time of the assay falls within the linear range of accumulation of Pi over time (i.e., during the initial velocity of Pi accumulation and not within the plateau of the reaction). Under these conditions, the end-point concentration of Pi reflects the rate of accumulation of Pi, which enables detection of partial or weak inhibition affecting either step of the pathway.
Optimization of the concentration of AlaRS was performed in the presence of non-rate-limiting amounts of AlaDAGS (2 µM) and PPase (2 U/mL; see above). Various concentrations of AlaRS (ranging from 8–64 nM) were assayed, and Pi accumulation was determined kinetically by removing aliquots of the reaction mixture over a period of 40 min at room temperature ( Fig. 3A ). We selected 32 nM AlaRS based on the capacity of the enzyme provided at this concentration to (1) allow for sensitive detection of Pi accumulation and (2) produce a linear accumulation of Pi over the 40-min incubation period.
The amount of AlaDAGS required to maintain the tRNA-recycling step as the rate-limiting step for Pi synthesis was determined empirically. Figure 3B shows that at concentrations of AlaDAGS greater than 0.5 µM, the rate-limiting step of the pathway is the tRNA aminoacylation step, and the apparent rate of Pi synthesis remains constant regardless of the amount of AlaDAGS. Concentrations of AlaDAGS below 0.5 µM resulted in decreased rates of Pi accumulation proportional to the amount of AlaDAGS added, demonstrating that the tRNA recycling step is the rate-limiting step in the overall process. AlaDAGS at a concentration of 0.375 µM allowed for sufficient levels of Pi to be generated for sensitive detection and provided a satisfactory proportionality between the amount of enzyme and the rate of accumulation of Pi to ensure that inhibition of AlaDAGS during an HTS assay would be observable. These combined data demonstrate optimized concentrations of AlaRS and AlaDAGS suitable for an HTS format.
Validation of the Assay Using an Inhibitory Model and Z′ Factor
To produce a lipid aminoacylation system suitable for detection of potential inhibitors in an HTS assay, it is necessary to ensure that enzyme concentrations are sufficiently optimized and that inhibition of either enzyme will result in a significant decrease in Pi accumulation. To do this, an inhibitory model was generated to simulate 50% inhibition of each enzyme in the pathway by using either optimal amounts or half of optimal amounts of each enzyme. HTS assay conditions were mimicked, and accumulation of Pi was determined at a single point (after 40 min of incubation) during the initial velocity timeframe of the reaction in 15 µL of reaction mixture incubated in a 96-well microtiter plate. Fifty percent reduction of either enzyme resulted in decreased accumulation of Pi by approximately 50%, confirming that the enzymes are present in appropriate amounts to allow for detection of inhibition of either step of the overall reaction ( Fig. 4A ). The reproducibility and reliability of our assay were assessed by calculation of the Z′ factor 25 ( Fig. 4B ). The optimized assay exhibited a Z′ factor of 0.67, which indicates a separation between background noise and the signal of the assay of 18 times the average standard deviation of the two controls. This value validates the suitability of the assay for HTS investigations.
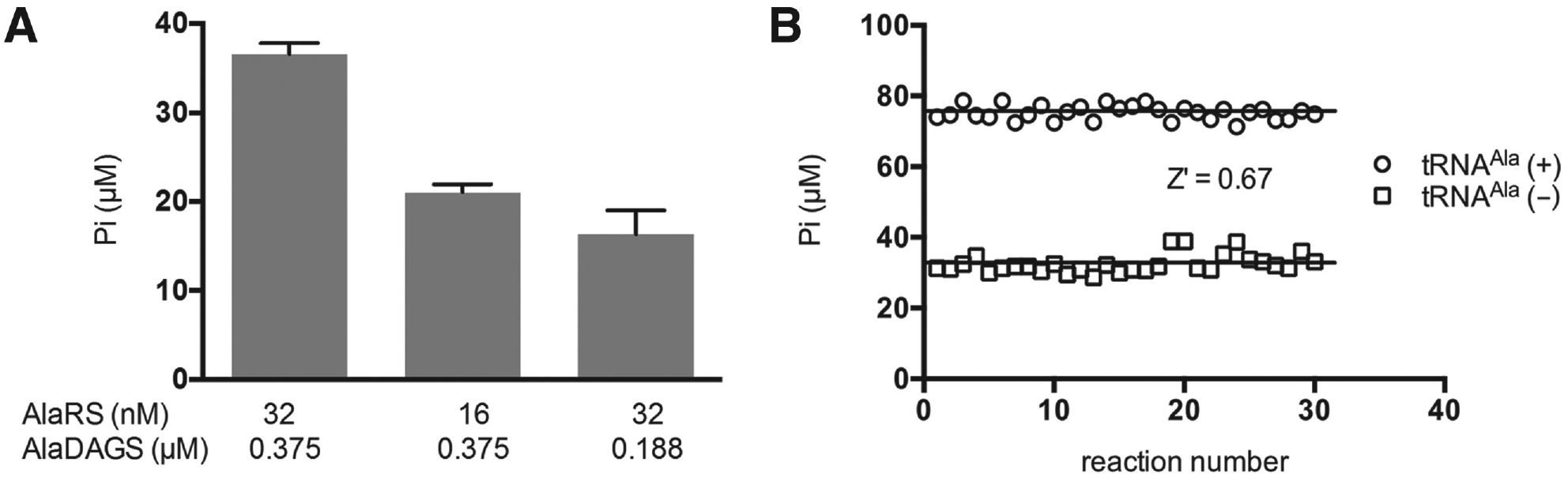
Inhibitory model and assay validation. (
Discussion
The tRNA-dependent pathway for lipid aminoacylation contributes to antimicrobial resistance and requires the activity of both aaRSs and aaPGSs, representing an appealing target for HTS to identify novel inhibitors of each of these enzymes. The malachite green assay exploits the large amount of Pi accumulation that occurs during charging and subsequent recycling of tRNA in the reaction to quantify pathway activity. Our assay represents the first nonradioactive method for quantifying the activity of the lipid aminoacylation pathway in vitro. Furthermore, we optimized the components of the pathway, using DAG/Triton X-100 (1:9) mixed micelles as substrates, as well as the AlaRS (32 nM) and AlaDAGS (0.375 µM) as enzymes, to catalyze the charging of tRNAAla and transfer of Ala to DAG in levels amenable to HTS. The inhibitory model and high Z′ factor score (Z′ = 0.67) described here validate the suitability of this assay for use in HTS. It is worth noting that following the primary screening of inhibitory compounds the specific mode of inhibition of lead compounds will need to be further explored. This will be necessary to determine which enzyme is being inhibited and to rule out the possibility of interference with other components of the assay (e.g., PPase, malachite green reagent, pH conditions, aa-tRNA stability).
Reconstitution of the lipid aminoacylation pathway allows for screening of inhibitory compounds targeting two enzymes (aaRSs and aaPGS/AlaDAGS) at once, whereas traditional HTS systems generally screen using a single target. This assay could be adapted to screen for inhibitors of other aaPGS and aaRS homologs, but the concentrations of each individual protein would need to be optimized. Use of AlaDAGS along with the DAG lipid substrate presented here will complement existing HTS methods for aaRS screening. The signal-to-background separation in our method provides a higher signal dynamic range than that of an assay targeting aaRS alone, which might allow for detection of weak inhibitors that would not elicit a detectable signal in an assay targeting just one enzyme.
Footnotes
Acknowledgements
The authors thank Dr. A. M. Smith (University of Central Florida) for critical reading of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institute of General Medical Sciences grant R15-109404.