
Abstract
The enzyme glucose-6-phosphate dehydrogenase (G6PDH) catalyzes the first step of the oxidative branch of the pentose phosphate pathway, which provides cells with NADPH, an essential cofactor for many biosynthetic pathways and antioxidizing enzymes. In Trypanosoma cruzi, the G6PDH has being pursued as a relevant target for the development of new drugs against Chagas disease. At present, the best characterized inhibitors of T. cruzi G6PDH are steroidal halogenated compounds derivatives from the mammalian hormone precursor dehydroepiandrosterone, which indeed are also good inhibitors of the human homologue enzyme. The lack of target selectivity might result in hemolytic side effects due to partial inhibition of human G6PDH in red blood cells. Moreover, the treatment of Chagas patients with steroidal drugs might also cause undesired androgenic side effects. Aiming to identify of new chemical classes of T. cruzi G6PDH inhibitors, we performed a target-based high-throughput screen campaign against a commercial library of diverse compounds. Novel TcG6PDH inhibitors were identified among thienopyrimidine and quinazolinone derivatives. Preliminary structure activity relationships for the identified hits are presented, including structural features that contribute for selectivity toward the parasite enzyme. Our results indicate that quinazolinones are promising hits that should be considered for further optimization.
Keywords
Introduction
Human American Trypanosomiasis constitutes a relevant socioeconomic and health problem in the Americas. Chagas disease is endemic in Latin America, but because of immigration, it has spread to other continents. Seven to eight million people worldwide are estimated to be infected with Trypanosoma cruzi, and about 25 million people are at risk. 1 Benznidazole and nifurtimox, prodrugs that are activated by trypanosomal type I nitroreductase,2,3 were introduced more than 40 years ago and remain the only available therapeutic alternatives. However, these drugs lack a desirable safety profile and are effective only when administered in the early acute phase. Furthermore, the occurrence of naturally resistant strains 4 and the development of drug resistance 5 have been reported. In this way, there is an urgent need for new drugs to challenge this unmet medical need. 6
Currently, there are few drugs in the development pipeline against Chagas disease. 7 Azole-based antifungals such as posaconazole were shown to inhibit the sterol 14α-demethylase (CYP51) from the lipid ergosterol biosynthetic pathway and are in clinical development. 8 Amiodarone and dronedarone kill the parasites in cell culture and animal models by disrupting calcium homeostasis and also inhibiting ergosterol biosynthesis. 9 In addition, reversible cruzipain inhibitors showed tripanocidal activity in both in vitro assays and in vivo murine model of infection. 10 Despite the development of the aforementioned molecules, it is noteworthy that the number of targets being explored in drug discovery initiatives against Chagas disease is still limited. Thus, target validation and early-stage drug discovery projects are desired.11,12
Glucose-6-phosphate dehydrogenase (G6PDH; EC. 1.1.1.49) catalyzes the first and rate-limiting step of the pentose-phosphate pathway, converting glucose-6-phosphate (G6P) to 6-phosphogluconolactone and reducing NADP+ into NADPH. G6PDH is a validated target in Trypanosoma brucei, the causative agent of Human African Trypanosomiasis. In the bloodstream form of T. brucei, both RNAi-mediated reduction of G6PDH levels
13
and specific inhibition of the enzyme by dehydroepiandrosterone (DHEA)
14
are able to kill parasites in vitro. In T. cruzi, DHEA derivatives such as epiandrosterone (EA) and 16α-bromo-epiandrosterone (16BrEA) were also shown to inhibit G6PDH and kill epimastigote forms in vitro.
15
Despite the promising effects of DHEA derivatives against T. cruzi, steroidal drugs might have androgenic side effects in humans. In addition, steroidal compounds, especially DHEA derivatives, are potent inhibitors of human G6PDH
16
(HsG6PDH), and this lack of selectivity toward trypanosomatid G6PDH (
In the present work, we describe the discovery and characterization of new uncompetitive G6PDH inhibitors belonging to two novel chemical classes, quinazolinones and thienopyrimidines. Among these new hits there were compounds with a high selectivity toward the parasite G6PDH and also having trypanosomal activity in vitro against T. cruzi epimastigote forms.
Materials and Methods
Expression and Purification of Recombinant G6PDH
The TcG6PDH was overexpressed and purified as previously described. 15 Briefly, Escherichia coli BL21 (DE3) transformed with the expression vector pET28-TcG6PDH was incubated at 25 °C in ZYM-5052 autoinduction media containing kanamycin 50 µg.mL−1. Cells were harvested after 48 h and disrupted, and the enzyme was purified by metal affinity chromatography (Ni-NTA; Invitrogen, Carlsbad, CA) following the fabricator’s instructions. The purified protein was stored in a buffer solution containing 25% glycerol at −80 °C.
The HsG6PDH gene cloned in the pET30b was kindly provided by Professor Paul Engel (University College of Dublin, Ireland). The primers 5′-AGGAATTCTCGG-ATACACACATATTCATCATCATG-3′ and 5′-AGCTCG-AGTTAGTTCACCCACTTGTAGGTGCC-3′ were used to amplify a gene fragment corresponding to the G6PDH ranging from amino acids 29 to 511 (HsΔG6PDH). This fragment was subcloned into the expression vector pET SUMO, using as a restriction site EcoRI and XhoI. HsΔG6PDH was overexpressed using the same protocol adopted for TcG6PDH. The cells were lysed and the soluble extract was loaded onto a pre-equilibrated nickel charge column (Ni-NTA, Invitrogen). The resin was extensively washed and the His-SUMO tag was cleaved with the incubation of ULP-1 protease at 4 °C overnight. The cleaved protein was eluted from the resin with buffer A, concentrated (Amicon 10 kDa; Milipore, Billerica, MA), and further purified by gel filtration using the column HiLoad Superdex 200 16/60 (GE Healthcare), pre-equilibrated with 50 mM Tris-HCl, pH 8.0, and 150 mM NaCl. The purified protein was stored in a buffer solution containing 25% glycerol at −80 °C.
Compound Libraries and Reagents for HTS
TcG6PDH primary screening was done using 30,000 compounds from the DIVERSet library of Chembridge (San Diego, CA). The compounds were received preplated in 384 well microplates at the stock concentration of 10 mM, in 100% DMSO. Before screening, compounds were transferred to daughter plates and diluted to 1 mM, in 100% DMSO. Columns 1, 2, 23, and 24 from daughter plates were filled only with DMSO to be used as positive and negative controls. In addition, hits selected for resupply after the primary screening were also purchased in powder form from Chembridge. EA, DHEA, and 16BrEA were acquired from Steraloids Inc. (Newport, RI) also in powder form.
Clostridium kluyveri Diaphorase (E.C. 1.8.1.4), resazurin, NADP+, G6P, and DMSO were supplied from Sigma-Aldrich (St. Louis, MO). Tris-HCl and NaCl were acquired from Merck (Whitehouse Station, NJ). Triton X-100 was purchased from Serva (Heidelberg, Germany). Microplates were bought from Greiner Bio-One (Monroe, NC).
HTS Primary Assay
For the primary screening assay, TcG6PDH’s direct reaction was coupled with the reduction of resazurin to resorufin, catalyzed by Diaphorase ( Fig. 1A ). Briefly, compounds were assayed at the final concentration of 20 µM, and TcG6PDH activity was correlated to resorufin relative fluorescence intensity after 3 h incubation at room temperature. Experimental conditions were adjusted to ensure linearity during the entire assay. The assay was prepared on 384 wells, black-, v-bottom microplates in a final volume of 25 µL per well. Pipetting steps were performed by a JANUS-MDT (PerkinElmer, Waltham, MA) liquid handler equipped with a 384 tip head.
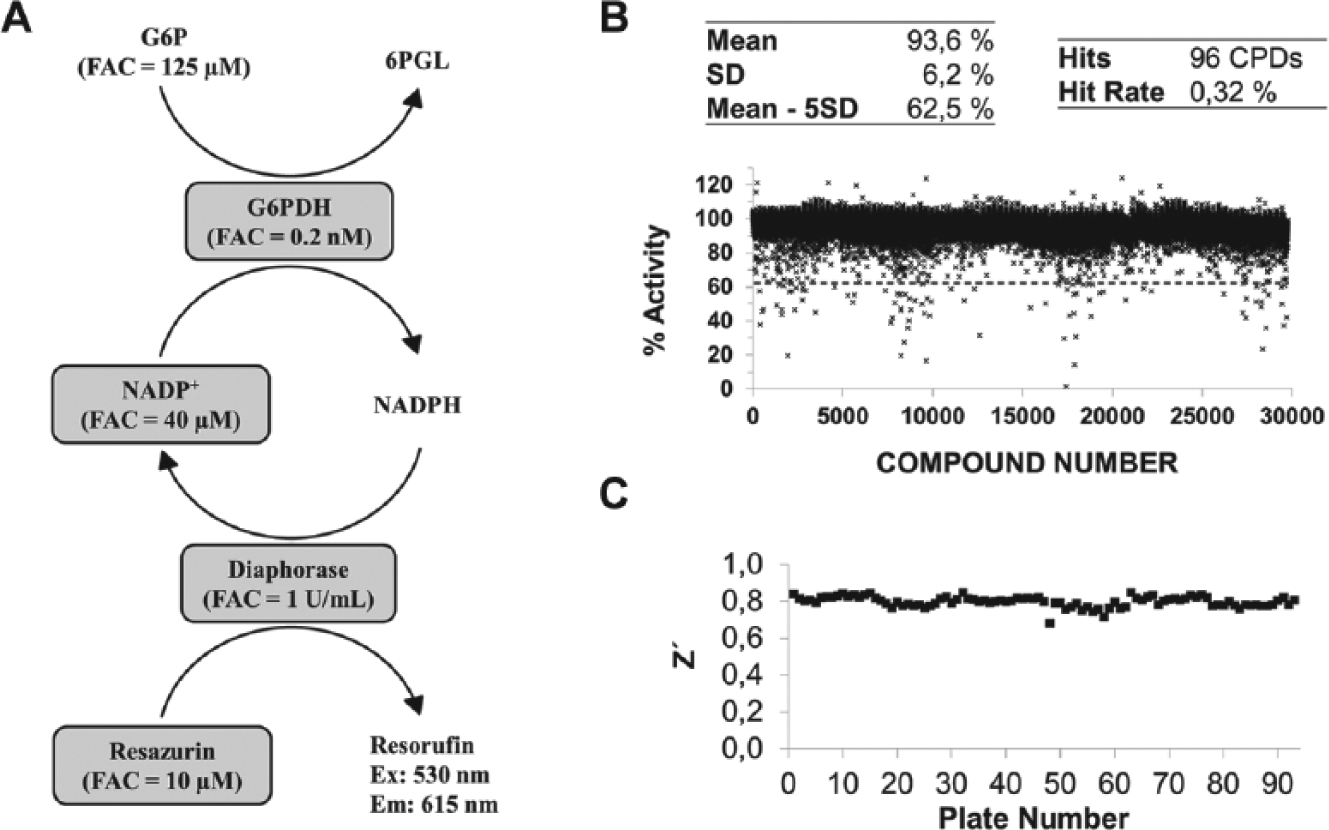
High-throughput screening (HTS) assay and statistics. (
Compounds that inhibited TcG6PDH by 37.5% or more in the primary assay were considered hit candidates.
Orthogonal Assays
Hit candidates were tested in a direct assay against TcG6PDH, measuring the NADPH production rates by fluorescence intensity readout at different inhibitor concentrations. Compound plates (384 wells) were prepared in serial dilution (1:1) ranging from 60 to 0.47 µM in reaction buffer (50 mM Tris-HCl, pH = 7.6, 25 mM NaCl, and 0.01% triton X-100). The assay was prepared on 384-well, black- and v-bottom microplates (Greiner Bio-One) in a final volume of 20 µL per well. For this, each well received 13.3 µL of compound and 4 µL of Enzyme-NADP+ mix (10 nM TcG6PDH, 200 µM NADP+, in reaction buffer), and the reaction was initiated by addition of 2.7 µL G6P at 940 µM. The final concentrations were 2 nM TcG6PDH, 40 µM NADP+, 125 µM G6P, and 40 to 0.31 µM for each compound. Pipetting steps were performed by a JANUS-MDT liquid handler equipped with a 384 tip head, and NADPH fluorescence intensity was measured in the microplate reader ENVISION from PerkinElmer (ex 340 nm; em 485 nm). These measurements were done in duplicate. Initial velocities were calculated in the linear phase of the reaction and normalized by controls to obtain IC50 curves. Compounds with confirmed activity and concentration-response profile were selected for resupply.
Hits Confirmation
Confirmed hits and commercially available analogs from the Chembridge catalog were purchased in powder form. Each purchased compound was dissolved in DMSO to a 10 mM stock solution and stored in individual vials at −20 °C. Stock solutions were diluted to 1 mM using DMSO and reformatted to a 96-well plate (daughter plates). First, all of the compounds were tested in quadruplicate against TcG6PDH using the diaphorase-resorufin coupled assay. To do so, the JANUS-MDT liquid handler equipped with a 96-tip head was used to transfer 44 µL of a reagent mix (0.25 nM TcG6PDH, 45.44 µM NADP+, 1.14 IU.mL−1 diaphorase, and 11.36 µM resazurin), 1 µL of compounds from the daughter plate, and 5 µL of G6P 1.25 mM to a 384-well plate. The final assay concentrations (FAC) were 0.22 nM TcG6PDH, 40 µM NADP+, 1 IU.mL−1 diaphorase, 10 µM resazurin, 20 µM compound, and 125 µM G6P. The reaction was monitored by measuring the formation of resorufin over time, and the calculated velocities were normalized using controls. Second, the same reaction scheme was used to assess the activity of the compounds against the human enzyme by substituting TcG6PDH by 0.45 nM HsΔG6PDH in the reagent mix (FAC = 0.4 nM). Lastly, the influence of the purchased molecules at 20 µM on the diaphorase-resorufin coupled system was evaluated. Similarly, the JANUS-MDT equipped with a 96-tip head was used to transfer, quadruplicated to a 384-well black plate, 44 µL of a reagent mix containing resazurin 11.36 µM and NADPH 22.72 µM, 1 µL of compounds from daughter plates, and the reaction was started with 5 µL of diaphorase 125 mIU.mL−1. FACs were diaphorase 12.5 mIU.mL−1, NADPH 20 µM, and resazurin 10 µM. Compounds that inhibited TcG6PDH by 40% or more and did not inhibit diaphorase were used for further study.
Determination of IC50 Values
IC50 values for confirmed hits were determined using the G6PDH-diaphorase-resorufin coupled assay by measuring the reaction velocities at varying inhibitor concentrations. The chosen compound’s solutions were prepared by serial dilutions in 96-well plate using DMSO, leaving the first column with the solvent to be used as controls and diluting the tested compounds from columns 2 to 12. Using the JANUS-MDT equipped with a 96-tip head, the reactions were prepared in quadruplicate in a 384-well plate transferring 44 µL of a reagent mix (0.25 nM TcG6PDH or 2 nM HsΔG6PDH; 45.44 µM NADP+, 1.14 IU.mL−1 diaphorase, and 11.36 µM resazurin), 1 µL of the compound solutions, and 5 µL of G6P 1.25 mM. The enzymatic activity was followed measuring the formation of resorufin using the Envision plate reader in the fluorescent mode (ex 530 nm; em 615 nm). The velocities obtained were normalized by the controls, and the IC50 values were calculated by nonlinear regression of the data using GraphPad Prism 6.0.
Compound Clustering
The program SARANEA 17 was used to cluster the active compounds. The input file was prepared including molecules’ smile, identification, name, potency, and fingerprints. Smiles and fingerprints were generated from structure data format files using the program Open Babel. 18 Molecules were clustered based on MACCS molecular fingerprints using a similarity index of 0.65.
Mechanism of Inhibition
The reversibility of the inhibition was assessed in triplicate by preincubating the inhibitors with the enzyme at high concentration and then measuring the enzyme activity using a G6PDH direct assay. Thus, 100 nM of TcG6PDH was incubated for 30 min with the test compound at 10-fold the IC50 concentration or DMSO as control. Then, 0.5 µL of this solution was transferred to a well containing 49.5 µL of a G6P/NADP+ solution (125 and 40 µM, respectively). The reaction velocity was obtained by measuring the rate of NADPH formation using the Envision plate reader in the fluorescent mode (ex. 340 nm; em. 485 nm).
The mechanisms of inhibition were determined evaluating the effect of inhibitors on Vmax and Km values for both substrate and cofactor. The Km value for G6P was determined by measuring the initial reaction velocity at substrate concentrations ranging from 700 to 5.47 µM with a fixed concentration of NADP+ at 80 µM. Similarly, the Km of NADP+ was calculated varying the NADP+ concentration from 80 to 0.625 µM and keeping the G6P at 700 µM. Fluorescence of NADPH was measured in the Envision Multilabel Reader (PerkinElmer) in quadruplicate. Km and Vmax values were calculated by nonlinear regression of the data using GraphPad Prism 6.0.
Concurrent Effect of Two Inhibitors on TcG6PDH
The concomitant effects of steroids and the new inhibitors on TcG6PDH were evaluated by using various concentrations of the tested compound, with fixed concentrations of EA, while keeping constant the concentrations of the substrate, cofactor, and enzyme. EA was chosen as representative of the steroidal uncompetitive inhibitors. The reactions in the presence of two inhibitors were performed in a 96-well black plate (Greiner) adding 2 µL of a serial dilution (3:4) of the first inhibitor starting at 100 times the IC50, 38 µL of G6P/NADP+ solution (328.9 and 105.3 µM, respectively), 20 µL of EA (5, 3.33, 1.67, and 0.0 µM), and 40 µL of TcG6PDH 2.5 nM. The reaction progress was followed by measuring the formation of NADPH by fluorescence (ex. 340 nm and em. 485 nm) using the Envision plate reader, in triplicate.
T. cruzi Viability Assay
The trypanocidal activity of the hits with IC50 ≤10 µM for TcG6PDH were assessed by incubating T. cruzi epimastigotes (Y strain) with the compounds at 80 µM and measuring the cellular viability after 96 h. Each well of a 96-well plate was seeded with 100 µL of LIT medium containing 1 × 105 parasites in the exponential growth phase. Cell viability was measured using the CellTiter96 Aqueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) following the manufacturer’s recommendations. The EC50 were determined using the above described conditions but varying the concentration of the tested hits from 80 to 2.5 µM or 40 to 1.25 µM. All experiments were done in triplicate, and the data were fitted to a sigmoidal semi-logarithmic curve to obtain the EC50 values using GraphPad Prism 6.0.
Results and Discussion
In 1960, androstanes and pregnanes were shown to inhibit G6PDH from mammals but not from plants, bacterium, nor yeast, and neither did they inhibit other NADPH-producing enzymes. 19 Characterization of the G6PDH-steroids interaction revealed an uncompetitive mechanism of inhibition for both substrate and cofactor,20,21 which established steroidal G6PDH inhibitors as a promising chemical class for the development of new drugs against cancer and obesity. 22
Our group was the first to demonstrate that trypanosomatid G6PDHs are also uncompetitively inhibited by mammalian steroids and that DHEA derivatives are able to kill both T. brucei and T. cruzi in in vitro experiments.13–15 These results encouraged us to test DHEA derivatives in the animal model for the acute phase of Chagas disease. Despite all the promising results in in vitro assays, the treatment of T. cruzi–infected mice with DHEA, EA, and 16BrEA was very disappointing. None of the steroids were able to reduce the blood parasitemia in the infected animal groups, in contrast to control groups that received benznidazol (data not shown), most probably due to rapid metabolism by enzymes able to modify steroidal molecules.
Recently, novel inhibitors of the human G6PDH 23 and of the bifunctional glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase from Plasmodium falciparum (PfGluPho) 24 were identified by HTS. However, no uncompetitive inhibitors were reported from those experiments. To prioritize the discovery of hits that bind to the same site occupied by steroids, we set up the primary screening condition to favor uncompetitive inhibitors by using concentrations of G6P and NADP+ at least twofold their Km values. Our intention was to discover new uncompetitive G6PDH inhibitors with distinct chemical scaffolds from the well-known steroids. We hypothesized that working with new scaffolds, it would be possible to achieve the desired selectivity toward the parasite G6PDH, reduce drug metabolism, avoid androgenic side effects, and improve the efficacy of G6PDH inhibitors against human trypanosomiasis.
Primary Screening
In the primary screening, the DIVERSet library (~30,000 compounds) was screened at the concentration of 20 µM against TcG6PDH using a previously reported diaphorase coupled assay ( Fig. 1A ).23–27 The primary screening assay resulted in the identification of 96 hit candidates, which diminished the enzyme activity to at least 62.5%, which is equivalent to the mean percentage of enzyme activity less 5 standard deviations ( Fig. 1B ). The mean Z′ factor 28 over 93 plates was 0.80 ± 0.03 ( Fig. 1C ). Next, hit candidates were cherry-picked from the library compound plates and retested against TcG6PDH using the NADPH production direct assay. In this confirmatory assay, hit candidates were serially diluted from 40.0 to 0.3 µM (FAC). Twenty-two (out of 96) hit candidates confirmed inhibitory activities, and from these, 10 compounds showed good concentration-response profiles, thus being selected for follow-up studies.
Hits Resupply and Characterization
The 10 selected hits and additional 77 analogs, all resupplied by Chembridge, could be grouped into seven chemical classes, with the following distribution: 59 quinazolinones, 3 quinazolines, 15 quinolines, 7 thienopyrimidines, 1 benzoquinone, 1 chromenoindole, and 1 benzoxazole. These compounds were tested at 20 µM against TcG6PDH using the diaphorase coupled assay, and the effect of compounds in modulating diaphorase activity was also evaluated. Diaphorase inhibition was observed for three compounds (
Compounds that inhibited the TcG6PDH from 40% to 100% of its normal activity were used in the concentration-response studies to determine IC50 values. Among the resupplied compounds, 32 new TcG6PDH inhibitors were identified with IC50 values between 32 and 0.48 µM. These compounds were clustered by SARANEA
17
into two chemical classes: thienopyrimidines and quinazolinones (
Fig. 2
). The thienopyrimidines cluster contains four active compounds,
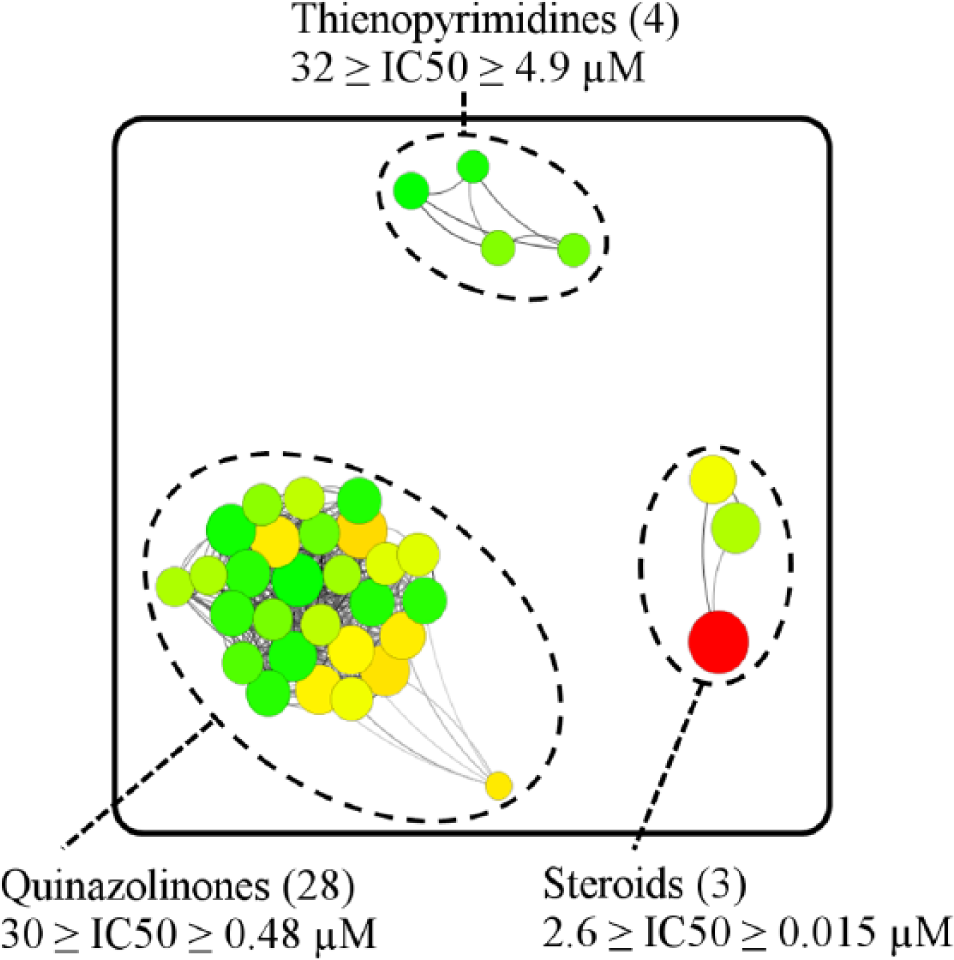
Clustering of T. cruzi glucose-6-phosphate dehydrogenase inhibitors. In addition to the cluster with previously known steroidal inhibitors (dehydroepiandrosterone, epiandrosterone, and 16-α-bromo-epiandrosterone), two new clusters were identified, one with 28 quinazolinones and another with 4 thienopyrimidines. IC50 values and 99% confidence intervals for thienopyrimidines and quinazolinones are reported in
Table 1
(data for steroids is in
Activity of thienopyrimidine (
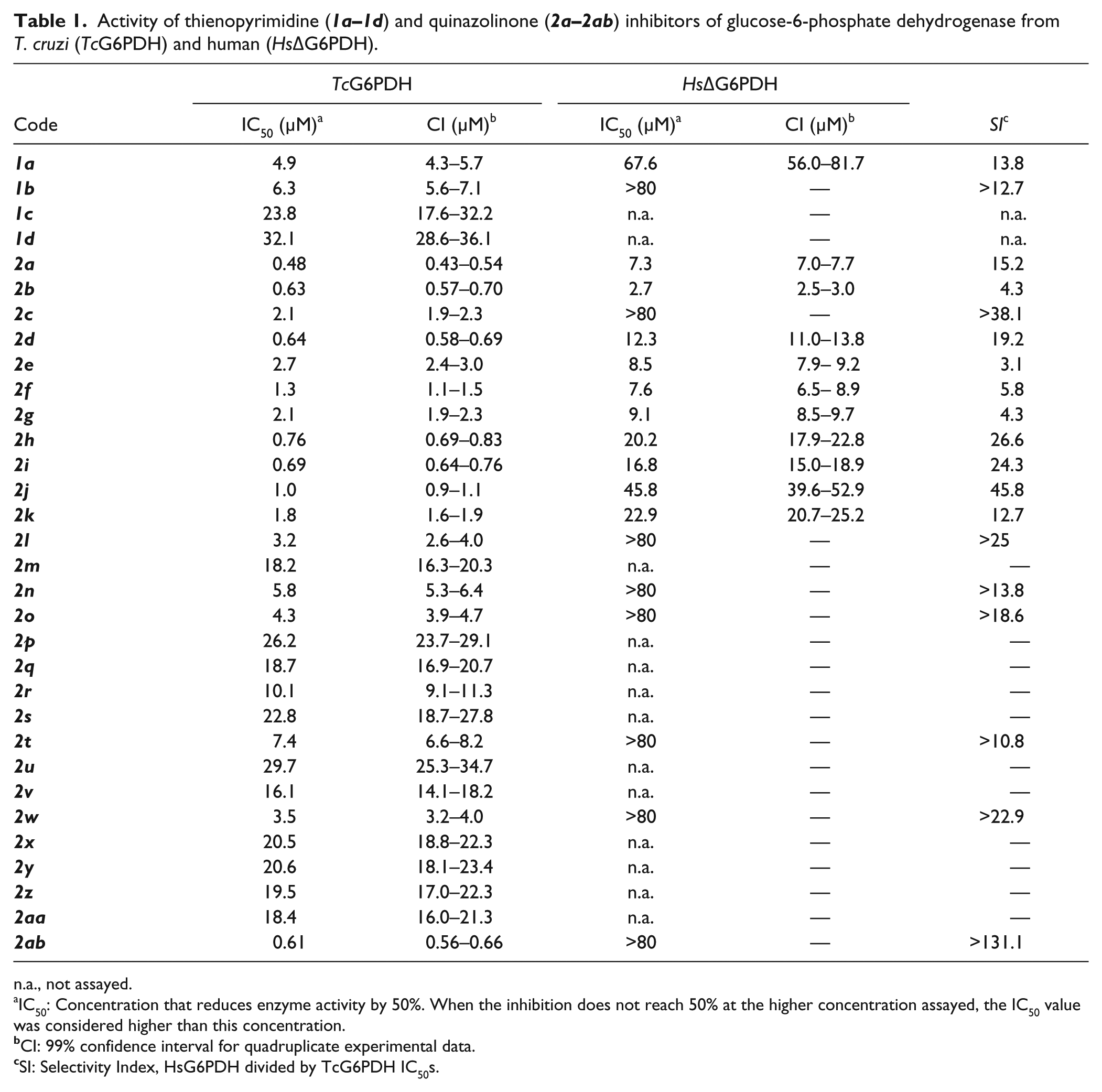
n.a., not assayed.
IC50: Concentration that reduces enzyme activity by 50%. When the inhibition does not reach 50% at the higher concentration assayed, the IC50 value was considered higher than this concentration.
CI: 99% confidence interval for quadruplicate experimental data.
SI: Selectivity Index, HsG6PDH divided by TcG6PDH IC50s.
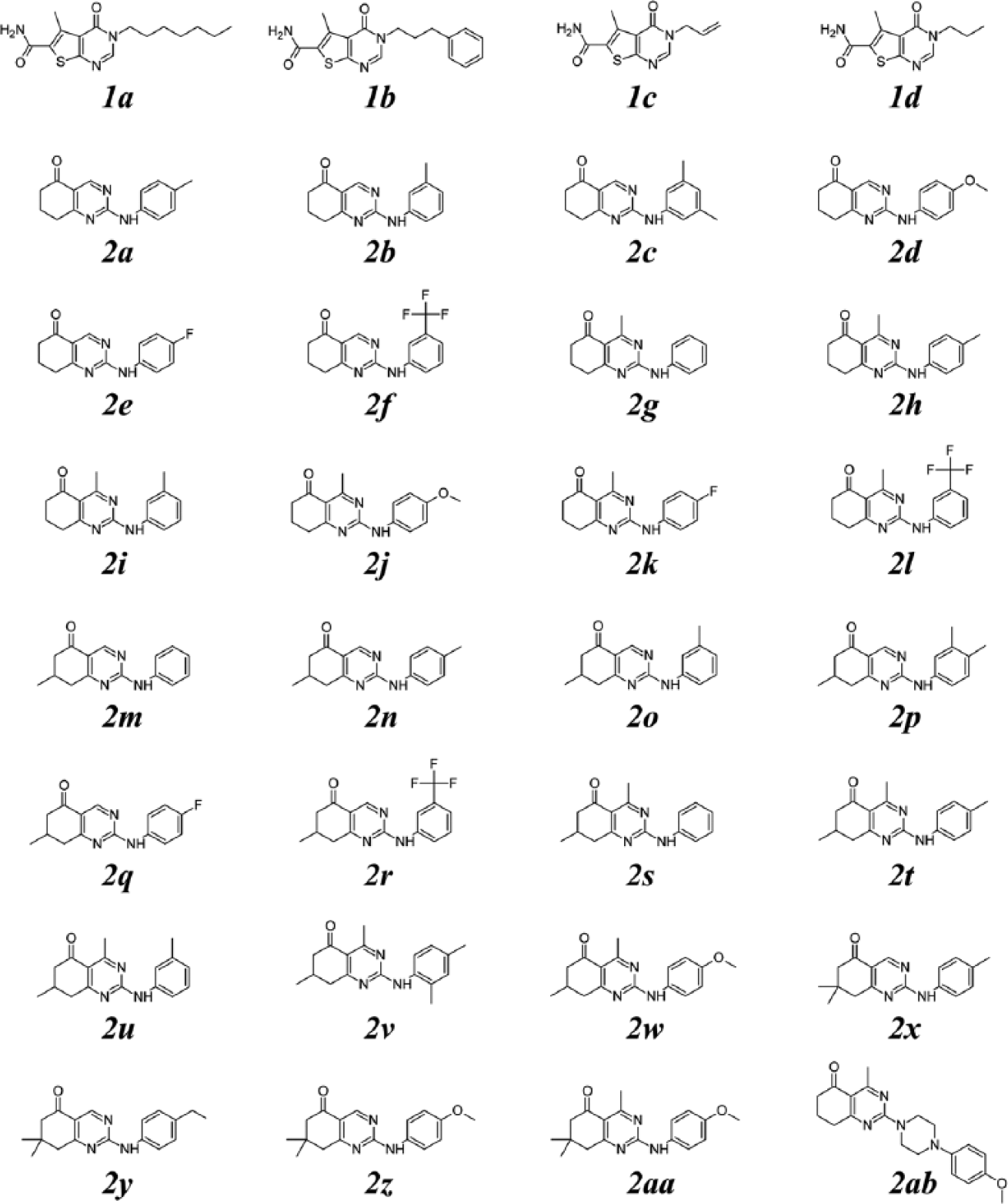
Chemical structure representation of thienopyrimidine (
Confirmed hits with IC50 less than 10 µM for TcG6PDH were assayed against the HsΔG6PDH to address the selectivity for the parasite enzyme (
Table 1
). In both thienopyrimidine and quinazolinone clusters, selective compounds were identified (
Table 1
;
Mechanism of Inhibition
The most potent compounds inside thienopyrimidine (
Structure Activity Relationship
Thienopyrimidine derivatives
The structure activity relationship (SAR) analysis of thienopyrimidine derivatives indicates that the presence of a carboxamide group at C6 and a hydrophobic group at N3 (R1) are relevant for TcG6PDH inhibition (
Fig. 4
). This becomes evident when the carboxamide group is removed or even replaced by N-(dimethylphenyl)-carboxamide in compound
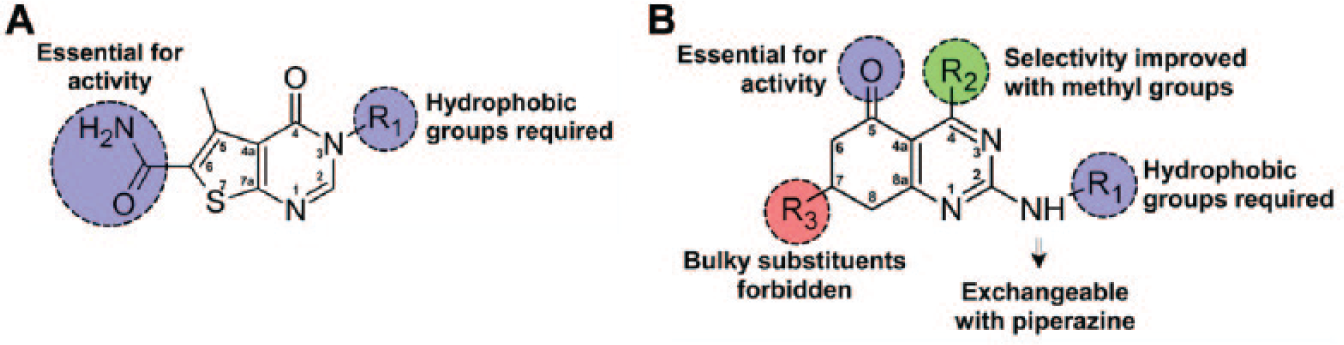
Illustration showing the main structure activity relationship observed for thienopyrimidine and quinazolinone derivatives.
Quinazolinones
The set of quinazolinones was composed of 59 derivatives, and the analysis of this data set gave the following SAR patterns (or rules): the addition of bulky substituents at C7 (R3) is forbidden because it reduces G6PDH inhibition, C5 ketone is essential for activity, methylation at C4 (R2) can improve selectivity toward the parasite enzyme, and finally, hydrophobic substituents at the amine group at C2 (R1) are essential for activity, and variations of these groups have effects on potency and selectivity ( Fig. 4 ).
Incorporation of R3 substituents at the C7 position of the quinazolinone moiety is deleterious to activity. Bulky substituents such as phenyl, furyl, or thienyl groups are forbidden (see compounds
Removal of C5 ketone from quinazolinones produces an inactive quinazolinamine (compound
The substitution of quinazolinones at C4 (R2) has a pronounced effect on the inhibition of HsΔG6PDH but not on TcG6PDH, and thus, this modification has a direct impact on the selectivity index. For instance, C4 methylation of compound
Concerning position R1 of the quinazolinones, it was observed that all active derivatives, except compound
Comparing the set of compounds
For compounds bearing a methyl aniline, the addition of a second methyl group reduces the inhibition on TcG6PDH (see compounds
T. cruzi Viability Assay
Trypanocidal activities of 19 hits—with IC50s less than 10 µM for TcG6PDH—were evaluated against epimastigote forms of T. cruzi (Y strain) at a fixed concentration of 80 µM. According to the percentage of remaining viable cells (RVC), four compounds were classified as inactive (RVC > 75%), six as moderately active (75% > RVC > 25%), and nine as active (RVC < 25%;
Fig. 5A
;
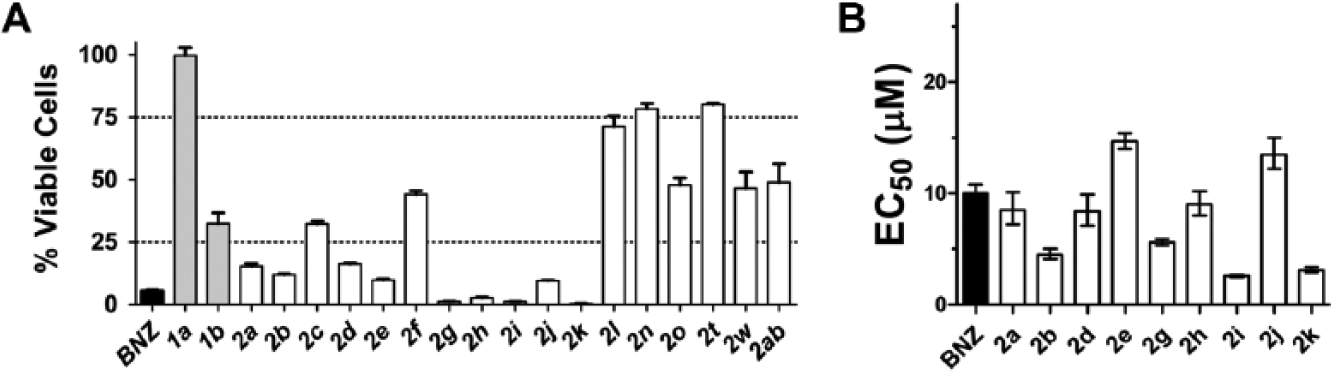
In vitro trypanocidal activity of the T. cruzi glucose-6-phosphate dehydrogenase inhibitors. Thienopyrimidines are in gray, quinazolinones in white, and benznidazole (BNZ) that was added as control in black. (
In conclusion, new uncompetitive inhibitors of G6PDH with selectivity for the T. cruzi enzyme have been discovered, and the preliminary SARs for thienopyrimidines and quinazolinones are presented. In vitro trypanocidal activities against T. cruzi epimastigotes were observed for quinazolinones, with some derivatives showing a trypanocidal effect as potent as benznidazole. Consequently, these molecules open new opportunities for follow-up and development of new drugs targeting G6PDHs.
Footnotes
Acknowledgements
We thank Prof. Dr. Paul Engel (University College of Dublin, Ireland) and Prof. Dr. Otavio H. Thiemann (University of São Paulo, Brazil) for supplying the human G6PDH gene and the T. cruzi cells, Dr. Marjorie Bruder for critical review of the manuscript, and the Brazilian National Laboratory of Biosciences (LNBio) from the Brazilian National Center for Research in Energy and Materials (CNPEM) for the infrastructure and technical assistance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: São Paulo Research Foundation (FAPESP) research grant for A.T.C. (2013/03983-5) and PhD fellowships for G.F.M. (2010/17849-0) and A.T.R. (2012/23682-7).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.