
Abstract
Plasmodium falciparum causes severe malaria infections in millions of people every year. The parasite is developing resistance to the most common antimalarial drugs, which creates an urgent need for new therapeutics. A promising and attractive target for antimalarial drug design is the bifunctional enzyme glucose-6-phosphate dehydrogenase-6-phosphogluconolactonase (PfGluPho) of P. falciparum, which catalyzes the key step in the parasites’ pentose phosphate pathway. In this study, we describe the development of a high-throughput screening assay to identify small-molecule inhibitors of recombinant PfGluPho. The optimized assay was used to screen three small-molecule compound libraries—namely, LOPAC (Sigma-Aldrich, 1280 compounds), Spectrum (MicroSource Discovery Systems, 1969 compounds), and DIVERSet (ChemBridge, 49 971 compounds). These pilot screens identified 899 compounds that inhibited PfGluPho activity by at least 50%. Selected compounds were further studied to determine IC50 values in an orthogonal assay, the type of inhibition and reversibility, and effects on P. falciparum growth. Screening results and follow-up studies for selected PfGluPho inhibitors are presented. Our high-throughput screening assay may provide the basis to identify novel and urgently needed antimalarial drugs.
Keywords
Introduction
Malaria remains a major health concern. Each year, more than 200 million people contract malaria with around one million people dying from the disease. Plasmodium parasites of the species falciparum are the main cause of severe malaria infections.1,2 The P. falciparum parasite is transmitted to the human host by Anopheles mosquitoes and undergoes a complex life cycle. 3 The parasite is capable of adapting to environmental changes in the vector and the human host, which increases its survival and transmission rates and enhances its resistance to most antimalarial drugs.4,5 Searches for new targets for antimalarial drug development draw attention to the pentose phosphate pathway (PPP). Emerging evidence suggests that both human and P. falciparum PPP play an important role in malaria pathogenesis. Most intriguingly, deficiency in glucose-6-phosphate dehydrogenase (G6PD), the key enzyme of the PPP and the most common human enzyme deficiency worldwide, 6 protects the carrier from malaria infections.7,8 Plasmodium parasites, developing within red blood cells (RBCs), and the human host both possess a complete PPP, which consists of an oxidative and a nonoxidative branch. G6PD is located in the oxidative branch, converting glucose-6-phosphate to 6-phosphogluconolactone, which generates nicotinamide adenine dinucleotide phosphate (NADPH). In a second reaction, 6-phosphogluconolactonase (6PGL) transforms the product into 6-phosphogluconate, which is further converted into ribulose-5-phosphate by the enzyme 6-phosphogluconate dehydrogenase, which also produces NADPH. 9 NADPH plays a key role as a reducing agent in the antioxidative defense of cells. Since RBCs lack mitochondria, the PPP is the only NADPH-generating pathway in these cells and of special importance during malaria infections, which induce oxidative stress. 10 Moreover, the P. falciparum PPP is suggested to be the major source of NADPH for the parasite, since other putative NADPH sources such as isocitrate dehydrogenase and glutamate dehydrogenase most likely play a minor role.11,12 Plasmodium parasites possess a variety of antioxidant defense mechanisms, which are located in several subcellular compartments 13 and protect the parasites, which are highly sensitive to oxidative stress. 10 The Plasmodium redox system includes a complete glutathione 14 and thioredoxin system, 15 which are both NADPH dependent, highlighting the importance of the parasitic PPP.
In contrast to the human isofunctional G6PD, P. falciparum contains a bifunctional enzyme consisting of G6PD and 6PGL (PfGluPho), thus catalyzing the first and second reactions of the PPP. 16 Being 107 kDa in size, PfGluPho is rather large compared with the 59-kDa human enzyme. 17 Previous studies showed that glucose utilization through glycolysis and PPP increases ~70-fold when RBCs are infected with P. falciparum.18,19 Remarkably, ~80% of the increased PPP activity is caused by the P. falciparum PPP. 19 The importance of the parasitic PPP and the structural dissimilarity compared with human G6PD make PfGluPho an ideal target for drug development. 20 This notion is supported by RNAi-based PfGluPho silencing, which leads to growth arrest at the trophozoite stage, enhanced gametocyte formation, and increased transcription of thioredoxin reductase. 21 However, these studies must be considered cautiously based on recent reports that question the functionality of RNAi in Plasmodium. 22 Access to small-molecule inhibitors that specifically target PfGluPho would be a valuable tool to confirm the importance of the parasitic PPP in general and PfGluPho in particular.
In the past, studies on PfGluPho were restricted due to limited availability of the enzyme purified from Plasmodium parasites. Most recently, our group succeeded in the cloning and overexpression of full-length PfGluPho, 20 which has made the enzyme available for analyses in a high-throughput format as well as for follow-up studies to characterize potential inhibitors.
On the basis of a previously described high-throughput screening (HTS) assay for G6PD from Leuconostoc mesenteroides (PubChem AID 1020), we developed and optimized an HTS assay for recombinant PfGluPho and screened three small-molecule compound libraries. We identified and characterized several inhibitors, which could provide a basis for the discovery of novel antimalarial drugs.
Materials and Methods
Reagents
PfGluPho and hG6PD were produced according to Jortzik et al. 20 PfGluPho aliquots were stored with 50% glycerol at −80 °C and hG6PD was stored with 1.8 M ammonium sulfate at 4 °C. NADP+ was obtained from Amresco (Solon, OH); G6P, resazurin, and diaphorase were obtained from Sigma-Aldrich (St. Louis, MO).
HTS Assay Development
Km determination
The Km value for the substrate G6P was determined by titrating G6P (0–1600 µM) while keeping NADP+ constant at 1.5 mM. The G6P concentration was constant at 1.5 mM, and NADP+ was varied (0–400 µM) for Km determination of the cosubstrate. Fluorescence of the reaction was measured at excitation 340 nm and emission 460 nm (ex340/em460) with a SpectraMax M5 (Molecular Devices, Sunnyvale, CA) multiwell plate reader. The initial slope was determined with the SoftMax Pro 5.2 software (Molecular Devices), and Km and Vmax values were calculated by nonlinear regression using GraphPad Prism 5.0 software (GraphPad, San Diego, CA).
Resazurin concentration
For determination of the optimal resazurin concentration, the G6PD assay was performed including G6P at Km concentration, 1.5 mM NADP+, 1 U/mL diaphorase, 3.3 mM MgCl2, 0.05 M Tris (pH 7.5), 0.005% Tween-20, PfGluPho in an appropriate concentration, and varying resazurin concentrations (0–1 mM). Fluorescence of resorufin was monitored over 30 min at ex530/em580 using the Analyst HT (LJL BioSystems, Sunnyvale, CA) multiwell plate reader in combination with the Criterion Host software (LJL BioSystems).
Enzyme titration and linearity of the reaction
Enzyme titration was performed using the screening protocol (as described in “HTS Screening”) including varying PfGluPho concentrations (0–4 µg/mL). The reaction was started by addition of the enzyme, and the fluorescence signal of resorufin was monitored over 30/60 min (LOPAC and Spectrum/ChemBridge screening) at ex530/em580 using the Analyst HT (LJL BioSystems) multiwell plate reader. The appropriate enzyme concentration was chosen based on linearity over 30/60 min of the reaction curves created by the Criterion Host software (LJL BioSystems). Furthermore, the initial slope of each curve was plotted against the enzyme concentration to see up to which concentration the enzyme concentration was linear. Enzyme titrations were performed for every protein batch that was used.
Determination of the signal-to-background ratio, Z factor, and coefficient of variation
The signal-to-background ratio (S/B), the Z factor (Z), and the percent coefficient of variation (% CV) were determined based on formulas described elsewhere.23,24
DMSO tolerance test
The screening assay was performed including varying DMSO concentrations (0%−4%). The reaction was linear up to 30 to 40 min, and the final relative fluorescence units (RFUs) of the signal and the background were plotted as average and standard deviation of 4 replicates against the DMSO concentration using GraphPad Prism software.
Compound libraries
Compound libraries screened in this study were the LOPAC library (Sigma-Aldrich; 1280 drug-like molecules in the fields of Cell Signaling & Neuroscience), the Spectrum collection (MicroSource Discovery Systems, Gaylordsville, CT; 1969 biologically active and structurally diverse compounds, experimental bioactives, and pure natural products), and the DIVERSet (ChemBridge, San Diego, CA; 49 971 drug-like compounds with maximum pharmacophore diversity). The LOPAC and Spectrum libraries, containing a compound concentration of 10 mM, were screened at a concentration of 20 µM. The DIVERSet was present at a concentration of 0.5 mg/mL in 2 µL DMSO and screened at 5 µg/mL.
HTS Screening
LOPAC and Spectrum libraries
The screening assay for the LOPAC and Spectrum libraries was performed in 384-well plates for small volume (Greiner Bio-One, Monroe, NC). In brief, 9 µL of a substrate mix (final assay concentration [FAC] 0.05 M Tris, 3.3 mM MgCl2, 15 µM G6P, 1 U/mL diaphorase, 22 µM resazurin) was added to the wells of rows 3 to 24 using the Multidrop Combi reagent dispenser (Thermo Scientific, Hudson, NH). Then, 9 µL of the mix without G6P was added to rows 1 and 2 as positive controls. Columns 23 and 24 served as negative controls and did not contain compounds. The plates were centrifuged, and either 36 nL of the compounds or DMSO for the controls was added with the Echo 555 liquid handler (Labcyte, Sunnyvale, CA), resulting in a final compound concentration of 20 µM and a DMSO concentration of 0.2%. Finally, 9 µL of an enzyme mix (FAC 10 µM NADP+, 0.005% Tween-20, 0.22 µg/mL PfGluPho) was added to start the reaction. After 30 min of incubation, fluorescence of resorufin was measured at ex530/em580 (end point determination) using the multiwell plate reader Analyst HT (LJL Biosystems). The data were analyzed using CBIS (Chemical and Biological Information Systems, www.cheminnovation.com).
Compounds that showed ≥50% inhibition were included in a secondary screening. The assay was performed as described before with addition of varying compound concentrations (0–40 µM). Data were analyzed and IC50 values determined using CBIS.
ChemBridge library
Primary and secondary screening of the ChemBridge library was performed as described for the LOPAC and Spectrum libraries with minor modifications. Due to slight differences between protein batches, the G6P concentration in the substrate mix was 12.5 µM and the resazurin concentration 16 µM. For the enzyme mix, the NADP+ concentration was 8 µM, and the enzyme was used at a concentration of 0.125 µg/mL. In total, 157 compound plates containing 2 µL compounds at a concentration of 0.5 mg/mL were stored at −20 °C. Every day, 10 to 15 plates were thawed and the compounds were diluted 10-fold with H2Odd using the WellMate (Matrix; Thermo Scientific) liquid dispenser. Then, 5 µg/mL of the compounds or DMSO for the controls was transferred to 384-well plates by the BioMek FX (Beckman Coulter, Brea, CA) automation workstation. The plates were incubated in the dark for 60 min, after which the fluorescence of resorufin was measured. Data of the primary screening were analyzed directly, and hit compounds (≥50% inhibition) were subjected to a secondary screening on the same day. For this purpose, hit compounds were manually cherry-picked and transferred to 384-well plates, in which serial dilutions were prepared. The assay was then performed as described before.
Follow-Up on Hit Compounds
IC50 determination
Several hit compounds were reordered in powder form, dissolved in DMSO to a concentration of 5 mg/mL, and retested in an orthogonal assay without resazurin and diaphorase. For IC50 determination, 6 µL of a substrate mix (FAC 0.05 M Tris, 3.3 mM MgCl2, 11 µM NADP+, 18 µM G6P) was added to a 384-well plate. Then, 6 µL of varying compound concentrations (0–5 µg/mL) was added and the reaction was started with 6 µL of an enzyme mix (FAC 0.005% Tween-20, 0.39 µg/mL PfGluPho). Fluorescence of NADPH was detected at ex340/em460 using the multiwell plate reader Infinite M2000 combined with the Magellan 6 software (Tecan, Männedorf, Switzerland). Reaction curves were monitored over 20 to 30 min, and the end point of the linear reaction was used to calculate IC50 values with GraphPad Prism. No addition of substrate was used as 100% inhibition.
Mechanistic characterization
Mechanistic characterization of inhibitors was performed by titrating either G6P or NADP+ against different compound concentrations. The assay was composed of a substrate mix (FAC 3.3 mM MgCl2, 1 mM tris(2-carboxyethyl)phosphine [TCEP], 0.05 M Tris; for G6P titration: 5 µM NADP+; for NADP+ titration: 25 µM G6P) in which varying G6P or NADP+ concentrations were prepared, varying compound concentrations (starting around 8× IC50) titrated in DMSO, and an enzyme mix (FAC 0.2 µg/mL PfGluPho, 0.005% Tween-20). All mixes were added in equal amounts to a 384-well plate, and the fluorescence of the reaction was monitored at ex340/em460 with a SpectraMax M5 (Molecular Devices) multiwell plate reader over 10 min. For CB68 and CB103, the enzyme mix containing slightly higher enzyme concentrations was incubated with the inhibitor for 60 min before the substrate mix was added. The initial slope was determined with SoftMax Pro 5.2 software (Molecular Devices), which was then plotted as 1/slope for each inhibitor concentration against 1/substrate (Lineweaver-Burk plots). In addition, Km and Vmax values were calculated with the GraphPrad Prism software, and α from the rate equation shown below, which is used for general description of inhibitors 25 (equation (1); v = velocity, S = substrate, I = inhibitor), was calculated with SigmaPlot (Systat Software, San Jose, CA).
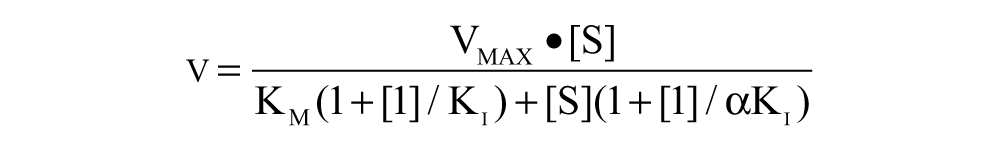
Based on the following assumptions, 26 the most likely mechanism of inhibition was determined:
Competitive inhibitor: Lineweaver-Burk plots intersect at the y-axis, Km increases, and Vmax stays constant with increasing inhibitor concentrations, α = ∞.
Noncompetitive inhibitor: Lineweaver-Burk plots intersect at the x-axis, Km stays constant, and Vmax decreases with increasing inhibitor concentrations, α = 1.
Uncompetitive inhibitor: Lineweaver-Burk plots produce parallel lines, and Km and Vmax decrease with increasing inhibitor concentration, α = 0.
Mixed type: parameters between those of competitive and noncompetitive inhibitors.
Reversibility and time-dependent inhibition
For determination of time-dependent effects and reversibility, the enzyme was incubated with different inhibitor concentrations for different times in the following way. First, 50 µL of an enzyme mix (0.1 M Tris, 1 mM TCEP, 37.5 µg/mL PfGluPho [= 37× final concentration]) was pipetted in three columns of a 96-well plate, and 50 µL of varying inhibitor concentrations (0–0.15 mg/mL inhibitor [= 37× final concentration or max. 3% DMSO]) was added to the first column for preincubation. After 1 h, 8 µL of the enzyme-inhibitor mix of column 1 was added to 92 µL buffer (0.05 M Tris, 1.635 mM TCEP, 0.0082% Tween-20 [= 1.635× final concentration]) for postincubation, and 50 µL of varying inhibitor concentrations was added to the second column. After 2 h, 8 µL of the enzyme-inhibitor mix of column 2 was added to 92 µL buffer. Moreover, 50 µL inhibitor was added to the third column, and subsequently 8 µL of the enzyme-inhibitor mix of column 3 was added to 92 µL buffer. In this way, the following pre-/postincubation conditions were tested: 1 h/1 h, 1 h/0 h, and 0 h/0 h. Then, 12 µL of each enzyme-inhibitor-buffer mix was transferred to a 384-well plate, and 6 µL of a substrate mix (9.9 mM MgCl2, 15 µM NADP+ [= 3× final concentration], 0.05 M Tris, 1 mM TCEP, 75 µM G6P [= 3× final concentration]) was added to start the reaction. For control, the substrate mix without G6P was added, representing 100% inhibition. Fluorescence of NADPH was measured at ex340/em460 over 20 to 30 min, and the initial slope data were analyzed using the GraphPad Prism software. If IC50 values were lower for pre-/postincubation 1 h/0 h compared with 0 h/0 h, the compound showed time-dependent inhibition. In addition, if the IC50 for 1 h/1 h was lower or similar to that of 0 h/0 h, the compound was assigned to be irreversible. If IC50 values were lower for pre-/postincubation 0 h/0 h compared with 1 h/1 h and 1 h/0 h, the compound was assigned to be reversible. For analysis of human G6PD, 1 mg/mL bovine serum albumin (BSA) instead of TCEP was added due to stability issues. In addition, NADP+ and G6P concentrations were adjusted to Km values for hG6PD. No addition of substrate was used as 100% inhibition.
IC50 determination in P
falciparum. The P. falciparum 3D7 strain (choroquine sensitive) was grown in continuous culture as described previously. 27 For IC50 determination of the best inhibitors in P. falciparum, isotopic drug sensitivity assays using a semiautomated microdilution technique 28 were performed according to the modifications of Fivelman et al. 29 The method is based on incorporation of radioactive 3H-hypoxanthine, a precursor of purine deoxynucleotides for DNA synthesis, which is taken up by the parasite. Twofold serial dilutions of the inhibitors were prepared in 96-well plates (Nunc, Rochester, NY). Parasites were incubated with the inhibitors and chloroquine as control at a parasitemia of 0.25% (>70% ring forms) and 1.25% hematocrit in hypoxanthine-free medium. Then, 0.5 µCi 3H-hypoxanthine was added to the wells after 48 h, and the plates were incubated for a further 24 h. The cells of each well were harvested on a glass fiber filter (PerkinElmer, Waltham, MA), washed, and dried. Afterward, radioactivity was measured in counts per minute, which is expected to be proportional to P. falciparum growth, and IC50 values were determined.
Cytotoxicity
Immortalized human hepatocytes, Fa2N-4 cells (XenoTech, Kansas City, KS), were resuspended in MFE Plating medium (XenoTech), seeded in collagen-coated plates (VWR, Radnor, PA) at ~50 000 cells/well, and incubated in a humidified CO2 incubator at 37 °C. After 4 h, the medium was replaced with MFE Support medium (XenoTech). On the third day, the cells were incubated with the test compound at a range of concentrations (0.01–50 µM). After 24 h, cell viability was determined by cellular adenosine triphosphate (ATP) levels using the ATP-Lite Luminescence live cell Detection Assay System (PerkinElmer), according to the protocol provided by the manufacturer. Luminescence was measured using an Infinite M200 plate reader (Tecan) and the data plotted as a function of the raw luminescence signal versus the log of the concentration of the compound. Curves superimposed upon the data were generated by nonlinear regression analysis with a 4-point logistic using Prism5 software (GraphPad).
Structure-activity relationship analysis
Structure-activity relationship (SAR) analysis was performed for the compounds included in the secondary screening using the SARvision 2.7 software (Altoris, San Diego, CA). This analysis allowed subdivision of the compounds in chemical groups based on their structure and thus identification of core structures.
Results
HTS Assay Development
The basic principle for the HTS assay is shown in Figure 1 . G6PD catalyzes the conversion of G6P to 6-phosphoglucono-δ-lactone and generates NADPH. 9 The standard G6PD assay, 30 which directly measures NADPH production, is now coupled to diaphorase, which uses NADPH to reduce resazurin to form the highly fluorescent molecule resorufin. The generated resorufin is stoichiometrically proportional to the converted G6P and allows for the determination of G6PD activity. Coupling of the assay to diaphorase and resazurin shifts detection to higher wavelengths (ex540/em590) to increase assay sensitivity and to avoid the high degree of fluorescence interference from test compounds that is common in the UV portion of the spectrum. We modified and optimized this HTS assay for PfGluPho. PfGluPho was prepared according to our previously described protocol 20 and stored in aliquots at −80 °C. Km and Vmax values remained constant over several weeks, indicating high enzyme stability. Aliquots of the assay components G6P, NADP+, resazurin, and diaphorase were also stored at −80 °C and were stable over the entire period of assay development and HTS as assessed by similar Km values and titration patterns of freshly purified enzyme (data not shown).
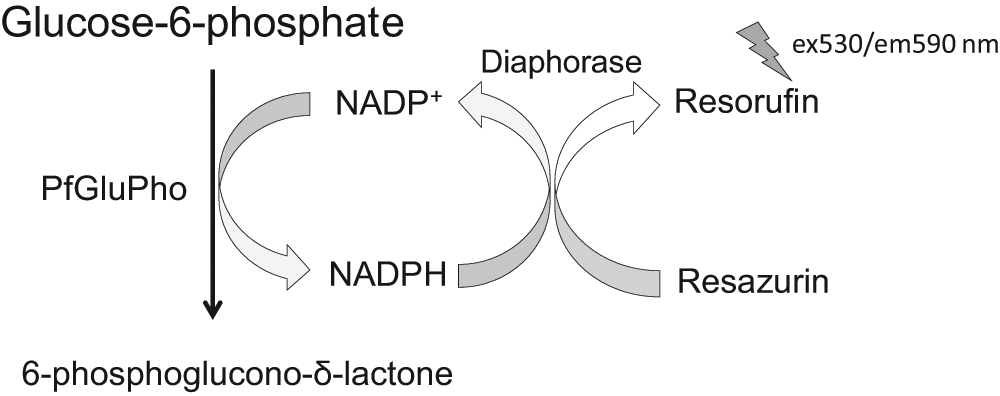
High-throughput screening assay for PfGluPho. The G6PD part of PfGluPho catalyzes the conversion of G6P to 6-phosphoglucono-δ-lactone (6PGδL), whereby NADPH is generated. The assay was coupled to diaphorase, which catalyzes the reduction of resazurin using NADPH to form the highly fluorescent resorufin.
We used different enzyme batches for screening of the different compound libraries and follow-up studies and determined the Km values for the substrate G6P and the cosubstrate NADP+ for each enzyme batch. The Km for G6P was around 14.5 µM (batch for LOPAC and Spectrum screening; Fig. 2A ) and 12.5 µM (batch for ChemBridge screening). The Km for NADP+ was around 10.0 µM (batch for LOPAC and Spectrum screening) and 8.6 µM (batch for ChemBridge screening; Fig. 2B ).
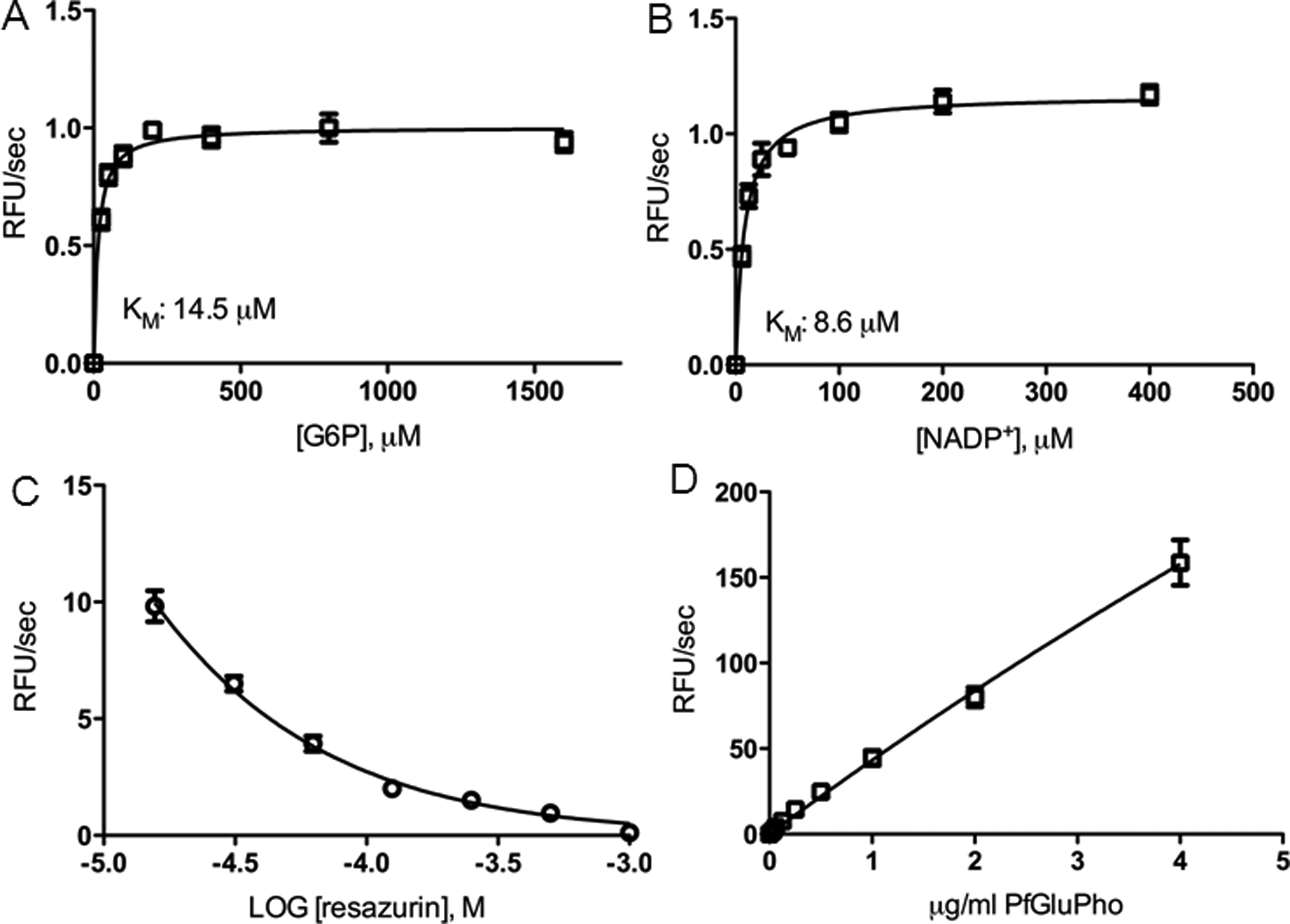
High-throughput screening assay development and optimization. (
To determine the optimal resazurin concentration for the coupled G6PD assay, we titrated the resazurin concentration in the presence of G6P at Km concentration. Fluorescence detection of resorufin revealed that the signal at ex530/em580 was negatively correlated with the resazurin concentration ( Fig. 2C ). Based on this observation, a resazurin concentration, which was slightly higher than the G6P concentration, was chosen.
We titrated the enzyme to determine the optimal concentration for the screening assay. The LOPAC and Spectrum screening was set up for an incubation time of 30 min, whereas screening of the ChemBridge library involved a 60-min incubation step to allow better handling of a higher number of plates. For the 30-min reaction, a PfGluPho concentration of 0.22 µg/mL was chosen based on linearity of the reaction curves over this period of time, whereas a concentration of 0.125 µg/mL was chosen for the ChemBridge screening assay. In addition, by plotting the initial slope of each curve against the enzyme concentration, we determined that PfGluPho shows linear activity up to a concentration of 4 µg/mL ( Fig. 2D ). Therefore, the final screening assay concentrations of 0.22 and 0.125 µg/mL were in an acceptable range.
The optimized assay was proven suitable for HTS and met accepted HTS standards24,31 with a Z factor of 0.79, a % CV of 4.1 for the negative control and 4.6 for the positive control, and an S/B ratio of 4.2 (
Screening of LOPAC and Spectrum Libraries
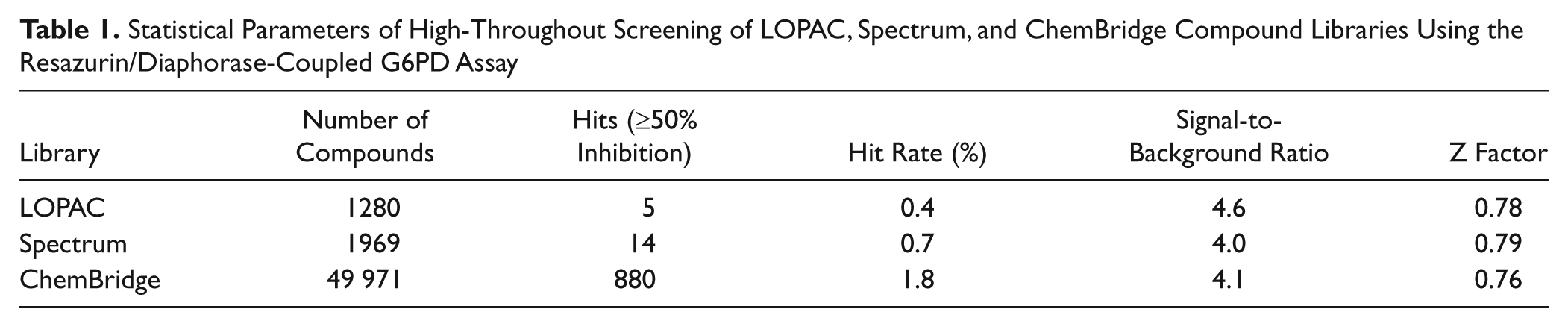
The LOPAC and Spectrum libraries were screened in a 384-well plate format at a final compound concentration of 20 µM. Primary screening of the LOPAC library included four 384-well plates with a Z factor of 0.78 ± 0.05, an S/B ratio of 4.6, and a hit rate of 0.39% ( Table 1 ). Screening of the Spectrum library, which consisted of seven 384-well plates, showed a Z factor of 0.79 ± 0.04, an S/B ratio of 4.0, and a hit rate of 0.71% (≥50% inhibition) ( Table 1 ). Compounds that showed ≥50% inhibition were later retested in varying concentrations, and IC50 values were determined based on the data of the resazurin/diaphorase-coupled assay.
Statistical Parameters of High-Throughout Screening of LOPAC, Spectrum, and ChemBridge Compound Libraries Using the Resazurin/Diaphorase-Coupled G6PD Assay
Screening of ChemBridge DiverSet
To prevent several thawing cycles of the compound plates, primary and secondary screening of the ChemBridge library was performed within 1 day. Approximately 10 to 15 plates out of the 157 total library plates were screened per day at a final compound concentration of 5 µg/mL. The primary screening ran well, resulting in a Z factor of 0.76 ± 0.07, an S/B ratio of 4.1, and a hit rate of 1.76% (≥50% inhibition) ( Table 1 ). On the same day, compounds showing ≥50% inhibition were retested in dose response, resulting in a hit confirmation of 72.6%.
Follow-Up on Hit Compounds
Mechanistic characterization and reversibility
Based on their drug-like properties and availability, the best 106 compounds were reordered for more detailed investigation. First, IC50 values of the compounds were determined using an orthogonal assay without the coupled resazurin/diaphorase system to exclude compounds that interfere with either resazurin or diaphorase. IC50 values of the most potent PfGluPho inhibitors (IC50 <12 µg/mL, <33 µM) are shown in Table 2 .
Characterization of Selected Inhibitors for Recombinant PfGluPho
Determination of IC50 values and type of inhibition and of reversibility were based on at least three independent experiments. Presented IC50 values were determined using the orthogonal G6PD assay without resazurin and diaphorase and are given as average with standard deviation. C, competitive; MT, mixed type; NC, noncompetitive; IR, irreversible; R, reversible.
Based on the point of intersection of Lineweaver-Burk plots, the variation of Vmax and Km with increasing compound concentrations, and the determination of α, the most likely inhibition type was determined for the five most potent compounds, which are shown in
Table 2
. All five compounds showed mixed-type inhibition for G6P. Four of the compounds (CB61, CB63, CB70, CB104) showed noncompetitive inhibition for NADP+, and one compound (CB22) was found to be competitive versus NADP+.
Figure 3
shows representative Lineweaver-Burk plots (
Fig. 3A
,
B
) and data distributions (
Fig. 3C
,
D
) for compound CB70. The respective data for CB22, CB61, CB63, and CB104 are shown in
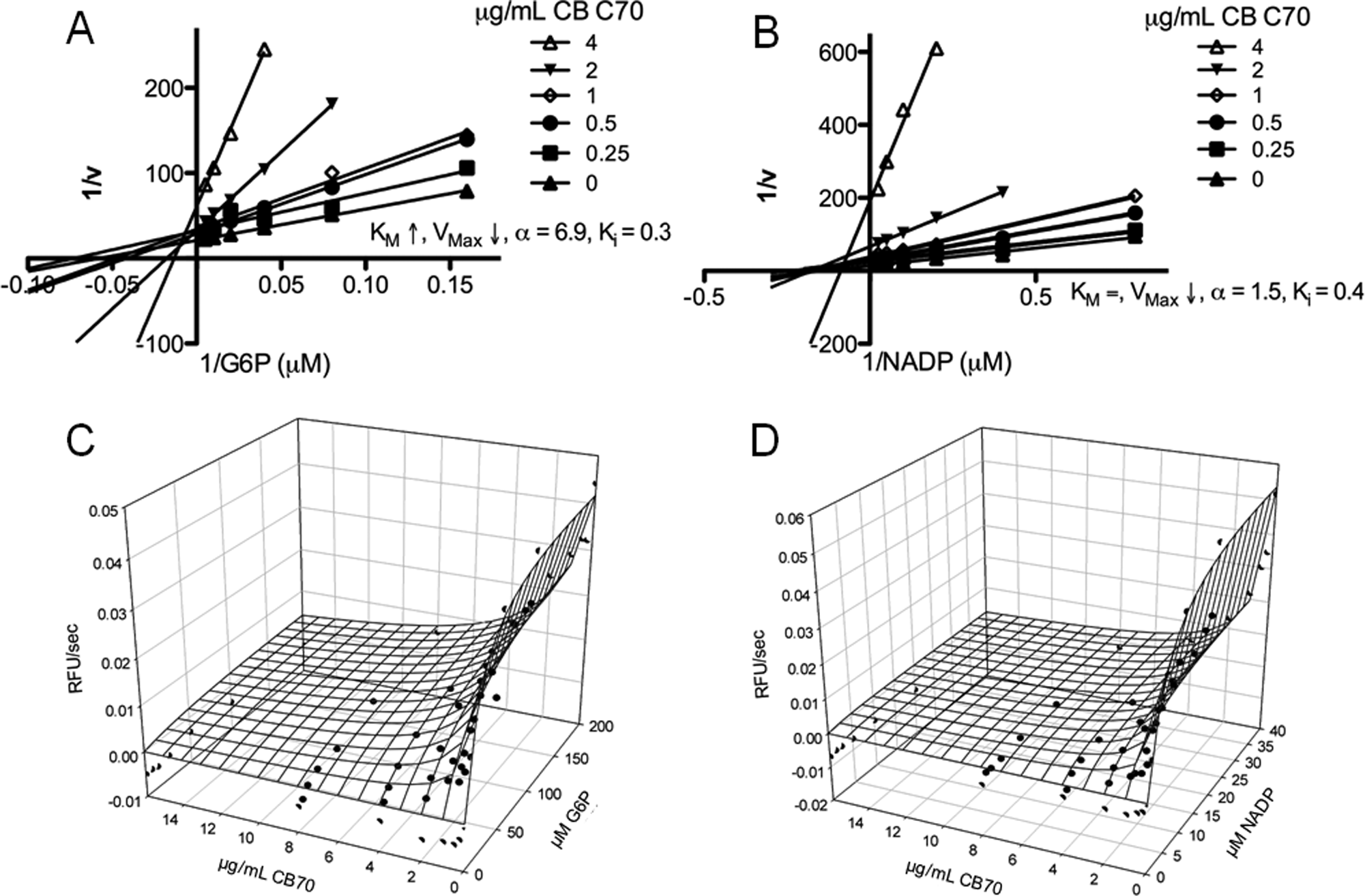
Mechanistic characterization of CB70. (
Time-Dependency and Reversibility of PfGluPho Inhibitors
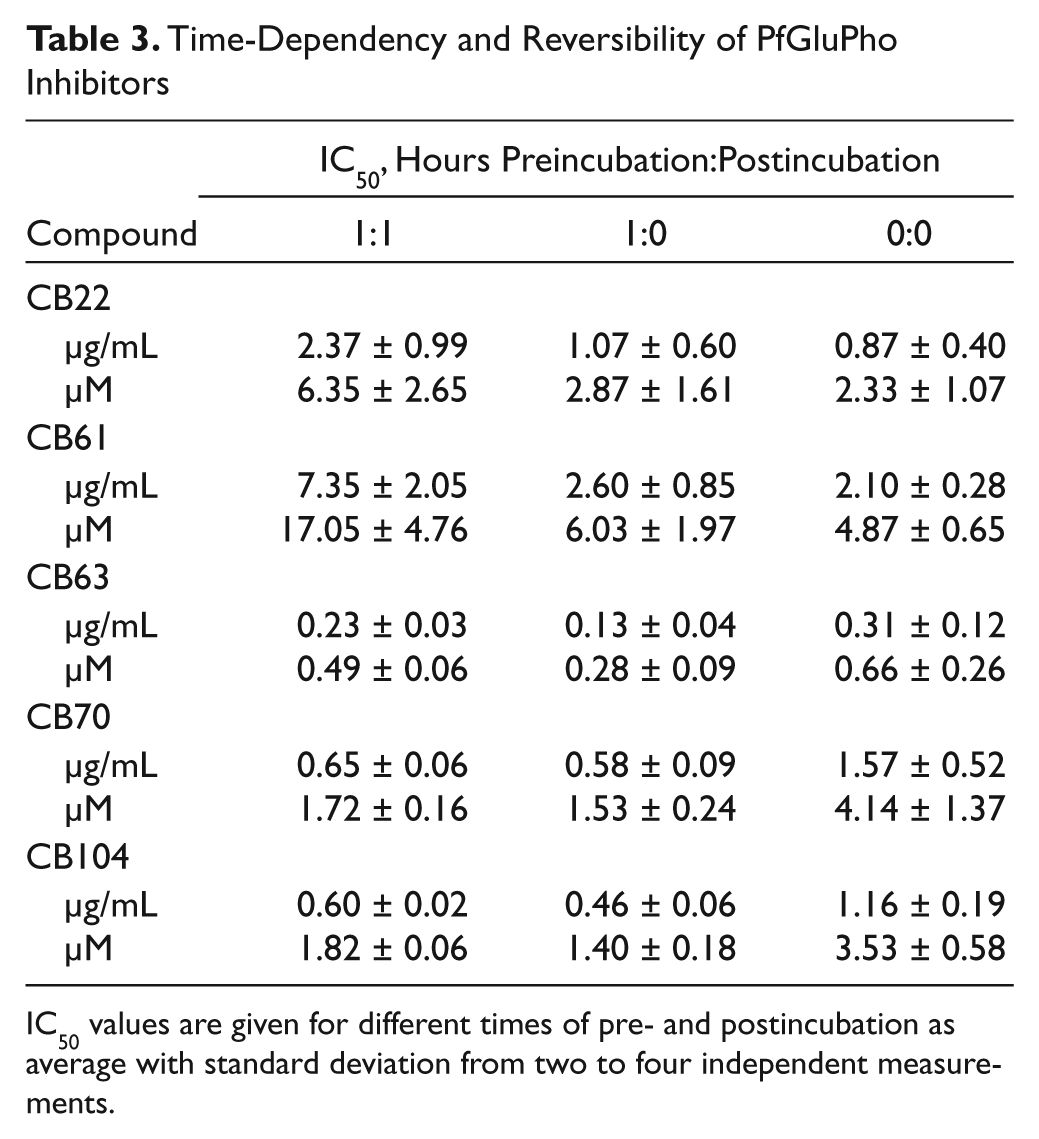
IC50 values are given for different times of pre- and postincubation as average with standard deviation from two to four independent measurements.
P. falciparum culture
In addition to testing the compounds with recombinant PfGluPho, we also investigated their effects on P. falciparum growth. P. falciparum parasites (3D7 strain) were incubated with varying concentrations of compounds that showed inhibition of recombinant PfGluPho, and parasitic growth was determined after 72 h. All 106 reordered compounds were tested, and the results for the four most potent growth inhibitors (CB68, CB83, CB90, CB103) are given in Table 4 (IC50 value <1.8 µg/mL, <5.5 µM). These four compounds, however, were not among the most potent inhibitors of recombinant PfGluPho. In the parasite growth assay, the parasites were incubated with the compounds for 72 h, whereas IC50 determination for recombinant PfGluPho in the orthogonal assay included a 20- to 30-min incubation. To determine if the compounds needed more time to bind to the enzyme and were therefore not identified as potent inhibitors in the orthogonal assay, time-dependent inhibition experiments with the recombinant enzyme were performed. The studies showed that CB68 indeed had an effect after longer incubation, whereas CB83 and CB90 only had minor effects on recombinant PfGluPho. CB103 was originally identified as a potent PfGluPho inhibitor in the primary screening. It was, however, excluded from follow-up studies in the orthogonal assay as it showed autofluorescence at ex340/em460 at higher concentrations. After seeing effects on parasite growth, CB103 was reevaluated by measuring the initial slope of the reaction, which allowed IC50 determination in the orthogonal assay.
Characterization of Selected Inhibitors of Recombinant PfGluPho and Plasmodium falciparum Growth
Time-dependent inhibition studies and mechanistic characterization were performed using recombinant PfGluPho (Pf) and human G6PD (h) in at least three independent experiments. IC50s for P. falciparum growth were determined in at least two independent experiments. ND, not determined; C, competitive; MT, mixed-type; NC, noncompetitive.
To assess selectivity, compounds were tested against recombinant human G6PD (hG6PD). CB68 and CB83 inhibited hG6PD time-dependently (
Table 4
) and more potently than PfGluPho. On the other hand, CB103 inhibited PfGluPho more strongly than hG6PD, without showing a time-dependent effect (
Table 4
). Overall, CB68 and CB103 inhibited PfGluPho and hG6PD and presented an IC50 <2 µM in the parasite culture, whereas CB83 and CB90, which inhibit only hG6PD at lower concentrations, presented IC50 values >2 µM (
Table 4
). Thus, inhibitory effects on parasite growth were enhanced if PfGluPho and hG6PD were inhibited at the same time. Mechanistic characterization was performed for CB68 and CB103 with recombinant PfGluPho, revealing that CB68 follows noncompetitive or mixed-type inhibition for G6P and noncompetitive inhibition for NADP+ (
Table 4
and
Structure-activity relationship analysis
SAR analyses revealed that the compounds included in the follow-up studies shared two common scaffolds with regard to the 880 compounds of the secondary screening. Of the 880 compounds, 14.3%, including CB22, CB61, CB62, and CB64, belong to a group of pyrimidine triones. Compounds of this group showed good activity against recombinant PfGluPho with IC50s ranging from 54 ng/mL to >5 µg/mL. Table 5A shows the core structure of this group, the follow-up compounds found in this group, and some compounds with low and high potency. Moreover, 1.2% of the 880 compounds are part of the group of chromen-2-ones. CB103, the most potent PfGluPho inhibitor with antiplasmodial activity, was found in this group. IC50s of the presented compounds with this scaffold against recombinant PfGluPho ranged between 61 ng/mL and >5 µg/mL ( Table 5B ).
Structure-Activity Relationship Tables of PfGluPho Inhibitors
(A) IC50 values of confirmed hit compounds with a pyrimidine trione scaffold. (B) IC50 values of confirmed hit compounds with a chromen-2-one scaffold. Presented data are based on a single IC50 determination as part of the secondary screening. CB22, CB61, CB62, CB64, and CB103 were included in follow-up studies.
Discussion
The bifunctional enzyme PfGluPho presents a promising target for antimalarial drug design.16,20 Previous studies were restricted in enzyme quantities,17,32 and to date, only two compounds with antimalarial activity binding to PfGluPho have been reported.20,33 The recombinant enzyme became available in milligram quantities only recently,
20
which allowed us to develop an HTS assay to identify PfGluPho inhibitors for the first time. For this purpose, the standard G6PD assay
30
was coupled with resazurin and diaphorase, shifting detection to higher wavelengths (ex540/em590) to increase assay sensitivity and boost resistance to optical artifacts, which are common at ex340/em460 (NADPH fluorescence) (
Fig. 1
). This coupled assay was previously developed and used for an HTS of G6PD of L. mesenteroides (PubChem AID 1020). Within the present study, this assay was modified for PfGluPho and used for HTS screening of three small-molecule libraries. To allow for the best sensitivity versus all possible inhibitor types—namely, competitive, noncompetitive, and uncompetitive inhibitors—the substrates were tested at their respective Km values. During the assay development process,
24
which included determination of Km values, optimal resazurin concentration, optimal enzyme concentration, DMSO tolerance, and Z factor, it was revealed that the signal of resorufin detection was negatively correlated with the resazurin concentration (
Fig. 2C
). Therefore, a resazurin concentration was chosen that was slightly higher than the G6P concentration. Overall, the finalized assay demonstrated excellent HTS statistics according to accepted HTS standards,24,31 represented by a Z factor of 0.79 and an S/B ratio of 4 (
CB68, CB83, CB90, and CB103 were found to inhibit P. falciparum growth at low concentrations (IC50 <1.8 µg/mL, <5.5 µM). Since these compounds did not show significant inhibition in IC50 determinations using the orthogonal assay, these compounds were subjected to time-dependent inhibition experiments. CB83 and CB90 did not inhibit recombinant PfGluPho at low concentrations. However, they inhibited human G6PD time-dependently. CB68 and CB103 inhibited PfGluPho and hG6PD time-dependently, resulting in lower IC50 values for parasite growth inhibition when compared with inhibition against hG6PD, indicating decreased parasite survival through parallel inhibition of hG6PD and PfGluPho compared with inhibition of only one of the enzymes. Under physiological conditions, the effects might even be stronger than what we discovered in our experiments, taken the above-mentioned mechanism of removal of Plasmodium-infected RBCs due to impaired G6PD activity
8
into consideration. CB68 and CB103 presented noncompetitive or mixed-type inhibition for G6P and noncompetitive inhibition for NADP+ (
SAR analyses revealed two classes of PfGluPho inhibitors based on 880 compounds that were included in the secondary screening and with regard to the compounds included in the follow-up. Of the compounds, 14.3% were classified as pyrimidine triones ( Table 5A ) and 1.2% as chromen-2-ones ( Table 5B ), respectively. So far, several screening approaches for identification of potential antimalarials have been made, which were whole organism based, 36 a combination of whole organism and target based, 33 or biochemical target based. 37 However, to our knowledge, the groups identified in the present study have not yet been reported in the context of malaria. Compounds with a pyrimidine-2,4,6-trione scaffold have been reported as matrix metalloproteinase inhibitors 38 and in the context of amyotrophic lateral sclerosis probe development. 39 Moreover, previous studies identified compounds with a chromen-2-ones scaffold as dopamine D4 antagonists. 40 We could not find reports connecting these chemical groups with antimalarial activity.
Unfortunately, we were not able to identify selective inhibitors for PfGluPho with antimalarial activity within this study. All compounds inhibiting Plasmodium growth showed parallel inhibition of hG6PD. However, combined inhibition of PfGluPho and hG6PD resulted in parasite growth inhibition with IC50s in the low µM range. Interestingly, in one of the previous screening approaches, one compound with antimalarial activity and assigned to target P. falciparum 6PGL 33 was later identified as an inhibitor for the G6PD part of PfGluPho. 20 However, this compound also showed parallel inhibition of PfGluPho and hG6PD. 20 Modification of side groups of the presented compounds might result in PfGluPho specificity, which can be addressed in future studies.
Within the presented study, we developed a target-based HTS assay for PfGluPho. Screening of three compound libraries resulted in the identification of several PfGluPho inhibitors for the first time. Some PfGluPho inhibitors showed antimalarial activity in the low µM range without being cytotoxic and thus provide a starting point for antimalarial drug design.
Footnotes
Acknowledgements
The authors thank Michaela Stumpf and Beate Hecker for their excellent technical assistance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by the National Institutes of Health (1R21AI082434-01) to LB as well as the Deutsche Forschungs-gemeinschaft (BE 1540/18-1).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.