
Abstract
Glucose-6-phosphate dehydrogenase (G6PD) is the key enzyme of the pentose phosphate pathway, converting glucose-6-phosphate to 6-phosphoglucono-δ-lactone with parallel reduction of NADP+. Several human diseases, including cancer, are associated with increased G6PD activity. To date, only a few G6PD inhibitors have been available. However, adverse side effects and high IC50 values hamper their use as therapeutics and basic research probes. In this study, we developed a high-throughput screening assay to identify novel human G6PD (hG6PD) inhibitors. Screening the LOPAC (Sigma-Aldrich; 1280 compounds), Spectrum (Microsource Discovery System; 1969 compounds), and DIVERSet (ChemBridge; 49 971 compounds) small-molecule compound collections revealed 139 compounds that presented ≥50% hG6PD inhibition. Hit compounds were further included in a secondary and orthogonal assay in order to identify false-positives and to determine IC50 values. The most potent hG6PD inhibitors presented IC50 values of <4 µM. Compared with the known hG6PD inhibitors dehydroepiandrosterone and 6-aminonicotinamide, the inhibitors identified in this study were 100- to 1000-fold more potent and showed different mechanisms of enzyme inhibition. One of the newly identified hG6PD inhibitors reduced viability of the mammary carcinoma cell line MCF10-AT1 (IC50 ~25 µM) more strongly than that of normal MCF10-A cells (IC50 >50 µM).
Keywords
Introduction
Glucose-6-phosphate dehydrogenase (G6PD) is the first and rate-limiting enzyme of the oxidative branch of the pentose phosphate pathway (PPP). G6PD catalyzes the conversion of glucose-6-phosphate (G6P) to 6-phosphoglucono-δ-lactone (6PGδL), which is accompanied by NADPH production. The product 6PGδL is hydrolyzed by 6-phosphogluconolactonase (6PGL) to form 6-phosphogluconate, which is converted by 6-phosphogluconate dehydrogenase (6PGD), resulting in ribulose 5-phosphate and NADPH. 1 The oxidative PPP thus yields NADPH essential to maintain the redox equilibrium 2 and adequate ribose-5-phosphate levels crucial for nucleotide biosynthesis in proliferating cells. 3
Several studies suggest an important role of G6PD in the pathology of various human diseases such as heart failure, type 2 diabetes, hypertension, 4 and cancer.3,5
In the context of cancer research, glucose metabolism has been of interest for more than 80 years because of initial observations that cancer cells take up high amounts of glucose and produce high amounts of lactate. 6 Because lactate production is pronounced even under aerobic conditions, a high aerobic glycolytic rate was associated with cancer cell metabolism, which is known as the Warburg effect. 7 At present, the increased glucose uptake by cancer cells is commonly used as a diagnostic tool for tumors. 8 However, the assumption that the increased glucose metabolism involves only glycolysis has been questioned. 7 Boros et al.3,9 observed that inhibition of the nonoxidative and/or oxidative PPP reduces cell proliferation of pancreatic carcinoma cells in vitro, suggesting an important role of the PPP in cancer cells. These findings are supported by the fact that mammary carcinoma cells present an increased glucose flux through the PPP in vitro. 10 Moreover, G6PD activity is increased in tumor tissues compared with normal cells,5,11 and silencing G6PD in tumor cells results in decreased proliferation and enhanced apoptosis in vitro. 12 Very recently, it was shown that the tumor suppressor p53 regulates the PPP activity by binding to and suppressing G6PD, highlighting the impact of G6PD in cancer cells. 13
Inhibitors with in vivo activity against human G6PD are 6-aminonicotinamide (6AN) and 17-ketosteroides such as dehydroepiandrosterone (DHEA). 6AN, a competitive G6PD and 6PGD inhibitor,14,15 was used in the past for chemotherapy of various tumors. 4 However, 6AN treatment had severe side effects, such as nerve damage and vitamin B deficiency. Disadvantages of the uncompetitive G6PD inhibitor DHEA are high oral doses and the generation of active androgens. 4 The recent finding that antiproliferative effects of DHEA are most likely not due to G6PD inhibition but rather to changes in mitochondrial gene expression 16 highlights the need for novel selective G6PD inhibitors with few side effects in order to investigate the impact of G6PD on human diseases.
Here we describe the discovery of novel G6PD inhibitors using a high-throughput screening (HTS) approach. Selected G6PD inhibitors were characterized and investigated in the MCF10 model for mammary carcinoma. 17 Our results could provide a basis for research on several human diseases and potentially for the development of novel antitumor therapies.
Materials and Methods
G6PD Overexpression and Purification
hG6PD was overexpressed and purified according to our previously described protocol. 18 In brief, a clone (ID 282264) from the National Institutes of Health (NIH) Mammlian Gene Collection was purchased from Invitrogen (Carlsbad, CA). The hG6PD sequence (GenBank accession number NP_001035810) was cloned into the pET24a expression vector (Novagen) and overexpressed with a C-terminal His tag in Escherichia coli BL21 cells (Invitrogen) containing pRAREII. Overexpression was performed at 23 °C in 2xYT medium (16 g tryptone, 10 g yeast, 5 g NaCl per liter medium) containing 50 µg/mL kanamycin and 12.5 µg/mL chloramphenicol. Protein expression was induced with 0.1 mM IPTG at an optical density at 600 nm of 0.1. Cells were harvested after 24 h, lysed, and purified by metal affinity chromatography using Ni-NTA. Buffer used for hG6PD lysis and purification was 50 mM Tris/HCl, 300 mM NaCl, 0.1 mM NADP+, pH 8.0. hG6PD was eluted from the Ni-NTA column with 150 and 300 mM imidazole and stored with 1.8 M ammonium sulfate + 0.1 mM NADP+ at 4 °C.
Assay Reagents
NADP+ (Amresco), G6P (Sigma-Aldrich, St. Louis, MO), diaphorase (Sigma-Aldrich), and resazurin (Sigma-Aldrich) were prepared as concentrated stock solutions, which were stored in aliquots at −80 °C. Stability of the assay reagents was checked by monitoring Km values and enzyme titration patterns of freshly purified hG6PD from time to time. Km and Vmax determinations for one hG6PD batch over several weeks revealed information about enzyme stability. DHEA (Sigma-Aldrich) and 6AN (Sigma-Aldrich) were prepared at 30 and 90 mM, respectively and stored at −80 °C.
Assay Development
The HTS assay development, including determination of Km values, appropriate hG6PD, and resazurin concentrations, DMSO tolerance, signal-to-background (S/B) ratio, and Z′-factor, was performed according to our previously described HTS assay development protocols for G6PD from Plasmodium falciparum. 19
Compound Libraries for HTS
The LOPAC library (Sigma) consists of 1280 druglike molecules in the fields of cell signaling and neuroscience, which were present at a concentration of 10 mM. The Spectrum collection (Microsource Discovery Systems, Gaylordsville, CT) includes 2320 biologically active and structurally diverse compounds, experimental bioactives, and pure natural products, which were also present at a concentration of 10 mM. The DIVERSet (ChemBridge, San Diego, CA) consist of ~50 000 druglike compounds with maximum pharmacophore diversity at a concentration of 0.5 mg/mL. LOPAC and Spectrum libraries were stored in a desiccator cabinet at 20 °C, whereas the DIVERSet was stored at −20 °C.
Primary and Secondary Screening
The LOPAC and Spectrum compound libraries were screened following the HTS procedure described previously. 19 In brief, 9 µL of a substrate mix (Tris [pH 7.5], MgCl2, G6P, diaphorase, resazurin) were transferred into 384-well plates (Greiner, Kremsmuenster, Austria) using the Multidrop Combi reagent dispenser (Thermo Scientific, Waltham, MA). A total of 36 nL of the compounds was then added with the Echo 555 liquid handler (Labcyte, Sunnyvale, CA) to columns 3 to 22. Columns 1 and 2 served as positive controls (100% inhibition) and contained the substrate mix without G6P. Columns 23 and 24 were negative controls (0% inhibition) without compounds. A total of 36 nL DMSO was added to positive and negative controls. Afterward, 9 µL of an enzyme mix (NADP+, Tween 20, hG6PD) was added, and the plates were incubated in the dark for 40 min. Finally, the plates were measured at ex 530/em 590 using the multiwell plate reader Analyst HT (LJL Biosystems), and the data were analyzed with CBIS (Chemical and Biological Information Systems, http://www.cheminnovation.com). The final assay concentrations were 0.05 M Tris (pH 7.5), 3.3 mM MgCl2, 96 µM G6P, 1 U/mL diaphorase, 0.1 mM resazurin, 11 µM NADP+, 0.005% Tween 20, and 0.51 nM hG6PD. The compounds were screened at 20 µM with an incubation time of 40 min. For screening of the DIVERSet, the substrate mix contained 122 µM G6P and 130 µM resazurin and the enzyme mix 8 µM NADP+ due to slight Km variations of different protein batches. Compounds of the DIVERSet were screened at a final concentration of 5 µg/mL. In addition, the incubation time was increased to 60 min. Linearity of the reaction was achieved over 60 min using an enzyme concentration of 0.51 nM. Compounds inhibiting hG6PD ≥50% were retested in dose response in a secondary screening.
Orthogonal Assay
Selected hit compounds were purchased from ChemBridge in powder form, dissolved in DMSO, and included in IC50 determinations in an orthogonal assay, which did not include resazurin and diaphorase. The orthogonal assay was performed according to a previously described protocol. 19 In brief, 6 µL of a substrate mix containing 0.15 M Tris (pH 7.5), 9.9 mM MgCl2, 78 µM NADP+, and 354 µM G6P were transferred into wells of a 384-well plate (Greiner). Six microliters of varying compound concentrations (0–15 µg/mL) were added. DMSO was added instead of compounds as negative control (0% inhibition); the substrate mix without G6P was used as positive control (100% inhibition). Afterward, 6 µL of enzyme mix composed of 0.015% Tween 20 and 1.1 nM hG6PD were added to start the reaction (enzyme concentrations were adjusted if necessary). The fluorescence of NADPH was monitored over 20 to 30 min at ex 340/em 460 nm using the multiwell plate reader Infinite M2000 combined with the Magellan 6 software (Tecan). IC50 values were calculated using the GraphPad Prism software based on the endpoint of linear reaction curves. Dose-response tests for DHEA and 6AN included final concentrations of 0 to 1 mM DHEA or 0 to 5 mM 6AN. Fluorescence of NADPH was detected for these compounds with the SpectraMax M5 (Molecular Devices, Sunnyvale, CA) multiwell plate reader.
Mechanistic Characterization
The mechanism of inhibition of selected compounds was investigated by titrating the compounds at varying substrate and co-substrate concentrations as described in the following. Six microliters of G6P (0–2400 µM) or NADP+ (0–264 µM) in 9.9 mM MgCl2, 3 mM TCEP, 0.15 M Tris (pH 7.5), and 33 µM NADP+ for G6P titration or 300 µM G6P for NADP+ titration were transferred to 384-well plates (Greiner). Six microliters of varying compound concentrations (final about 0–10× IC50) were added, after which 6 µL of an enzyme mix consisting of 0.015% Tween 20 and 0.41 nM hG6PD were added to start the reaction. Fluorescence of NADPH was measured at ex 340/em 460 nm using the SpectraMax M5 (Molecular Devices) multiwell plate reader for several minutes, and the initial slope of the reaction was determined with the SoftMax Pro 5.2 software (Molecular Devices). Based on these data, Km and Vmax values were determined with the GraphPad Prism software, and α from the following rate equation (equation 1) used for general description of inhibitors was calculated with SigmaPlot (Systat Software).
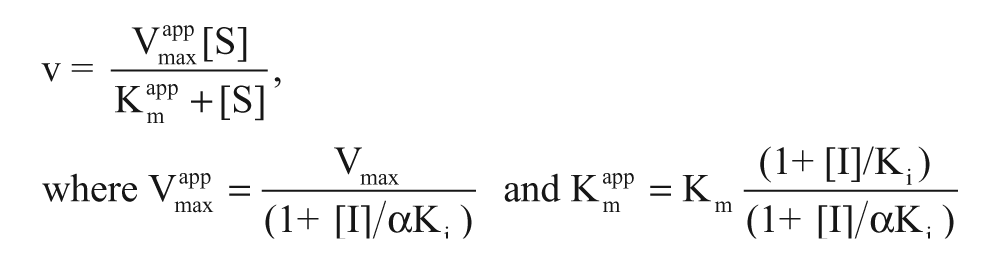
Determination of the mechanism of action was based on the following assumptions 20 : competitive inhibitors present increasing Kmapp values with increasing compound concentrations, whereas Vmaxapp values remain constant, α = ∞; noncompetitive inhibitors show decreasing Vmaxapp values with increasing compound concentrations, whereas Kmapp values stay constant, α = 1; uncompetitive inhibitors present decreasing Kmapp and Vmaxapp values with increasing compound concentrations, α << 1; mixed-type inhibitors are represented by parameters between those of competitive and noncompetitive inhibitors.
Time Dependency and Reversibility
To investigate whether compounds bind to hG6PD in a time-dependent manner, enzyme and inhibitor were preincubated at high concentrations in order to shift the binding equilibrium and promote time-dependent processes. The experiments were performed as described previously. 19 In brief, 50 µL of varying compound concentrations (0–0.15 mg/mL compound [=37× final concentration or max 3% DMSO]) were prepared in the first three columns of a 96-well plate (Greiner). At time point 0, 50 µL of an enzyme mix consisting of 0.1 M Tris (pH 7.5), 2 mg/mL BSA, and 0.12 µM hG6PD (=37.5× final concentration) was added to column 1. After 1 h, 8 µL of the enzyme/inhibitor mix of column 1 was added to 92 µL of buffer, which consisted of 0.05 M Tris (pH 7.5), 1 mg/mL BSA, and 0.0082% Tween 20 (=1.635× final concentration). At the same time, 50 µL enzyme mix was added to column 2. After another hour, 8 µL of enzyme/inhibitor mix of column 2 was added to 92 µL of buffer. Then, 50 µL enzyme mix was added to column 3, of which 8 µL was directly added to 92 µL of buffer. Finally, 12 µL of the enzyme/inhibitor/buffer mixes were transferred to 384-well plates, and 6 µL of a substrate mix (9.9 mM MgCl2, 30 µM NADP+ [=3× final concentration], 0.05 M Tris, 1 mg/mL BSA, 390 µM G6P [3× final concentration]) was added to start the reaction. The substrate mix without G6P was used as a control representing 100% inhibition. Fluorescence of NADPH was detected at ex 340/em 460 with the SpectraMax M5 (Molecular Devices) multiwell plate reader, and the IC50app values were calculated with GraphPad prism based on the initial slope of the reaction. In this way, 1 h preincubation and 1 h postdilution (1:1), 1 h preincubation and 0 h postdilution (1:0), and 0 h preincubation and 0 h postdilution (0:0) were determined. Condition 0:0 was used as the control. A decrease in IC50app values following the incubation suggests time-dependent binding and dissociation. If the IC50app after 1:0 h incubation was lower than the control, the compound was assigned to inhibit time dependently. If the IC50app after 1:1 h was lower than the control, the compound inhibited irreversibly; if the IC50app after 1:1 h was equal or higher than the control, the compound inhibited reversibly.
Cell Viability Assay for MCF10-A and MCF10-AT1 Cells
MCF10-A (ATCC, Manassas, VA) and MCF10-AT1 (The Barbara Ann Karmanos Cancer Institute, Detroit, MI) cell lines were cultured in DMEM medium (CellGro; 10% FBS, 1% penicillin-streptomycin, 100 ng/mL EGF) in an atmosphere of 37 °C and 5% CO2. A total of 15 000 to 30 000 MCF10-A cells and 10 000 to 15 000 MCF10-AT1 cells were seeded in the wells of a 96-well plate. After 24 h, the cells were ~50% confluent, and compounds were added in varying concentrations (0–100 µM) to the wells (final 0.98% DMSO). DMSO was used as negative control; the known G6PD inhibitor 6AN (max 325 µM) was used as a positive control. The plates were incubated for 24 h. Afterward, the cells were washed twice with phosphate-buffered saline (PBS), and the cells were fixed by adding 50 µL paraformaldehyde (PFA; 4%) for 10 min. PFA was removed, and the cells were incubated with 50 µL 0.1% methylene blue for 1 h. After washing with PBS, the cells were incubated with 0.1 M HCl. After 1 h, the absorbance was measured at 660 nm using a Synergy x Multiwell reader (Biotek, Winooski, VD), and IC50 values were calculated using GraphPad Prism. The background (absorbance of 0.1 M HCl) was subtracted from all values. To test growth inhibition at varying confluences, MCF10-A and MCF10-AT1 cells were seeded at varying concentrations.
Solubility and Permeability Analysis
Solubility analysis was performed using a direct UV kinetic solubility method in a 96-well format. All liquid dispense and transfer steps were performed with the Freedom Evo automated liquid handler (Tecan US). Solubility measurements were performed in an aqueous buffer solution (pION) at pH 5.0, 6.2, and 7.4 in duplicate. Samples were incubated at room temperature for 18 h to achieve equilibrium, then filtered to remove any precipitate. The concentration of the compound was then measured by UV absorbance.
Permeability was assessed using the Parallel Artificial Membrane Permeability Assay (PAMPA) in a 96-well format. Permeability measurements were performed in an aqueous buffer solution (pION) at pH 5.0, 6.2, and 7.4, in duplicate. A sandwich plate consisting of a donor bottom plate and an acceptor filter plate was used. The donor wells contained the compound in 180 µL system solution and magnetic stir bars. The filter on the bottom of each acceptor well was coated with GIT-0 liquid and filled with 200 µL of Acceptor Sink Buffer, pH 7.4, containing a surfactant to mimic the function of serum proteins. The permeation time was 30 min, and moderate stirring was applied. After the permeation time, the sandwich was disassembled and compound present in both the donor and acceptor wells was measured by UV absorbance.
Results
Assay Development
The HTS assay used in this study was developed based on a resazurin/diaphorase–coupled G6PD assay, the principle of which was described previously. 19 In brief, G6P is converted to 6-phosphoglucono-δ-lactone by the enzyme G6PD. 1 The reaction is accompanied by production of NADPH, which is used in a coupled reaction to reduce resazurin by means of the enzyme diaphorase (Clostridium kluyveri) to form the highly fluorescent resorufin. 21 The final amount of generated resorufin is proportional to the converted G6P and thus reflects G6PD activity.
We overexpressed and purified mg quantities of hG6PD according to our previously developed protocol 18 and stored the enzyme with 1.8 M ammonium sulfate and 0.1 mM NADP+ at 4 °C. Under these conditions, the enzyme was stable for several weeks. Only slight changes in Km and Vmax values occurred after longer storage periods, and all parameters were confirmed before using the enzyme. Assay reagents such as NADP+, G6P, resazurin, and diaphorase were stable over months, as judged by Km values and enzyme titration patterns comparable to freshly purified enzyme (data not shown).
Km values were determined for all protein batches that were used during the study by titrating the substrate and co-substrate. hG6PD presented a Km value of 95 to 120 µM for the substrate G6P ( Fig. 1A ) and about 10 µM for the co-substrate NADP+ ( Fig. 1B ).
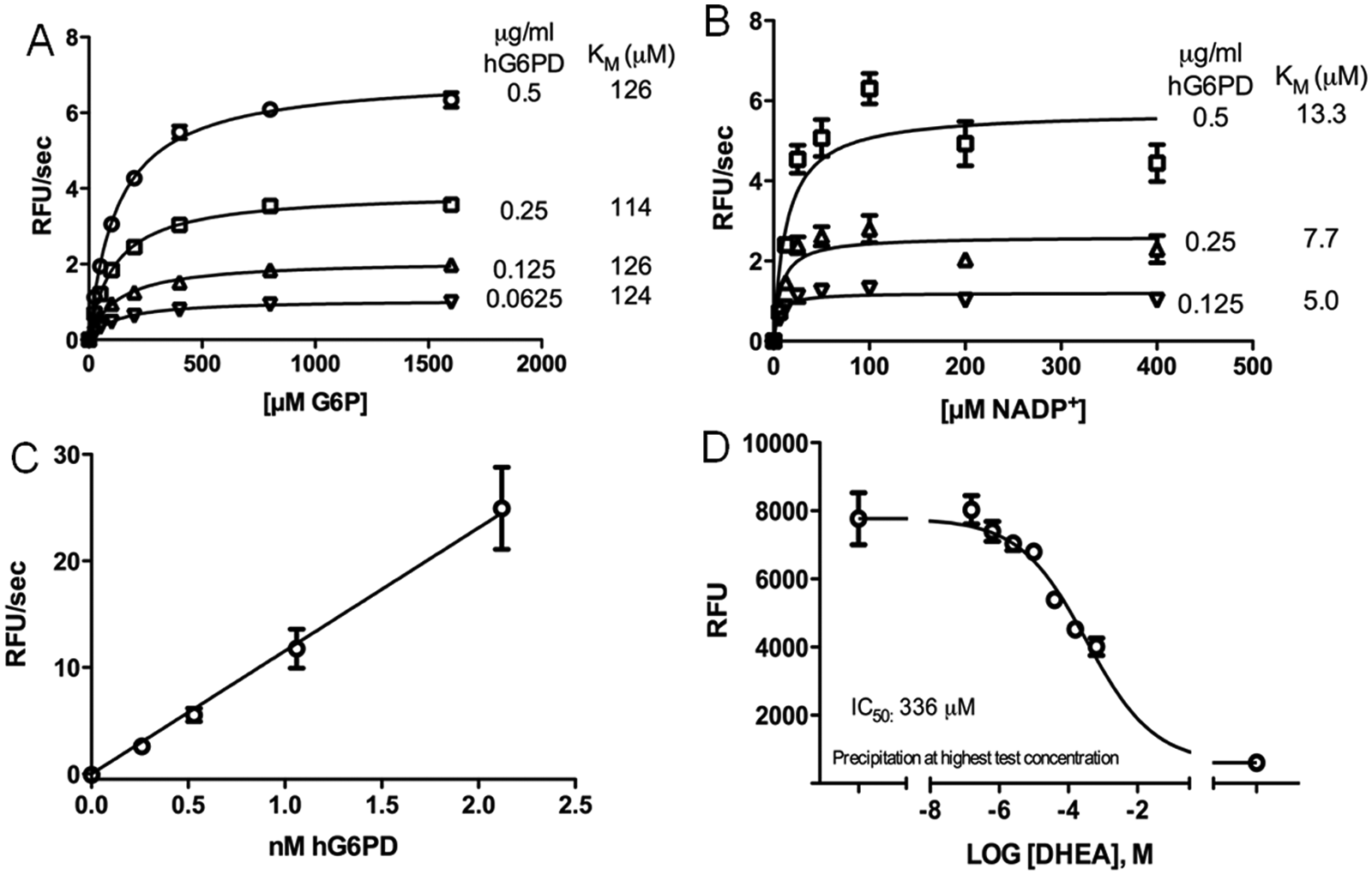
High-throughput screening assay development. Michaelis-Menten curves for G6P (
Previous development of a resazurin-coupled HTS assay for P. falciparum G6PD revealed that the fluorescence signal of resorufin was quenched at higher resazurin concentration,
19
consistent with the strong inner filter effect of resazurin. A similar effect of resazurin concentration was observed in the hG6PD assay (
hG6PD titration showed that the signal was linear for up to 60 min, including 0.51 nM hG6PD. Furthermore, the ratio of signal-to-enzyme concentration was linear up to a concentration of at least 2 nM (
Fig. 1C
), confirming that the included enzyme concentration was in an appropriate range. Based on these initial experiments, the screening assay was tested using 0.05 M Tris (pH 7.5), 3.3 mM MgCl2, 96 µM G6P, 1 U/mL diaphorase, 0.1 mM resazurin, 11 µM NADP+, 0.005% Tween 20, and 0.51 nM hG6PD. The test run presented an S/B ratio of 10 and a Z-factor of 0.84 (
Screening of Compound Libraries
The HTS assay was used to screen three small-molecule compound libraries. First, LOPAC and Spectrum libraries, which contain a smaller number of compounds (<3000), were screened for assay verification. Compounds of these libraries were included in the assay at a final concentration of 20 µM. By screening of the LOPAC library (1280 compounds), we identified seven compounds inhibiting hG6PD ≥50%, which corresponds to a hit rate of 0.55%. The average S/B ratio and Z-factor were 5.5 and 0.82, respectively ( Table 1 ).
Screening Results of Primary High-Throughput Screen of LOPAC (20 µM), Spectrum (20 µM), and DIVERSet Compound Libraries (5 µg/mL)
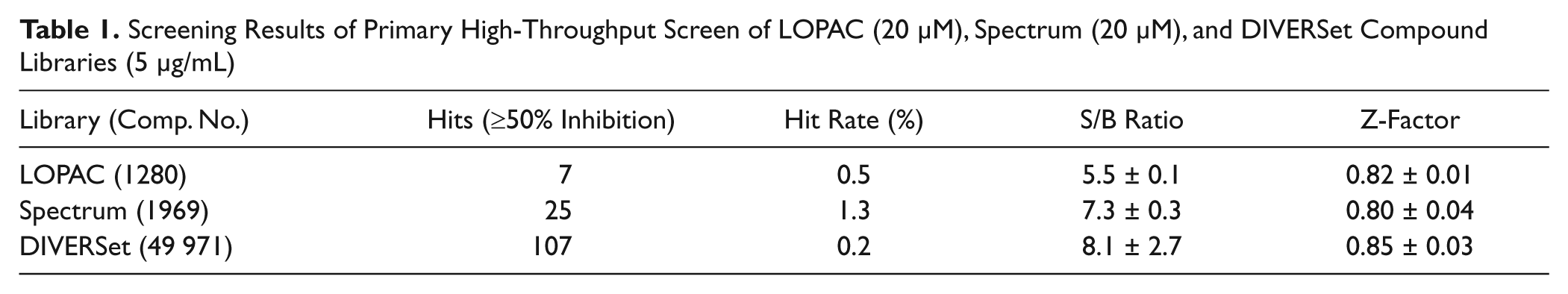
Twenty-five hG6PD inhibitors were found by screening the Spectrum library (1969 compounds), presenting a hit rate of 1.3% (≥50% inhibition), an S/B ratio of 7.3, and a Z-factor of 0.80 ( Table 1 ). Compounds showing ≥50% inhibition of hG6PD were later included in a secondary screening for hit confirmation and IC50 determination.
The DIVERSet (~50 000 compounds) was screened at a concentration of 5 µg/mL. Primary screening resulted in 107 hit compounds (≥50% inhibition), a hit rate of 0.21%, an S/B ratio of 8.1, and a Z-factor of 0.85 ( Table 1 ). Hit compounds were included in a secondary screening, resulting in a hit confirmation of 66%. The secondary screening was performed at the same day of the primary screening to prevent several thawing cycles of the DIVERSet library.
Orthogonal Assay
Four of the hit compounds, namely, CB63, CB70, CB72, and CB104, inhibited hG6PD with an IC50 value <4 µM in the orthogonal assay, the kinetic assay based on NADPH fluorescence intensity. Structures of these compounds and CB83, an hG6PD inhibitor identified in a previous study, 19 are shown in Figure 2 . Dose-response curves of CB63 (IC50 0.6 ± 0.0 µM), CB70 (IC50 2.6 ± 1.1 µM), CB72 (IC50 3.1 ± 0.8 µM), CB104 (IC50 3.0 ± 1.2 µM), and the previously identified inhibitor CB8322 are given in Figure 3 . For the known G6PD inhibitors DHEA and 6AN, we found IC50s of 483 ± 202 µM and >3 mM, respectively (uncoupled G6PD assay, data not shown). More accurate IC50 determinations for DHEA and 6AN were not possible because of solubility issues of DHEA at concentrations >350 mM and DMSO limitations greater than 5 µM of 6AN.
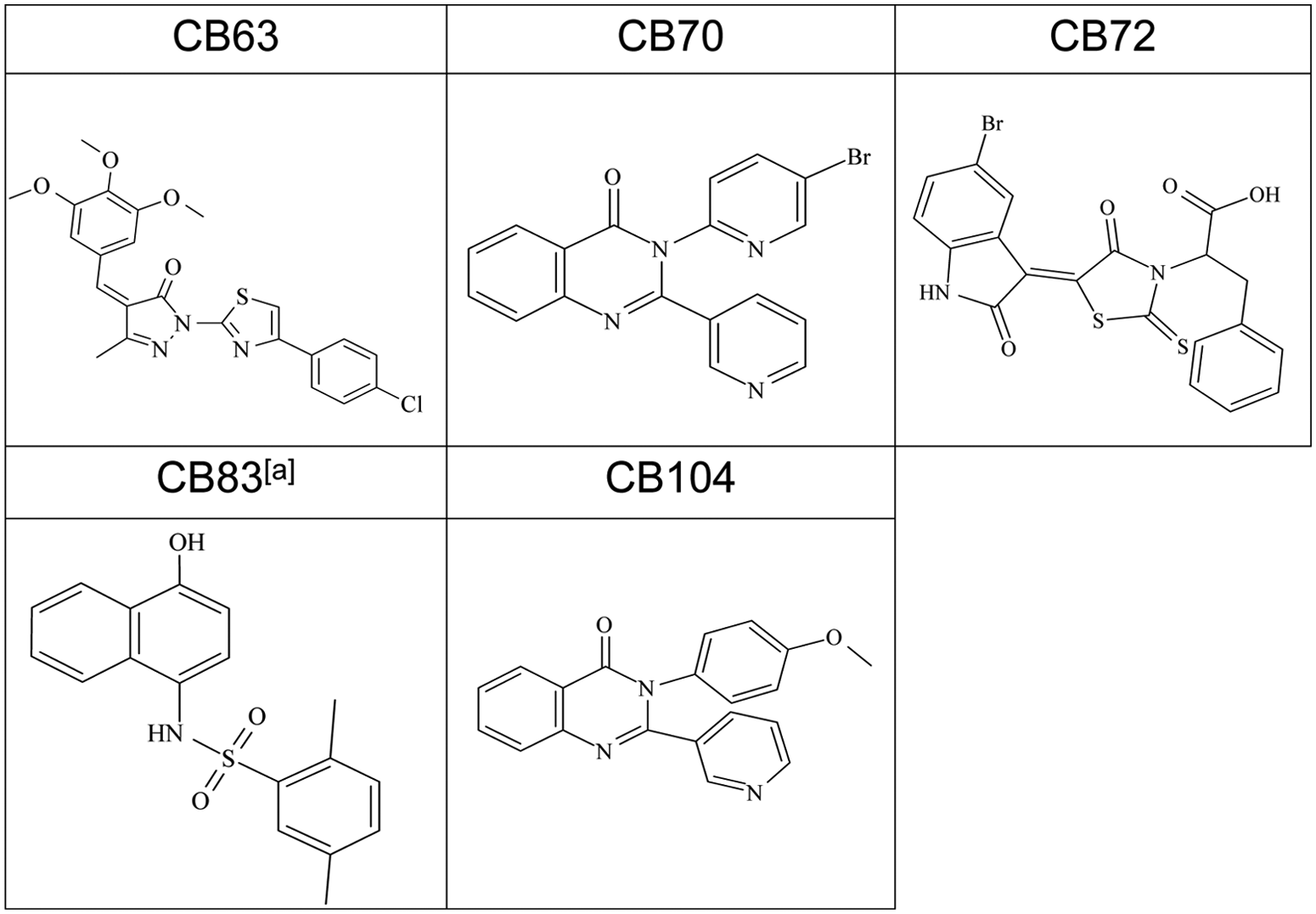
Structures of selected hG6PD inhibitors. (
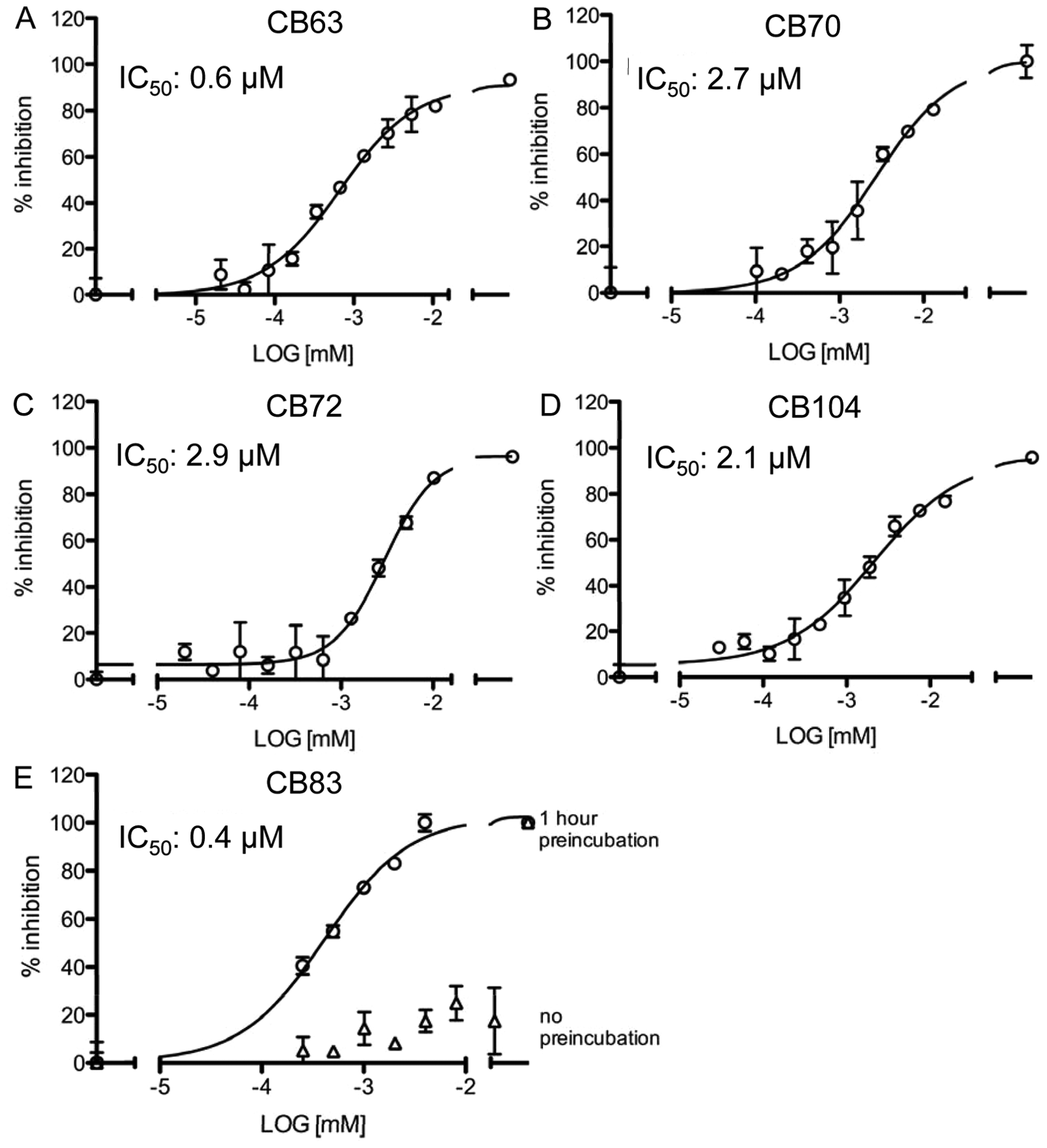
Dose-response curves for selected hG6PD inhibitors. Dose-response curves for CB63 (
Mechanistic Characterization, Time Dependency, and Reversibility
CB63, CB70, CB72, and CB104 were furthermore mechanistically characterized. All four compounds showed competitive inhibition to the substrate G6P (
Table 2
). CB63 showed noncompetitive or mixed-type inhibition against NADP+, CB70 and CB104 showed mixed-type inhibition against the cofactor, and CB72 demonstrated competitive inhibition with respect to NADP+.
Figure 4
shows representative data distributions for CB63 versus G6P (
Time Dependency and Reversibility of hG6PD Inhibitors a

IC50app values are given as average with standard deviation of two to three independent experiments, each performed in duplicate.
Values determined in a previous study.
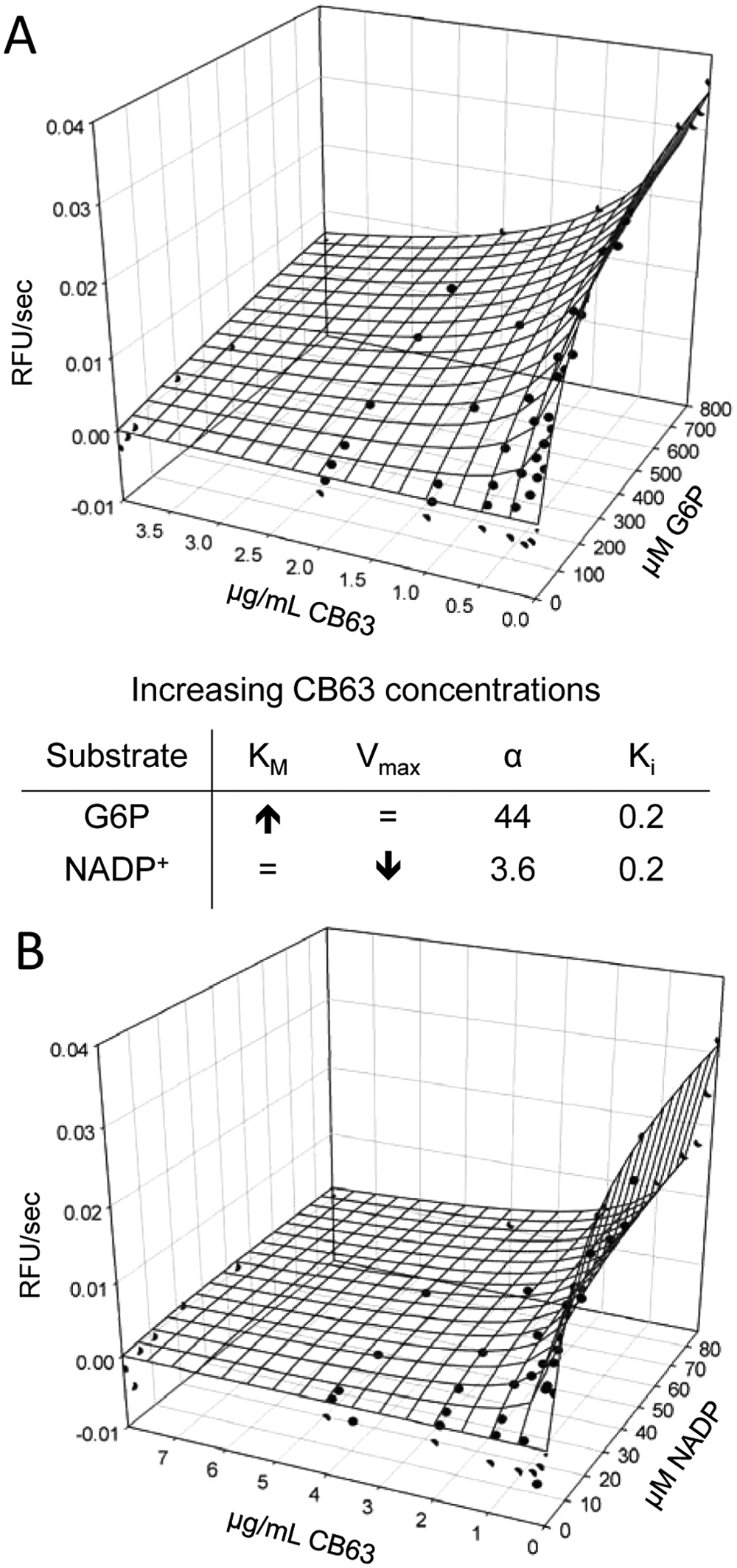
Mechanistic characterization of CB63 for hG6PD. Three-dimensional view of data distribution of G6P and CB63 titration (
Data for CB70, CB72, and CB104 are given in
Effect of hG6PD Inhibitors on Cancer Cells
In our study, we used the MCF10 model of mammary carcinoma 17 to investigate effects of hG6PD inhibitors on breast cancer cells. The spontaneously immortalized MCF10-A cells were used as a model for normal cells, 23 whereas the tumorigenic MCF10-AT1 cells served as a model for breast cancer cells. 24 It has been previously reported that the flux of G6PD to ribose is approximately four times higher in the MCF10-AT1 cells than in the MCF10-A cells. 10 We tested the effects of 0.1 to 100 µM of the most potent hG6PD inhibitors identified in our previous HTS approach on both cell lines. Of the tested compounds, CB83 showed cell growth inhibition that was stronger for MCF10-AT1 (IC50 around 25 µM) compared to MFC10-A cells (IC50 >50 µM; Fig. 5 ). CB63, CB72, and CB104 did not inhibit cell growth at concentrations ≤50 µM (data not shown).
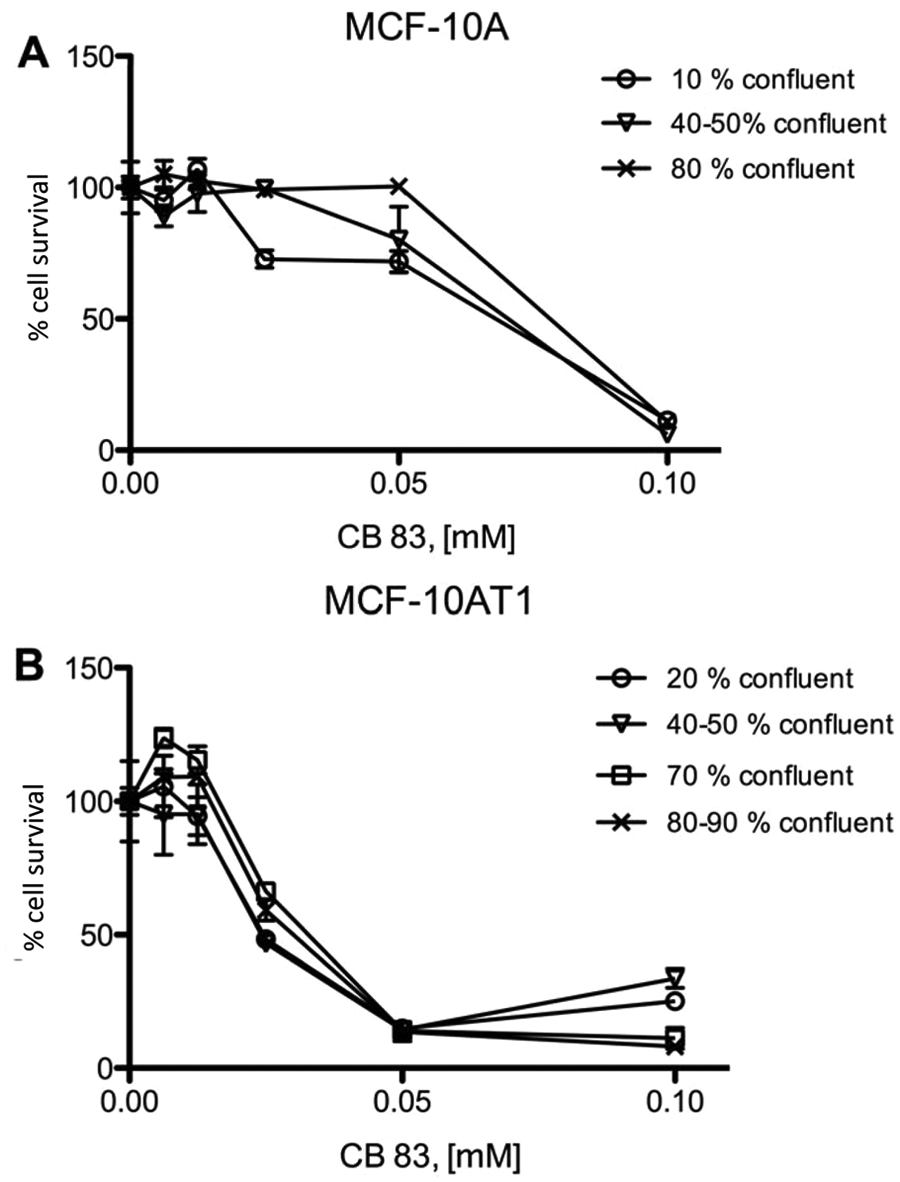
Effects of CB83 on MCF10-A (
Solubility studies for CB83 revealed an aqueous solubility in pION’s buffer of 13.2 µg/mL (pH 5.0), 6.4 µg/mL (pH 6.2), and 3.4 µg/mL (pH 7.2) and of 8.2 µg/mL in 1× PBS (pH 7.4). PAMPA assays presented at an acceptor pH of 7.4 values of 579/647/573 × 10−6 cm/s at a donor pH of 5.0/6.2/7.4.
Discussion
Assay Development and Screening
Several human diseases, including cancer,3,5 are associated with increased G6PD activity. However, only a few hG6PD inhibitors are available, and their use is limited because of adverse side effects. 4 In this study, we developed an HTS assay to identify new inhibitors for hG6PD. The assay development was based on previously established HTS assays for Leuconostoc mesenteroides (PubChem AID 1020) and P. falciparum G6PD, 19 using a resazurin/diaphorase–coupled detection system. 21 Detection of reduced resazurin (ex 530/em 590) instead of NADPH (ex 340/em 460) provides higher assay sensitivity and reduces the risk of detecting optical artifacts compared to the standard G6PD assay. 25 Determination of reagent and enzyme stability, Km values, optimal enzyme and resazurin concentrations, DMSO tolerance, dose response of a known inhibitor, and of the Z-factor built the assay development process.
Our HTS assay was set up to identify competitive, noncompetitive, and uncompetitive inhibitors and thus was run at G6P and NADP+ concentrations at their respective Km values. Based on our findings, hG6PD presented Km values of ~100 µM for G6P (
Fig. 1A
) and ~10 µM for NADP+ (
Fig. 1B
), which are similar to the ones determined in previous studies.
18
Slightly lower values were reported by other groups,26,27 which is most likely due to methodical differences (e.g., variations in co-substrate concentrations). A resazurin titration confirmed our previous finding
19
that high resazurin concentrations decrease the assay sensitivity by quenching the signal (
Moreover, the commercially available LOPAC and Spectrum libraries (<3000 compounds) were screened at a concentration of 20 µM to verify the established HTS assay. At a 50% cutoff, hit rates were with 0.5% for LOPAC and 1.3% for the Spectrum library in a normal range, and Z-factor (0.8) and S/B ratio (>5) were excellent ( Table 1 ). Subsequent screening of the DIVERSet library (49 971 compounds) presented similar HTS statistics and presented a hit rate of 0.2% ( Table 1 ), which is in the lower range for HTS assays. Because the compound composition of the libraries was different and consisted of different scaffolds and chemical classes, variations in hit rates are normal and usually expected to be about 0.5% to 1%. Furthermore, compared to screenings of the LOPAC and Spectrum collections, the final compound concentration was with 5 µg/mL (MW of compounds <500) slightly lower for screening of the DIVERSet. In a secondary screening, we were able to confirm 66% of the DIVERSet hits as hG6PD inhibitors.
Orthogonal Assay and Characterization of Inhibitors
Several hit compounds were retested in a G6PD assay that did not contain the resazurin/diaphorase–coupled system for identification of false-positives that were due to compound interference with the coupled system. We found four compounds (CB63, CB70, CB72, CB104) that inhibited recombinant hG6PD with IC50 values <4 µM (
Fig. 3
), which are 100- to 1000-fold lower compared to those of the known hG6PD inhibitors DHEA and 6AN. Mechanistic characterization studies revealed that all four compounds inhibited hG6PD competitively versus G6P. For the co-substrate, CB70 and CB104 showed mixed-type inhibition, whereas CB63 inhibited noncompetitively and CB72 competitively versus NADP+ (
Table 2
,
Fig. 4
,
Experiments to determine the mechanism of inhibition were limited for CB83. As discussed previously, adding BSA is necessary for assays run over several hours in order to stabilize hG6PD. However, adding BSA resulted in increased IC50app values, which was most likely due to the compound binding to BSA. Inclusion of 10xIC50 concentrations needed for mechanistic characterization was not possible because of autofluorescence and insolubility of the compound at these high concentrations. 19
CB63 and CB104 demonstrated time-dependent but fully reversible binding and dissociation to hG6PD. CB70 was found to be a reversible hG6PD inhibitor ( Table 2 ).
Based on our findings, CB72 caused improved hG6PD activity with increasing concentrations in the reversibility studies, because of which IC50 values could not be determined under the assay conditions. A possible explanation for this finding is that CB72 mimics NADP+ that serves as a structural component 34 and that binding of CB72 stabilizes hG6PD and prevents loss of enzyme activity over time.
In a previous study, CB63, CB70, and CB104 were found to inhibit P. falciparum G6PD (PfGluPho). 19 The sequence of the G6PD part of PfGluPho is very similar to hG6PD, except for a 62-amino-acid insertion, 35 which may explain this finding. However, all three compounds inhibit hG6PD two- to fourfold more potently than PfGluPho and present differences in the mechanism of inhibition. These variations may be caused by a combination of G6PD and 6PGL in the bifunctional PfGluPho, which most likely results in a slightly different folding compared to the isofunctional hG6PD.
Effects of hG6PD Inhibitors on MCF-10 Cells
Besides characterization of hG6PD inhibitors using the recombinant enzyme, we were further interested in effects of the compounds on human cells. Because several forms of cancer are associated with increased G6PD activity,5,11 we investigated the effects of hG6PD inhibitors on cell viability of mammary carcinoma cells. We used the MCF10 model, 17 including the spontaneously immortalized MCF10-A cells 23 as control (“normal” cells) and the tumorigenic MCF10-AT124 as cancer cells, which have the same genetic background. CB63, CB72, and CB104 presented no or very slight inhibition of MCF10-A and AT1 cell growth after a 24 h incubation with 0 to 100 µM of the compounds. Possible explanations for this finding are low membrane permeability or stability of the compounds as well as potential binding to other molecules (such as BSA, discussed above). These aspects will be addressed in structure-activity relationship studies in the future.
CB70 was not available at the time of the cell-based experiments. However, in a previous study, we observed inhibitory effects of another compound, CB83, against hG6PD. 19 This compound showed time-dependent effects with an IC50 of 0.37 µM after a 1 h incubation. Because CB83 showed antiplasmodial activity, 19 we assumed that it was membrane permeable and thus included it in our studies. CB83 indeed showed an effect on survival of MCF10 cells, with IC50 values of ~25 µM for the 10AT1 ( Fig. 5B ) and >50 µM for the 10A cell line ( Fig. 5A ). Permeability and solubility studies showed that CB83 has moderate membrane permeability, which might also be responsible for the relatively high concentrations needed and poor solubility. Solubility and permeability could potentially be improved by chemical alteration of functional side groups. Furthermore, detailed studies of CB83 on various metabolic pathways as well as on the underlying mechanisms that lead to reduced cell viablity should be addressed in the future.
Increased hG6PD activity is suggested to be involved in the pathogenesis of several human diseases. 4 Only a few hG6PD inhibitors are known,14,33 the use of which, however, is limited. 4 In this study, we developed an HTS assay for recombinant hG6PD and screened three small-molecule compound libraries. Overall, we were able to identify four new hG6PD inhibitors with IC50 values <4 µM. These new inhibitors are much more potent and follow different mechanisms of inhibition compared to previously known hG6PD inhibitors. Furthermore, one of the inhibitors reduced cell viability of mammary carcinoma cells. Thus, our study provides novel hG6PD inhibitors that could be used for research in several human diseases and potentially as a starting point for the development of new antitumor therapies.
Footnotes
Acknowledgements
The authors would like to thank Beate Hecker and Carolin Marx for their excellent technical assistance. We would also like to thank Layton Smith and Danielle McAnally of the pharmacology core at Sanford-Burnham Medical Research Institute for determining the solubility and permeability for CB83.
Abbreviations: 6AN, 6-aminonicotinamide; 6PGD, 6-phosphogluconate dehydrogenase; 6PGδL, 6-phosphoglucono-δ-lactone; 6PGL, 6-phosphogluconolactonase; DHEA, dehydroepiandrosterone; G6P, glucose-6-phosphate; G6PD, glucose-6-phosphate dehydrogenase; HTS, high-throughput screening; PAMPA, parallel artificial membrane permeability assay; P., Plasmodium; PPP, pentose phosphate pathway; S/B: signal-to-background.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by the National Institutes of Health (1R21AI082434-01 to L.B.), NIH Roadmap Initiative grant U54 HG005033 (A.P., M.H., E.S.), and the Deutsche Forschungsgemeinschaft (Be1540/18-1 to K.B., S.R.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.