
Abstract
One approach currently being developed in anticancer drug discovery is to search for small compounds capable of occupying and blocking the hydrophobic pocket of anti-apoptotic Bcl-2 family members necessary for interacting with pro-apoptotic proteins. Such an approach led to the discovery of several compounds, such as ABT-737 (which interacts with Bcl-2, Bcl-xl, and Bcl-w) or the latest one, ABT-199, that selectively targets Bcl-2 protein. The efficacy of those compounds is, however, limited by the expression of two other anti-apoptotic Bcl-2 members, Mcl-1 and Bfl-1. Based on the role of Bfl-1 in cancer, especially in chemoresistance associated with its overexpression in B-cell malignancies, we searched for modulators of protein–protein interaction through a high-throughput screening of a designed chemical library with relaxed drug-like properties to identify small molecules targeting Bfl-1 anti-apoptotic protein. We found two compounds that display electrophilic functions, interact with Bfl-1, inhibit Bfl-1 protective activity, and promote cell death of malignant B cells. Of particular interest, we observed a synergistic effect of those compounds with ABT-737 in Bfl-1 overexpressing lymphoma cell lines. Our results provide the basis for the development of Bfl-1 specific antagonists for antitumor therapies.
Introduction
Many of the cellular events that initiate malignant transformation of a normal cell (e.g., activation of oncogenes) also activate oncogenic stress pathways that usually cause the cell to enter apoptosis. For these cells to survive and cause cancer, they typically must have acquired changes in other cellular pathways to prevent apoptosis. Through their capacity to interact within each other, pro- and anti-apoptotic proteins of the Bcl-2 family are important regulators and executioners of the intrinsic apoptotic pathway. Expression of the Bcl-2 proteins is often deregulated in cancer and thus dictates whether many current cancer therapies are effective, because most of these treatments result in stress signals that ultimately must activate the apoptosis machinery of cancer cells. Increased expression of pro-survival members is recognized as a hallmark of many cancers, and drugs that can prevent their action would be very valuable. One approach currently being developed in anticancer drug discovery is to search for BH3 mimetics capable of occupying and blocking the hydrophobic pocket of anti-apoptotic Bcl-2 family members necessary for interacting with pro-apoptotic proteins. Illustrations of such a strategy are the identification of ABT-737, a small molecule with high affinity for Bcl-2, Bcl-xl, and Bcl-w, 1 and more recently the development of ABT-199, a selective inhibitor of Bcl-2. 2 Navitoclax, an orally bioavailable derivative of ABT-737, is currently being evaluated in phase 2 clinical trials.3–5 Besides a dose-limiting toxicity of navitoclax toward platelets, a limitation for the use of both ABT-199 and navitoclax is the overexpression by tumor cells of Mcl-1 or Bfl-1 (BCL2A1) anti-apoptotic proteins that were shown to confer resistance to ABT-737,6–8 thus emphasizing the need for BH3 mimetics specific for Mcl-1 or Bfl-1.
Since its discovery in 1995 as a gene overexpressed in stomach cancer, 9 high expression of Bfl-1 has been documented in various types of cancers (for a review, see reference 8 ), particularly in lymphoid malignancies such as a subgroup of diffused large B-cell lymphoma (DLBCL), 10 mediastinal B-cell lymphoma (MLBCL), 11 and mantle cell lymphoma. 12 Bfl-1 was also implicated in the emergence of resistance of B-cell chronic lymphocytic leukemia (B-CLL) patients to fludarabine treatment.13,14 Further studies using RNA interference strategies demonstrated that inhibition of Bfl-1 sensitizes fresh B-CLL or malignant B-cell lymphoma cell lines to chemotherapeutic agents such as cisplatin and fludarabine, and therefore validated Bfl-1 as a therapeutic target in B-cell malignancies.13–15 We recently identified peptide aptamers that specifically interact with the hydrophobic groove of Bfl-1 and disrupt the interaction of Bfl-1 with its pro-apoptotic partners such as the pro-survival protein Bax. As for the RNA interference strategy, we demonstrated that anti-Bfl-1 aptamers sensitize malignant B-cell lymphoma cell lines to chemotherapeutic agents. 16 Thus, disrupting interactions of Bfl-1 with pro-apoptotic partners appears to be an efficient strategy to overcome its pro-survival activity in malignant cells. Here, we describe the discovery of small molecules targeting Bfl-1 anti-apoptotic protein using a high-throughput screening (HTS) approach based on a fluorescence polarization (FP) method. High speed, robustness, simplicity, and relatively low cost combine to make FP an attractive technology for a primary screening. We have screened a 25,000-compound chemical library to identify Bfl-1 antagonists. Selected compounds were characterized for their antitumor activity in cellular models relevant for lymphoma. Our results provide the basis for the development of Bfl-1 specific antagonists as antitumor therapeutic agents.
Materials and Methods
Chemical Library Selection and Preparation
The compounds used in the experimental screening were selected from the Enamine HTS collection (September 2009) and processed with the program Pipeline Pilot™ version 7.5 (Accelrys, San Diego, USA). The selection was based on a hierarchical procedure combining the compliance with physicochemical thresholds (molecular weight, logP, etc.) and the absence of chemical moieties commonly associated with toxicity and an increase in false-positive rates. The physicochemical thresholds used for the compound selection were the following: number of hydrogen bond acceptors < 13; number of hydrogen bond donors < 8; 150 < molecular weight < 700; logP < 6; topological polar surface area (TPSA) < 160; and number of rotatable bonds < 13. Later in the process, a fingerprint-based criterion [using a similarity threshold of 0.8 for the Tanimoto index (Tn) and a Ward classification] was applied to ensure the chemical diversity of the resulting collection, followed by a random selection, to end up with 25,000 compounds for purchasing.
The chemical library was formatted in barcoded racks of 96 tubes and stored at room temperature or −20 °C for dried or 10 mM DMSO-dissolved copies, respectively. The workstation to dilute and aliquot the library was a CyBI-Well® (CyBio, Savigny Le Temple, France) liquid handler. The sample management system avoids repeated freeze–thaw cycles and ensures the longest possible lifetime for all of the samples. Structures, compound ID, and library manipulations were recorded in MDL ISIS and Microsoft Access databases.
Protein Preparation
Glutathione-S-transferase (GST) fusion proteins containing Δ-Cter-Bfl-1, Δ-Cter-Bcl-2, Δ-Cter-Bcl-XL, or Δ-Cter-Mcl-1 were expressed from pGEX4T-1 plasmid in XL1-Blue cells (Agilent Technologies, Les Ulis, France). The cells were grown in 1 L of TY media [1.6% (w/v) tryptone, 1% (w/v) yeast extract, and 85 mM NaCl] with 50 µg/mL ampicillin at 37 °C to an A600 nm of 0.8, followed by the addition of isopropyl-ß-D-thiogalactopyranoside (0.4 mM) and incubated at 25 °C for 7 h. The cells were recovered in 10 mL lysis buffer,” [50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Tween 20, 0.1% 2-ß-mercaptoethanol supplemented with protease inhibitor mixture (RocheDiagnostics, Meylan, France), 0.5 mg/mL lysozyme, and benzonase (250 units/µL)] 30 min at 30 °C, followed by three freezing cycles. The cellular debris were removed by centrifugation at 6000× g for 20 min, and the resulting supernatants were incubated with 2 mL of Glutathione Sepharose (GE Healthcare, Limonest, France) at 4 °C for 3 h. The resin was washed three times with buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% Tween 20, and 0.1% 2-ß-mercaptoethanol), followed by elution of GST fusion proteins in 10 mM of reduced glutathione dissolved in 50 mM Tris-HCl (pH 8.0).
Recombinant proteins in solution were thereafter dialyzed in phosphate-buffered saline (PBS) buffer containing 1 mM dithiothreitol using Amicon Ultra-4 10K columns (Millipore, Saint Quentin en Yvelines, France), and glycerol was added to a final concentration of 10% to stabilize proteins.
Fluorescence Polarization (FP) Assays
FP assays were performed in a buffer containing 10 mM PBS pH 7.4, 100 mM NaCl, 2.7 mM KCl, 0.01% Triton X-100™, and 1% DMSO in black 96-well microplates (Corning, Amsterdam, The Netherlands). Twenty-five µL of Bfl-1 or the other Bcl-2 family proteins were incubated with 25 µL test compound or vehicle at room temperature for 30 min. Then, 25 µL fluorescein isothiocyanate (FITC)–BH3 peptide were added. The BH3 peptides, FITC–Bim (FITC–DMRPEIWI-AQELRRIGDEFNAYYAR), FITC–Bax (FITC–VPQDAST- KKLSECLKRIGDELDSNMELQR), and FITC–Bak (FITC–KGGGQVGRQLAIIGDDINRRYDS), were synthesized by GeneCust (Luxembourg). After incubation at room temperature, direct fluorescence and FP were measured using a Victor3TM V1420 Perkin Elmer (Courtaboeuf, France) plate reader (excitation filter 485 nm and emission filter 535 nm). The polarization signal was defined in eq 1, in which Sc and Pc were the signals corrected in plan S and P by subtracting the buffer (without the labeled ligand in the well) signal. The g factor was a correction factor depending on the plate reader and fluorophore used.
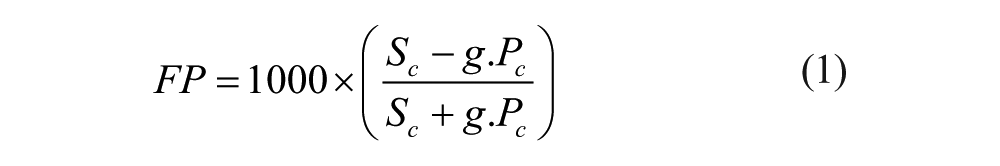
Saturation Binding Assays
Fixed concentrations of FITC–BH3 peptides were incubated for 1 h with increasing concentrations of Bcl-2 family proteins before FP measurement. Equilibrium dissociation constants (Kd) were calculated by fitting the sigmoidal FP increase as a function of protein concentrations (log M) using the four-parameter Hill equation in Graphpad Prism 5.02 software (San Diego, USA).
Competitive Binding Assays
Serial dilutions of competitors were incubated with fixed concentrations of FITC–BH3 peptides and Bcl-2 family proteins. Minimal (FPmin) and maximal (FPmax) polarization values were obtained by incubating FITC–BH3 alone (positive control) or with the Bcl-2 family protein (negative control), respectively. Percentages of inhibition were calculated by eq 2:
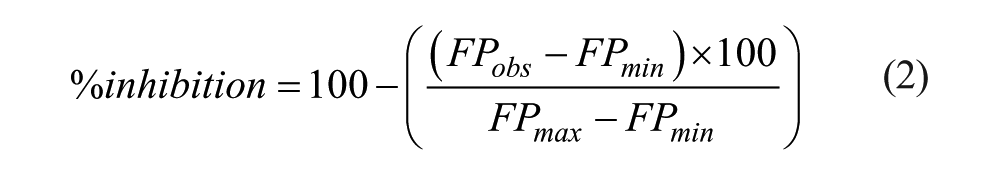
IC50 values were calculated from concentration–response curves by a nonlinear regression analysis at four parameters using GraphPad Prism.
Ki values were calculated as described in reference 17 using the measured IC50 values, the Kd value, and the concentrations of the protein and of the fluorescent peptide in the competitive assay.
Screening Assay
The selected screening conditions are summarized in
Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) Assay
The TR-FRET assay was conducted in black 384-well microplates (Corning, Amsterdam, The Netherlands). Five µL recombinant human Bfl-1 protein (120 nM) was incubated with 5 µL compounds at room temperature for 30 min. Then, 5 µl LanthaScreenTM Tb-anti-GST antibody (Invitrogen) and 5 µL FITC–Bim peptide were added at 20 nM and 40 nM, respectively. Buffer was PBS pH 7.4, 100 mM NaCl, 2.7 mM KCl, with 0.01% Triton X-100™ and 1% DMSO. After 60 min of incubation at room temperature, the plates were read using a Mithras LB 940 multimode microplate reader (Berthold Technologies, Thoiry, France) with a TR-FRET program (excitation at 340 nm, emission for terbium signal at 485 nm, and emission for FITC signal at 520 nm). Minimal TR-FRET value (Rmin) was obtained by incubating FITC–Bim and Tb-anti-GST antibody in the absence of Bfl-1. Maximal TR-FRET value (Rmax) was obtained by incubating Bfl-1, Tb-anti-GST antibody, and FITC–Bim. Gambogic acid (IC50 2 µM) was used as a reference inhibitor at 5 µM and 100 µM in each plate. The TR-FRET signal was expressed as Delta F according to eq 3:
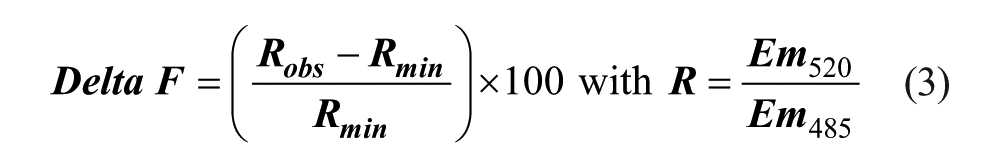
Percentage inhibition and IC50 values were calculated as described for the FP assay.
Bfl-1 Thermal Shift Assay (TSA)
The ligand-induced thermodynamic stabilization of proteins was explored in a TSA. 19 TSA monitors the fluorescence properties of an environmentally sensitive dye, which binds specifically to the hydrophobic surfaces of a target protein as they become exposed during thermal unfolding. Eighteen nL of compound at 10 mM was transferred in LightCycler® 384-well microplates (Roche, Meylan, France) using an Echo liquid handler (Labcytes, Sunnyvale, USA) and incubated with 6 µL of a mixture of Bfl-1 and SYPRO® Orange (Sigma, Saint-Quentin Fallavier, France). Final concentrations were 30 µM, 5 µM, and 10× for compound, protein, and dye, respectively. Buffer was 50 mM tris/HCl pH 7.5, 150 mM NaCl, 2 mM MgCl2, and 1% DMSO. After 30 min incubation at room temperature, the plates were loaded in a Real Time – PCR LightCycler® 480 and heated from 20 °C to 85 °C during a 8-min period. Melting temperatures (Tm) were calculated using the LightCycler® 480 protein melting analysis tool.
Glutathione Stability Experiments
Compounds and L-glutathione were incubated for 1 h at 37 °C under mixing, with final concentrations of 330 µM and 2.5 mM for compound and L-glutathione, respectively. After incubation, chemical analysis was performed by liquid chromatography mass spectrometry (LC-MS) (Alliance-ZQ2000, Waters, Guyancourt, France) under full scan detection (the substrate and the expected glutathione adduct masses were extracted from the full scan spectrum) using an XBridge™ C18 (50 × 4.6 mm, 5 μm) column (Waters). The mobile phase solvents used were (A) H2O 5 mM ammonium formate buffer pH 3.8 and (B) CH3CN 5 mM ammonium formate buffer pH 3.8. The following mobile phase gradient was applied: 2% B for 30 sec, 2% B to 98% B in 6 min, 98% B for 2 min, 98% B to 2% B in 10 sec, and 2% B for 1 min 20 sec. The injection volume was 20 µL and the flow rate of 1 mL/min.
Cell Lines and Products
All media and cell culture reagents were purchased from Life Technologies. BP3, IM9, and peripheral blood mononuclear cells (PBMCs) were cultured in RPMI supplemented with 10% fetal bovine serum, 2 mM glutamine, 10 mM HEPES, and 40 µg/mL gentamycin. PBMCs were collected from healthy donors (Etablissement Français du Sang, Lyon, France). PBMCs were obtained by standard Ficoll density gradient centrifugation. After two washes in PBS, PBMCs were cultured in RPMI with molecules as indicated. Mouse embryonic fibroblasts (MEFs), either wild-type (WT) or bak−/− bax−/− double knockout (DKO), were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 10 mM HEPES, and 40 µg/mL gentamycin. All cells were cultured at 37 °C in humidified atmosphere with 5% CO2.
ABT-737 and obatoclax were purchased from Euromedex (Souffelweyersheim, France), and gambogic acid was purchased from TEBU (Le Perray en Yvelines, France).
Cytotoxicity Assay
Cells were cultured at a density of 5.105 cells/mL with the indicated molecule concentrations. Adenosine triphosphate (ATP) monitoring was performed 24 h after treatment using the ATPlite™ assay (Perkin Elmer, Courtaboeuf, France) according to manufacturer instructions. This assay is based on the measurement of ATP levels, which is a marker for cell viability. Briefly, cells are lysed using the buffer provided with the kit and incubated 5 min with shaking. Then, substrate solution containing luciferase and D-luciferin (reacting with ATP to produce light) is added to the wells, and the plate is incubated 10 min in the dark. Luminescence is thereafter measured on a Tecan (Lyon, France) Infinite M200 plate reader. A luminescent signal is proportional to the number of viable cells in each well. Data are expressed as a percentage of signal compared to untreated wells.
Cell Death Assay
Cells were treated with molecules for 24 h. Cell death was evaluated by propidium iodide and Annexin V double staining (BD Pharmingen, Le Pont de Claix, France), and analyzed by flow cytometry with FlowJo software (TreeStar, Ashland, OR, United States). Viable cells are gated as propidium iodide and Annexin V double-negative cells. Drug combination effects with ABT-737 were measured using the Chou-Talalay method. 20 The resulting combination index (CI) calculated with the CompuSyn software (Paramus, USA) indicates an additive effect (CI = 1), synergism (CI < 1), or antagonism (CI > 1).
The 50% cytotoxic concentrations (CC50) were determined from the dose–response curves (DRCs) and the tumor-specificity (TS) index calculated with eq 4:
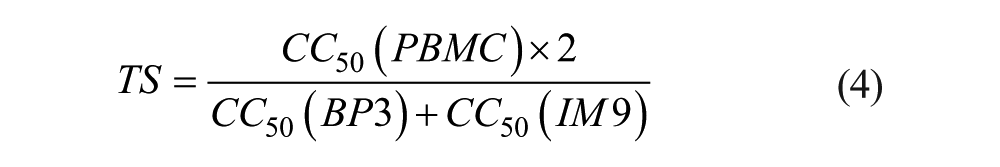
Docking Experiments
The two enantioners of BDM_53787 were docked into the binding pocket of Bfl-1 using the programs MOE-DOCK 2012.10 (Chemical Computing Group Inc., Montreal, Canada) and Autodock 4.2. 21 The structure of Bfl-1 (pdb code 2VM6) was prepared using MOE 2012.12 and the structure preparation protocol with default settings. MGL-Tools 1.5.6 was also used to prepare the protein structure prior to its use with Autodock 4.2. Figures were generated with MOE 2012.10.
The selected complex proposed by Autodock was also investigated within MOE 2012.10, and a full minimization of the complex was carried out. Then, the molecular surface of the protein in the vicinity of the binding site and the electrostatic map were calculated to picture the pocket of Bfl-1 and the polarity of the favored interacting regions with BDM_53787.
Results
Chemical Library Selection and Characteristics
The hierarchical protocol used for compound selection allowed us to prioritize the purchase of 25,000 compounds among the 1,100,307 compounds of the Enamine HTS collection (September 2009). The number of compounds along the selection process and the distribution of some major physicochemical properties for the resulting library are shown in
Development of the Bfl-1 FP Assay
A FP assay was set up to follow the interaction of the recombinant human GST–Bfl-1–ΔCter protein with FITC–Bim BH3. Preliminary experiments with various concentrations of Bim BH3 incubated in PBS buffer showed that reproducible and similar polarization values could be obtained with concentrations going from 15 nM to 50 nM (106 ± 8 mP at 15 nM to 99 ± 2 mP at 50 nM) (
Development of FP Assays for Compound-Selectivity Characterization
To characterize compound selectivity for Bfl-1–Bim interaction, we developed a panel of FP assays focused on Bcl-2 proteins (
HTS to Identify Bfl-1-Targeting Compounds Displaying Cytotoxic Properties
The 25,000 compounds of the library were screened at the concentration of 30 µM for their capacity to disrupt Bfl-1/Bim interaction (
Fig. 1
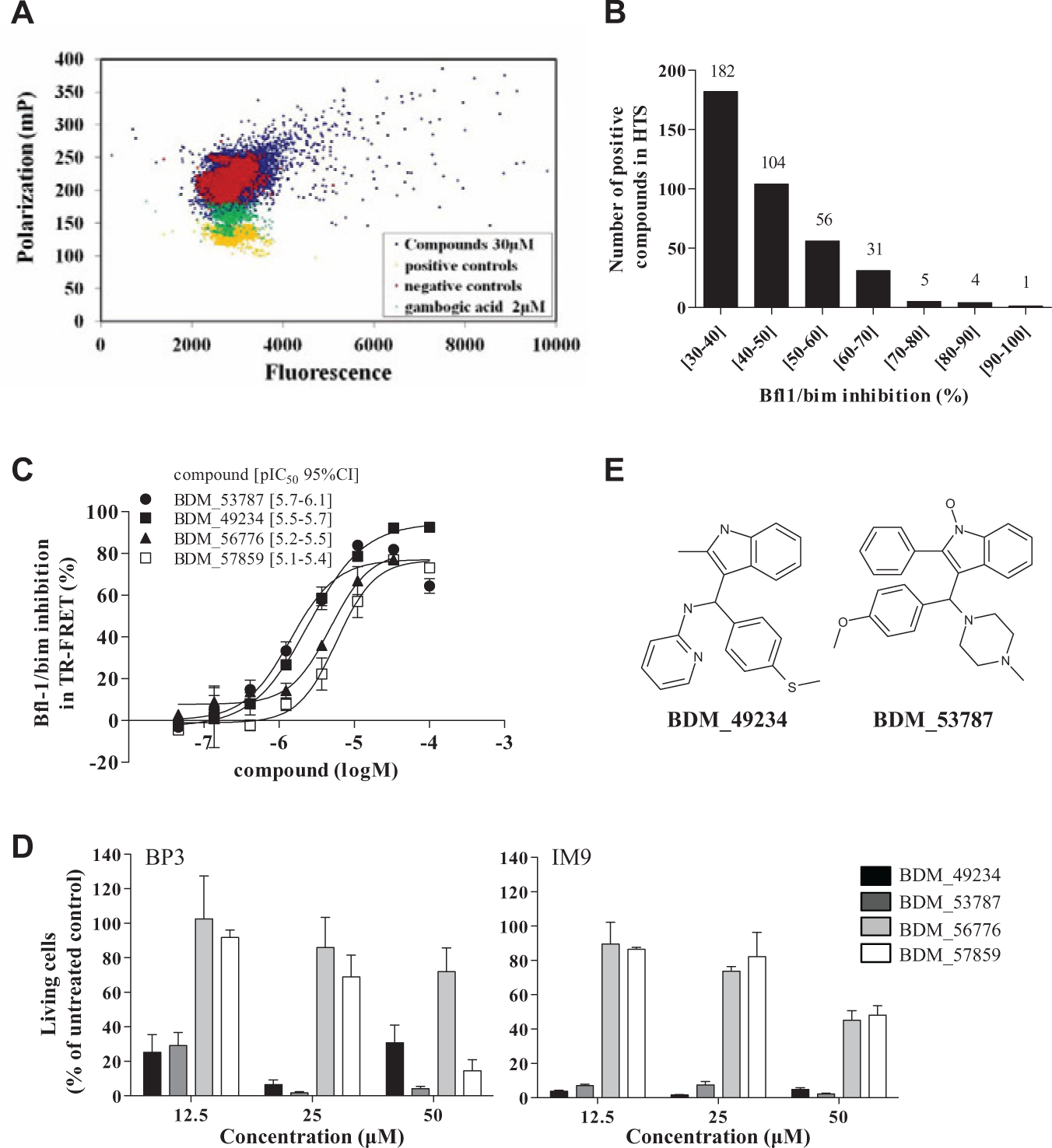
Screening to identify molecules targeting Bfl-1 and displaying cytotoxic properties. Twenty-five thousand compounds were screened at 30 µM to find inhibitors of Bfl-1 (30 nM) binding to Bim BH3 (15 nM). (
Compound Inhibition of Bfl-1 Interaction with Other Pro-Apoptotic Partners
It is known that Bfl-1 interacts with multiple pro-apoptotic proteins, potentially through the same BH3 domain binding site. As shown in
IC50 and Ki Values of BDM_49234 and BDM_53787 in Fluorescence Polarization Assays.
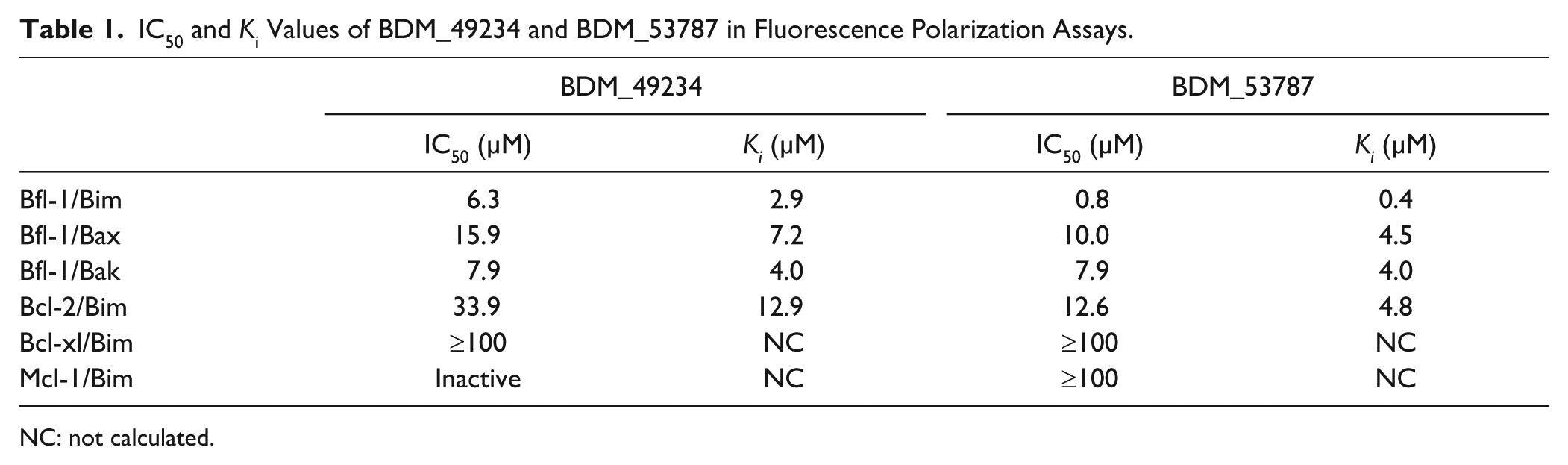
NC: not calculated.
Characterization of Compound Selectivity for Bfl-1 Anti-Apoptotic Protein
We next evaluated BDM_49234 and BDM_53787 for their capacity to inhibit FITC–Bim BH3 binding to Bcl-2, Bcl-xl, and Mcl-1 proteins (
Biological Activity of Selected Compounds toward B-Cell Lymphoma
To further characterize their biological activity, we tested the capacity of compounds BDM_49234 and BDM_53787 to induce apoptosis of B-cell lymphoma lines. For this purpose, BP3 and IM9 cells were cultured for 24 h in the presence of increasing concentrations of compounds BDM_49234 or BDM_53787, and apoptosis was assessed by propidium iodide–Annexin V double staining and flow cytometry analysis (
Pro-apoptotic effect of BDM_49234 and BDM_53787 compounds toward lymphoma cell lines. BP3 and IM9 cells were treated 24 h with increasing concentrations of (
Because competitive FP assays showed that BDM_49234 and BDM_53787 compounds preferentially interacted with Bfl-1, we next tested whether these two compounds could be combined with the selective Bcl-2/Bcl-xl ABT-737 molecule to exert an additive or synergistic pro-apoptotic effect. BP3 and IM9 cells were treated with suboptimal concentrations of ABT-737 and compound BDM_49234 or BDM_53787, and the percentage of viable cells was determined after 24 h (
Pro-apoptotic effect of BDM_49234 and BDM_53787 compounds in combination with ABT-737. (: slight and moderate antagonism (1.10 < CI < 1.45);  : nearly additive (0.90 < CI < 1.10);
: nearly additive (0.90 < CI < 1.10);  : slight and moderate synergism (0.70 < CI < 0.90);
: slight and moderate synergism (0.70 < CI < 0.90);  : synergism (0.30 < CI < 0.70); and
: synergism (0.30 < CI < 0.70); and  : strong and very strong synergism (CI < 0.30).
: strong and very strong synergism (CI < 0.30).
Anti-apoptotic Bcl-2 members suppress apoptosis by inhibiting Bax and Bak pro-apoptotic Bcl-2 members, and cells lacking both Bax and Bak proteins are resistant to apoptotic stimuli that act through Bax- and Bak-dependent disruption of mitochondrial function.
22
To assess the specificity of our anti-apoptotic Bcl-2 family inhibitors for this pathway, we used murine embryonic fibroblasts (MEFs), either wild-type (WT) or Bax–Bak DKO. Similarly to ABT-737, compound BDM_49234 did not induce death of WT or DKO MEFs for concentrations up to 6.25 µM (
As a first approach to assess the off-target toxicity of BDM_49234 and BDM_53787 compounds, we assessed their toxicity toward PBMCs in parallel to their effect on BP3 and IM9 cell lines (
Binding of BDM_53787 and BDM_49234 Compounds to Bfl-1
Direct binding of BDM_53787 and BDM_49234 molecules to Bfl-1 was then explored in a protein TSA. Such an assay is based on the stabilization of a protein conformation when it interacts with a ligand, which is reflected by a shift of the protein melting temperature. As shown in Figure 4A , we observed that both compounds increased the melting temperature of Bfl-1 with ΔTm values of 4.1 and 3.0 for BDM_49234 and BDM_53787, respectively, indicating that they directly bound Bfl-1 protein.
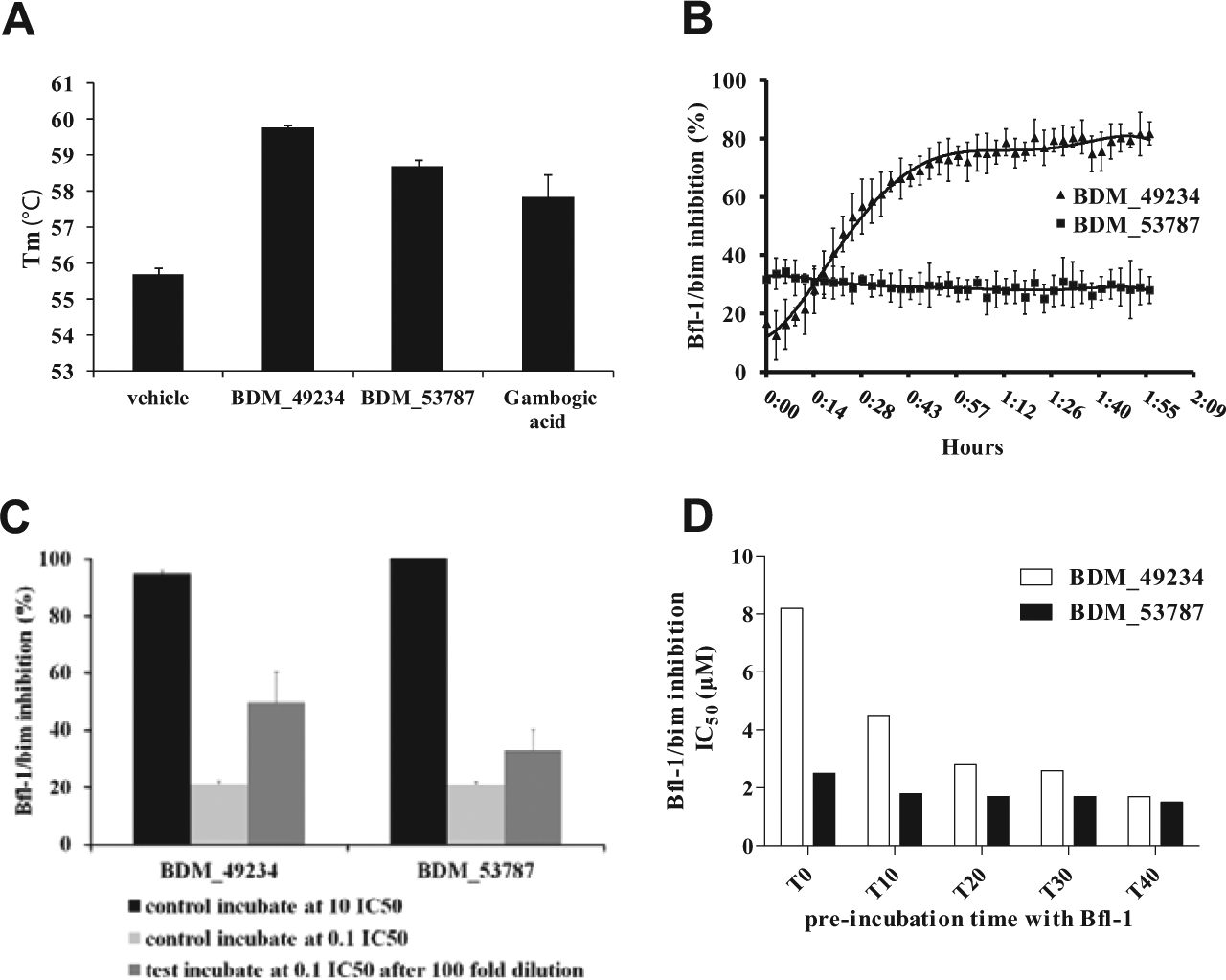
Mechanism of Bfl-1/Bim interaction inhibition by BDM_49234 and BDM_53787. (
Reactivity of BDM_53787 and BDM_49234 Compounds with Gluthathione
Because the screening had provided Bfl-1/Bim inhibitors displaying potentially electrophilic Mannich base functions, we investigated the reactivity of BDM_53787 and BDM_49234 with the thiol group of cysteine (Cys). Indeed, five Cys are present in the human full-length Bfl-1 protein, and the crystal structure of Bfl-1 in complex with Bim had revealed that Trp147 of the amphipathic helix of Bim BH3 stacks onto a surface patch formed by Leu52 and Cys55 in the Bfl-1 groove.
23
We thus incubated the compounds at 330 µM in PBS buffer, pH 7.4, with 2.5 mM excess of glutathione for 1 h at 37 °C, and we followed the compound addition reaction with LC-MS analysis. We observed that BDM_49234 reacts rapidly with glutathione, the glutathione adduct being identified at 4’21, with a mass-to-charge ratio (m/z) of 573 [M – N-pyridine + GSH]+ (
To dig deeper in the mechanism of inhibition of Bfl-1/Bim interaction by BDM_49234 and BDM_53787, we designed two types of experiments to assess the potential reversibility of compounds binding to Bfl-1, and consequently disruption of Bfl-1/Bim interaction. We first measured the inhibition of Bfl-1/Bim interaction throughout time. Incubations were performed with Bfl-1, Bim, and BDM_53787 or BDM_49234, and FP measurements were made at 90 s intervals for 2 h. We observed that the inhibitory effect of BDM_49234 is time dependent. This is consistent with the requirement for a slow but tight binding to the protein. Such an effect was not observed with BDM_53787 ( Fig. 4B ). A second assay was designed to assess the reversibility of compound binding to Bfl-1. This assay measures the reversibility of compound binding on dilution. As shown in Figure 4C , we observed that both compounds incubated at 10-fold their IC50 (in black) and completely inhibit Bim binding to Bfl-1, whereas only 20% of Bim-binding inhibition is observed at 0.1× IC50 (in pale gray). When the same slightly inhibitory concentrations are obtained from a 100-fold dilution of compound pre-incubated at 10× IC50, BDM_49234 showed a significantly higher residual binding to Bfl-1 than BDM_53787. This is consistent with the chemical reactivity of BDM_49234 and its slower binding to Bfl-1. Also, as shown in Figure 4D , the IC50 determined for BDM_49234, but not for BDM_53787, is heavily dependent on the pre-incubation time.
Docking of BDM_53787 Compound into the Bfl-1 Binding Pocket
For the docking procedure, we decided to only dock compound BDM_53787. The main reason is that because of its putative transient link to the Cys55 of Bfl-1, only the reaction product of BDM_49234 binds to Bfl-1, but not the parent compound. Although some docking programs, including Autodock 4.2, now allow docking of covalently bound ligands, we cannot rule out that the reactivity of this compound might involve another residue than Cys55. As such, it would be very speculative to propose a binding mode for this molecule bound to Cys55. On the contrary, the two enantiomers of BDM_53787 were docked into the binding pocket of Bfl-1 using two different programs (MOE-DOCK and Autodock 4.2). 21 In addition, to assist the selection of the binding pose of BDM_53787, the crystal structure of Bfl-1 in complex with the interacting α-helix of Bim (pdb code 2VM6) was used to predict the hotspot residues of Bim interacting with the binding pocket of Bfl-1 using the virtual alanine scanning DrugscorePPI server. 24 Several hotspot residues were predicted, including Bim-Ile148, Bim-Leu152, and Bim-Arg153, with predicted ΔΔG superior to 1.5 kcal.mol−1. The selection of the BDM_53787 docking pose was therefore made with the attempt to mimic the binding mode of Bim with regard to these three hotspot residues.
The proposed binding mode of BDM_53787 was selected among the Autodock poses of the docked ligand and corresponds to the enantiomer R of BDM_53787 ( Fig. 5A ). In this binding mode, the phenyl moiety of BDM_53787 occupies the hydrophobic subpocket of Bim-Leu152, the 4-methoxyphenyl moiety occupies the hydrophobic subpocket of Bim-Ile148, and the piperazine moiety occupies a region of the Bfl-1 binding pocket compatible with the type of interaction that Bim-Arg153 makes with Bfl-1, including a salt bridge with residue Glu80 of Bfl-1 ( Fig. 5B ).
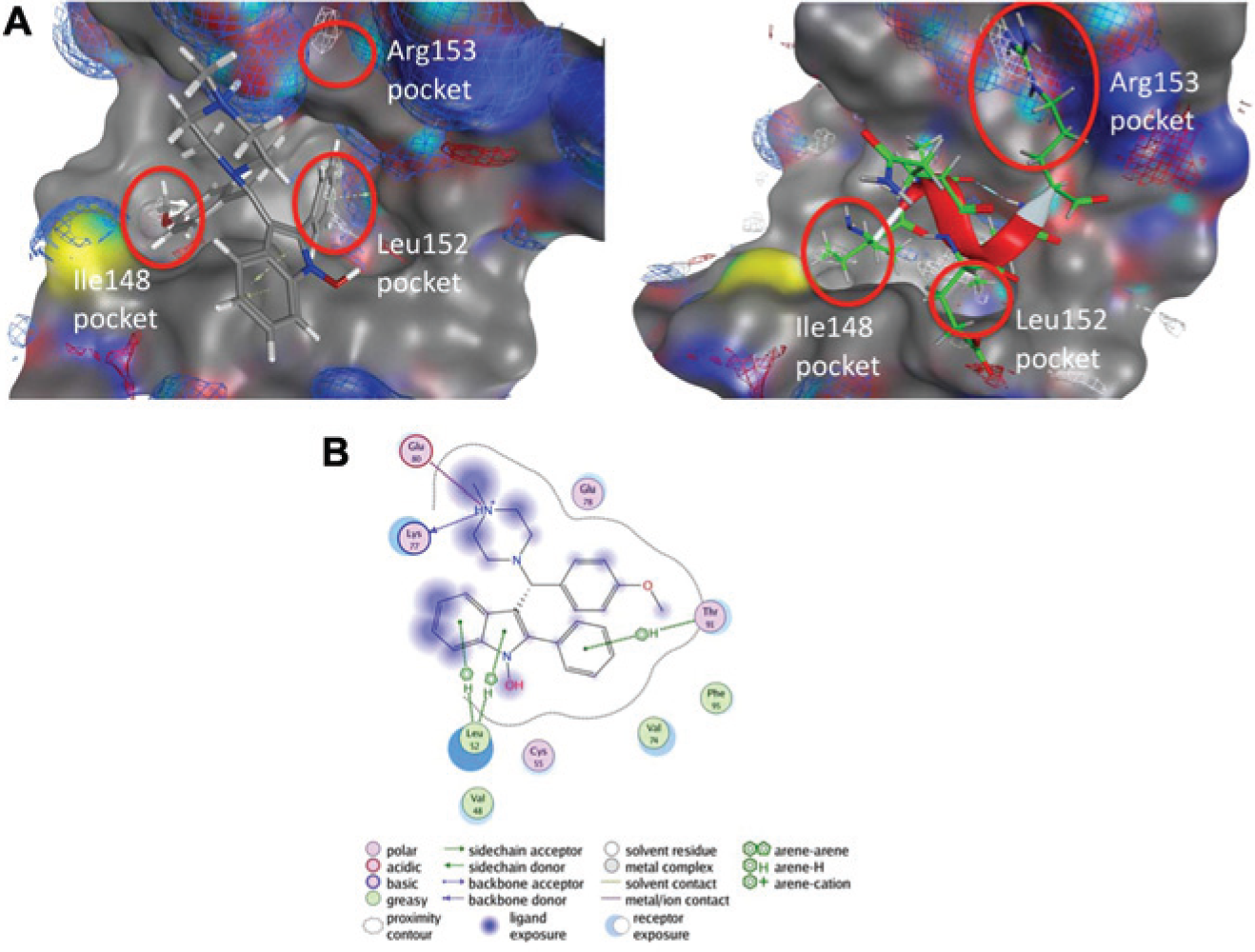
Docking of BDM_53787 compound into Bfl-1 binding pocket. (
Discussion
Novel small compounds targeting Bcl-2 and/or Bcl-xl anti-apoptotic protein of the Bcl-2 family such as navitoclax, an orally bioavailable derivative of ABT-737, or ABT-199 are now in clinical trials.3–5 It has been demonstrated that long-term exposure to ABT-737 results in increased levels of Bfl-1 and Mcl-1, and in the development of resistance in lymphoma cell lines that were initially sensitive. 6 Furthermore, the use of a highly sensitive and quantitative reverse transcription polymerase chain reaction assay to examine Bcl-2, Bcl-xl, Bcl-w, Bcl-b, Mcl-1, and Bfl-1 defined the (Bfl-1 + Mcl-1)/Bcl-2 ratio as the most predictive marker for the response of primary B-CLL and leukemic cell lines to ABT-737. 7 Thus, the efficacy of anti-Bcl-2 and Bcl-xl compounds is limited by the expression of Mcl-1 or Bfl-1, underlying the need for Mcl-1 and/or Bfl-1 chemical inhibitors. Based on the strong association between chemoresistance and the overexpression of Bfl-1 in DLBCL and B-CLL, in which it stands as an innovative target,13–16 we focused on the identification of Bfl-1 inhibitors. We described here two compounds that target the Bfl-1 hydrophobic BH3-binding groove and thereby efficiently disrupt its interaction with pro-apoptotic partners, such as Bim but also Bax and Bak. Both compounds inhibit Bfl-1 protective activity and promote cell death of malignant B cells, either alone or in combination with ABT-737. The synergistic effect of those compounds with ABT-737 is of particular interest, because Bfl-1 overexpression has been clearly identified as a limit to its efficacy.
The 25,000-compound chemical collection was designed mainly to favor chemical diversity. To this end, the choice of soft physicochemical thresholds like those described in the “Materials and Methods” section are inspired by several studies showing that indeed inhibitors of protein–protein interactions, and more precisely those of the Bcl-2 family, are characterized by significantly higher molecular weight and hydrophobicity,25,26 thus standing in sharp contrast with chemistry rules such as Lipinski’s RO5 and Veber’s rules.27,28 The other important step in the selection process deals with the detection of some problematic chemical moieties usually associated with toxicity or the observation of false positives or frequent hitters such as Michael acceptors or epoxides. This selection criterion was responsible for removing a more important number of compounds (~240,000 compounds).
Screening of such a library provided three structurally different series of Bfl-1/Bim inhibitors, among which two contained Mannich base function (Ar–CHR–NR1R2), in which the amine fragment can be substituted by a sulfur nucleophile. Bfl-1 contains at position 55 a Cys residue close to the binding groove, which could explain why we and others, using a similar screening approach (FPA using Bfl-1 and Bid BH3 domain, followed by a secondary TR-FRET assay)29,30 or through a multiplex flow cytometry–based HTS (using Bfl-1 and Bim BH3 domain), 31 described electrophilic Bfl-1 inhibitors. The indol series was progressed and tested in different cell lines. Positioning of the ligand in the groove is a prerequisite for activity: in the Mannich base family described here, one of the compounds is not thiol reactive and behaves as a reversible inhibitor, whereas its close analog is thiol reactive and a partially reversible binder. The reversible compound (BDM_53787) was more potent in the cell-free and cell-based assays, and molecular modeling clearly supports the experimental work and proposes a likely binding mode of Bfl-1. The partially reversible compound (BDM_49234) was less potent, but it was also less toxic, suggesting that chemical reactivity and irreversibility do not necessarily imply a nonspecific cell toxicity.
The thiol reactivity should not be by itself a reason to discard a hit before medicinal chemistry efforts to improve potency and explore chemical reactivity are done. In this context, careful attention should be given to incubation times when measuring activities on target and comparing compounds with very different kinetic properties. Later in development, at equivalent affinity to the target, binding kinetics and pharmacokinetics should be jointly analyzed: slow tight binders may be of interest if the Cmax and elimination rates are high. Therefore, having in hand compounds with various binding kinetics early in the drug discovery is favorable.
Most of the inhibitors that have been identified for the Bcl-2 family are characterized by a physicochemical profile that places them outside of the usual drug-like chemical space [i.e., with a higher molecular weight (>600 g/mol) or a higher lipophilicity (logP > 5–6)]. This can hamper their potential as clinically useful drugs, particularly if they are meant to be orally administrated. The present study demonstrates that it is indeed possible to identify inhibitors of Bfl-1 that possess reasonable physicochemical properties. As matter of fact, compounds BDM_49234 and BDM_53787 are both characterized by acceptable size (~400 g/mol) and lipophilicity (logP ~4).
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by institutional grants from INSERM, University of Lyon (to NB), University Paris Diderot (to BV), University of Lille 2 (to BD), and Institut Pasteur de Lille (to BD), and by a specific grant from the Association pour la Recherche sur le Cancer (ARC) (to NB, BV, and BD). The purchase of the 25,000-compound Enamine library was made possible thanks to funding from the French National Research Agency (ANR) Piribio Program PPIFocusDb (ANR-09-PIRI-0017) Edition 2009 du Programme Physique et Chimie du Vivant.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.