
Abstract
The p53 tumor suppressor is a potent transcription factor that regulates cell growth inhibition and apoptosis. The oncoprotein MDM2 suppresses p53 activity by direct inhibition of its transcriptional activity and enhances the degradation of p53 via the ubiquitin–proteosome pathway. Overexpression of MDM2, found in many human tumors, impairs p53-mediated cell death effectively. Inhibition of the p53–MDM2 interaction can stabilize p53 and may offer a novel strategy for cancer therapy. To search for new inhibitors of the p53–MDM2 interaction, the authors developed a cell-based high-throughput assay system based on mammalian two-hybrid technology. They also used a dual-luciferase reporter system to rule out false- positive hits due to the cytotoxic effect of compounds. Using this assay, they screened a library consisting of 3840 compounds and identified one compound that activates p53 pathway and induces growth arrest in tumor cells.
Introduction
T
Tight regulation of p53 function is critical for normal cell growth and development, and one mechanism by which p53 function is controlled is through interaction with the MDM2 protein. MDM2 is an oncoprotein, discovered by its overexpression in a spontaneously transformed mouse cancer cell line. 3 MDM2 suppresses p53 function either by binding to its transactivation domain and blocking its ability to activate transcription 4 or by enhancing the nuclear export and degradation of p53 via the ubiquitin–proteosome pathway. 5 Blocking interaction between p53 and MDM2 should lead to the accumulation of p53 and lead to cell growth arrest and apoptosis. So inhibitors of the p53–MDM2 interaction are considered attractive new anticancer agents by activating the p53 pathway in tumors expressing wild-type p53. 6
Many approaches have been employed to design and identify the small-molecule inhibitors of the MDM2–p53 interaction, including computational three-dimensional (3D) database screening, experimental screening of chemical libraries, and structure-based de novo design.7–10 For experimental high-throughput screening (HTS), in vitro methods such as fluorescence polarization–based binding assay, enzyme-linked immunosorbent assay (ELISA)–based assay, and reverse mammalian protein–protein interaction trap (MAPPIT) assay have been reported.11–13 The reverse MAPPIT assay is a well-designed bait-and-prey system with an inhibitory motif attached to the prey protein, which allows a positive readout upon interference with the designated protein–protein interaction. But the detection is dependent on the signal transduction of cytokine receptor and the JAK pathway. Anything that interferes with this signaling pathway would also affect the readout of the assay. In this aspect, a forward two-hybrid system with direct activation of the reporter gene would be more reliable.
The discovery of small molecules termed nutlins represents a breakthrough in the design of the potent small-molecule MDM2 inhibitor. 12 The nutlins are the first examples of potent and specific inhibitors of the p53–MDM2 interaction, and one of them, nutlin-3, can inhibit proliferation of tumor cells expressing wild-type p53.12,14 In an osteosarcoma cell line having MDM2 amplification (SJSA-1), nutlin-3 induces apoptosis and has an inhibitory effect in xenografts similar to doxorubicin. Another MDM2–p53 interaction inhibitor, MI-219, stimulates rapid but transient p53 activation in established tumor xenograft tissues, resulting in complete tumor growth inhibition. It activates p53 with minimal p53 accumulation in normal tissues and is not toxic to animals. 14 Analogs of nutlin-3 and MI-219 have progressed to advanced preclinical development or early-phase clinical studies. 15
Trying to identify inhibitors of the p53–MDM2 interaction in a more physiological condition, we developed a cell-based high-throughput assay system based on mammalian two-hybrid technology. A dual-luciferase reporter system was incorporated to rule out the cytotoxic effect of compounds. Using this assay, we screened a library consisting of 3840 compounds and eventually identified one compound that activates the p53 pathway and induces growth arrest in tumor cells.
Materials and Methods
Cell culture
HEK293, U2OS, and MDA-MB-435 cells were obtained from American Type Culture Collection (ATCC; Manassas, VA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) nutritional medium supplied with 10% fetal bovine serum (FBS), 100 mg/L penicillin, and 100 mg/L streptomycin at 37 °C in a humidified atmosphere of 5% CO2. For transient transfection, about 1×106 cells were mixed with 2 to 4 µg plasmids in 200 µL transfection buffer, and electroporation was carried out with a Scientz-2C electroporation apparatus (Scientz Biotech, Ningbo, China).
Mammalian two-hybrid assays
Full-length cDNAs encoding human p53 and MDM2 were cloned into the pCMV-AD and pCMV-BD vectors (Stratagene, La Jolla, CA) in-frame with AD or BD at the N terminus. The authenticity of the DNA sequences was confirmed by sequencing. For single-luciferase analysis, cells were co-transfected with pCMV-AD-p53 (or -MDM2), pCMV-BD-MDM2 (or -p53), and the pFR-Luc firefly luciferase reporter plasmid at a ratio of 1:1:1. For dual-luciferase analysis, the pRL-TK plasmid encoding a renilla luciferase was included as an internal control. After transfection, cells were seeded onto the 96-well plate at a density of 3×104 cells per well. Sixteen hours after transfection, compounds at various concentrations were added. DMSO at 1% was used as negative control. Another 24 h later, luciferase activities were measured using the Dual-Glo Luciferase system (Promega, Madison, WI) and EnVision (PerkinElmer, Waltham, MA) multiplate reader according to the manufacturer’s instructions.
Pilot screen of 3840 compounds
The compound library used for the screening was composed of 3840 compounds (provided by Chinese National Compound Resource Center, Shanghai, China). Compounds were screened at a concentration of 20 µM. In each 96-well plate, eight wells were used as positive controls (10 µM nutlin-3 in 1% DMSO) and another set of eight wells as negative controls (1% DMSO). The luciferase activity was normalized to 100% and 0 in the wells treated with DMSO and nutlin-3, respectively. The compounds causing the luciferase activity to reduce to less than 30% were considered “hits” in the primary screening.
Western blot
Cells were treated for 48 h with 10 µM nutlin-3 or other compounds. After removal of the culture medium, the cells were lysed with sample buffer (62.5 mM Tris-HCl, 2% sodium dodecyl sulfate [SDS], 10% glycerol, 50 mM dithiothreitol, and 0.01% bromophenyl blue, pH 6.8). After immediate scraping, the lysates were sonicated, and aliquots of samples were electrophoresed with 12% SDS–polyacrylamide gel electrophoresis (PAGE) gels. Then the proteins were transferred onto PVDF membranes (Millipore, Billerica, MA). After blocking, the membranes were probed with primary antibodies against MDM2 (SMP-14), p21 (SX118), p53 (FL-393), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) at 4 °C overnight. Next day, the membranes were incubated with anti-IgG antibodies conjugated to horseradish peroxidase (HRP) for 1 h at room temperature. After the final wash, the signal was developed with the ECL plus reagents (GE Healthcare, Piscataway, NJ), and images were captured and analyzed with the ChemiDoc Imaging Station (Bio-Rad, Hercules, CA).
MTT assay
Cancer cells with wild-type p53 (U2OS) or mutant p53 (MDA-MB-435) were seeded onto the 96-well plate at a density of 4000 cells/well. Twenty-four hours later, compounds at various concentrations were added to the cell culture. The culture medium with compounds was changed every other day. Five days later, cell growth was measured with an MTT kit according to manufacturer’s instruction (Beyotime, Haimen, China). The ratio (test/control) of the optical density at 570 nm was described as the cell proliferation value.
Coimmunoprecipitation
U2OS cells were lysed in lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 0.1% Chaps, 1 mM EDTA containing 1 mM NaF, 1 mM Na3VO4, and protease inhibitors) by sonication for 30 s on ice. Crude lysate was centrifuged at 13 000 rpm for 15 min at 4 °C and the supernatant was incubated with anti-MDM2(SMP14) antibody for 5 h. Then, protein A agarose (sc-2001; Santa Cruz Biotechnology, Santa Cruz, CA) was added and coincubated with gentle rocking at 4 °C overnight. The affinity gel was then collected by centrifugation and washed with lysis buffer six times. The protein complex was eluted with SDS sample buffer (62.5 mM Tris-HCl [pH 6.8], 3% SDS, 15% glycerol, 2% β-mercaptoethanol) and resolved by SDS-PAGE. Western blotting was performed as mentioned above.
Data analysis
Data were analyzed with GraphPad Prism software (GraphPad, San Diego, CA). Nonlinear regression analyses were performed to generate dose–response curves and calculate IC50 values.
Results and Discussion
Development of a cell-based assay for the detection of p53–MDM2 interactions
Using Stratagene’s mammalian two-hybrid system, we developed a cell-based assay for the measurement of the p53–MDM2 interaction. As shown in Figure 1a , full-length p53 or MDM2 were inserted at the C-terminus of the DNA binding domain (BD) of GAL4 or the transcriptional activation domain (AD) of NFκB. The interaction of p53 and MDM2 will bring the BD and AD in proximity and activate the downstream firefly luciferase reporter gene. Sometimes the fusion of extra domain interferes with the natural-occurring protein–protein interaction, so we tested different combinations of p53 and MDM2 tagged with AD or BD domain. The interaction between pBD–p53 and pAD-SV40T was used as a positive control, and the pair of pBD–p53 and pAD–TRAF was used as a negative control. The result indicated that pAD–p53 and pBD–MDM2 had strong interaction, whereas there was no interaction between pBD–p53 and pAD–MDM2 ( Fig. 1b ). To make sure that all the differences we observed were not due to variation in transfection efficiency or cell numbers, we included pRL–TK ( Fig. 2c ), a plasmid encoding a renilla luciferase under the control of the herpes simplex virus thymidine kinase promoter, as an internal control. The firefly luciferase activity was normalized to the renilla luciferase activity ( Fig. 2d ), and the pair of pAD–p53 and pBD–MDM2 still displayed the best interaction. So we decided to further validate the assay using the combination of pAD–p53 and pBD–MDM2 plasmids.
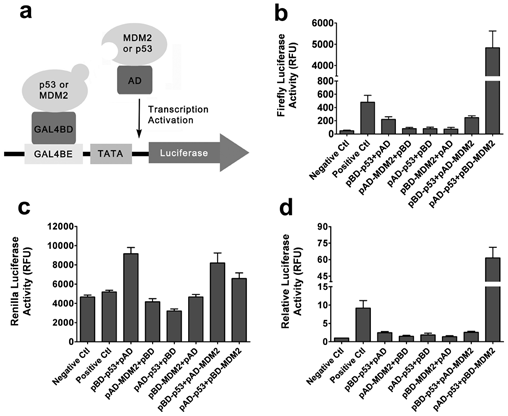
Detection of the p53–MDM2 interaction with the mammalian two-hybrid system. (
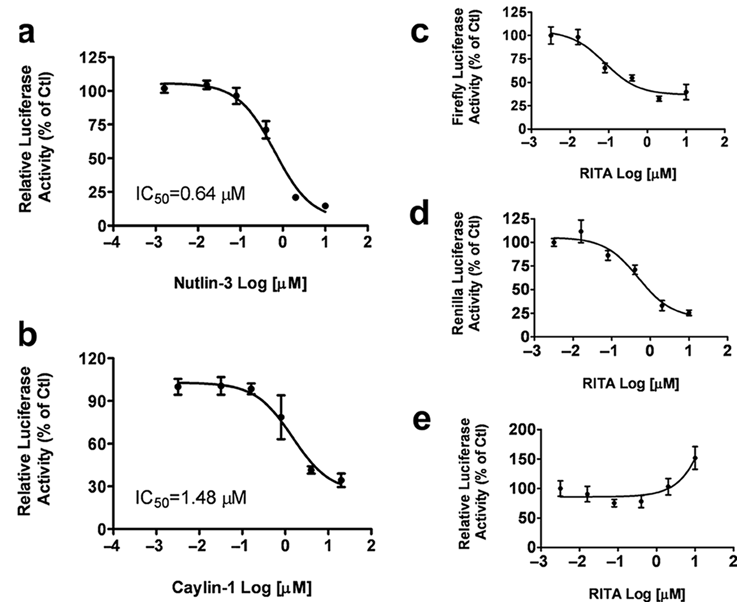
Inhibition of the p53–MDM2 interaction by nutlin-3 and caylin-1 but not RITA. Cells transfected with the mammalian two-hybrid constructs and the two luciferase reporters were treated with various compounds for 24h. (
Validation of the mammalian two-hybrid assay with known compounds
Nutlin-3, RITA, and caylin-1 (a nutlin-3 analog) are small molecules that have been reported to inhibit the interaction between p53 and MDM2,12,16so we tested these compounds in our system. Nutlin-3 and caylin-1 showed dose-dependent inhibition of firefly luciferase activity but had no effect on renilla luciferase activity. After normalization, the IC50 values of nutlin-3 and caylin-1 were approximately 0.64 µM and 1.48 µM ( Fig. 2a , b ), respectively, corresponding well with previous reports. 12 However, RITA not only blocked the firefly luciferase expression ( Fig. 2c ) but also significantly inhibited the expression of renilla luciferase ( Fig. 2d ). This indicates that the reduction of firefly luciferase expression by RITA is not due to the blockade of the p53 and MDM2 interaction but more likely is caused by a nonspecific cytotoxic effect of this compound. This agrees with a previous study that indicated that RITA does not block the formation of the complex between p53 and MDM2 in vitro. 17 These results demonstrate the value of the pRL–TK plasmid as an internal control reporter in the mammalian two-hybrid system to rule out certain false-positive compounds with cytotoxicity.
A pilot screen of 3840 compounds with the mammalian two-hybrid system
To identify compounds that inhibit interactions of p53–MDM2, we performed a small-scale screen of a library consisting of 3840 compounds with the two-hybrid system. To speed up the process, the initial screen was carried out with the single firefly luciferase reporter. DMSO (1%) was used as a negative control, and nutlin-3 (10 µM) was included as the positive control. Compounds were tested in duplicates at a concentration of 20 µM ( Fig. 3a ). The Z′ value is used to assess the robustness of an assay for screening and is the normalized 3 SD window between the negative and positive controls. The Z′ value for the assay was 0.57, and the signal-to-background ratio was 3.85, indicating that the system was adequately optimized for HTS ( Fig. 3b ). The compounds that reduced the firefly luciferase activity to less than 30% were picked and further validated with a dual-luciferase assay in quadruplicates ( Fig. 3c ). Finally, six compounds displaying consistent inhibitory effects were further tested to generate dose–response curves. The IC50 values of these compounds ranged from 1.18 to 6.82 µM ( Fig. 3d – i ).
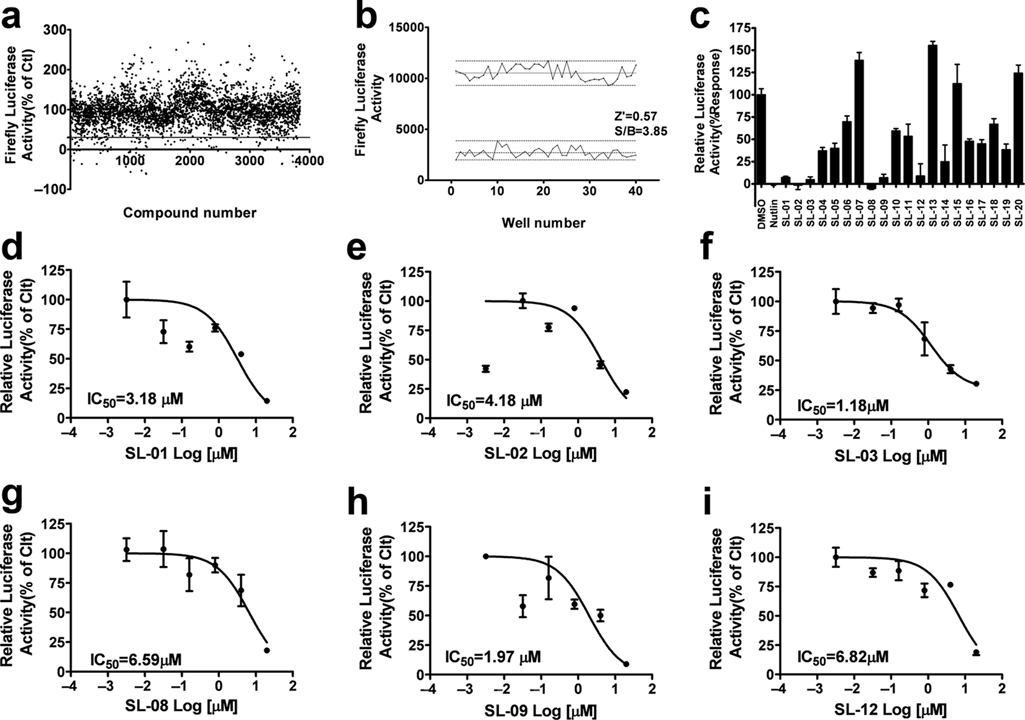
Results of a pilot screen of 3840 compounds with the mammalian two-hybrid system. (
Activation of the p53 pathway by compounds that block p53–MDM2 interaction
MDM2 is an E3 ubiquitin ligase known for its prominent role in regulating p53 degradation. 4 The p53 pathway is believed to be activated after the inhibition of the p53–MDM2 interaction in cells with wild-type p53,1,18 so we examined the effect of the p53–MDM2 interaction inhibitors identified with the two-hybrid system on the cellular levels of p53. U2OS cells with wild-type p53 were incubated with various compounds for 48 h. As shown in Figure 4a , nutlin-3, caylin-1, and three hit compounds (SL-01, 02, and 03) led to a significant increase of the p53 protein level. We also found that in U2OS cells, SL-01 and SL-02 caused the accumulation of p53 in a dose-dependent manner. They were also able to stimulate the expression of p21, a major transcription target of activated p53 19 ( Fig. 4b ). The expression of MDM2 is also regulated by p53.1,12 Nutlin-3 induced a significant upregulation of MDM2, but our compounds only displayed a very weak effect on MDM2 expression ( Fig. 4b ). In the p53 mutant cell line MDA-MB-435, SL-01, and SL-02 did not cause any change in p53, p21, and MDM2 protein levels, which is similar to the known p53–MDM2 interaction inhibitor nutlin-3 ( Fig. 4c ).
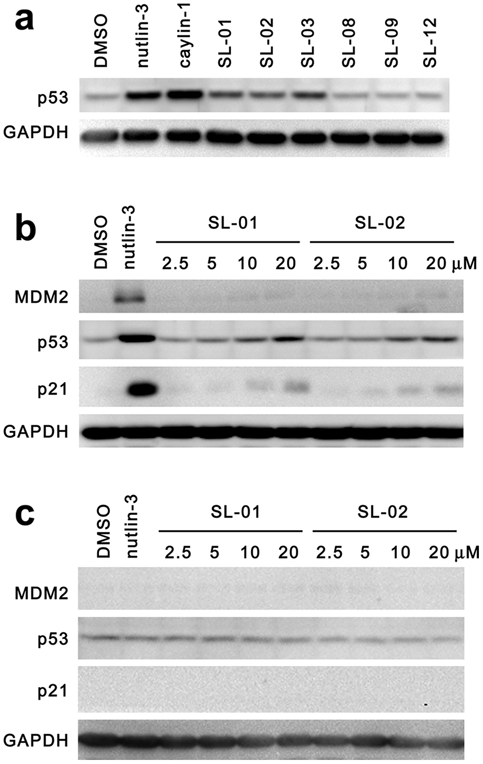
Activation of the p53 pathway by compounds that block p53–MDM2 interaction. Six compounds that displayed dose-dependent inhibition of the p53–MDM2 interaction were picked and analyzed in a Western blot analysis. (
We also used coimmunoprecipitation assay to test the direct inhibition of the p53–MDM2 interaction by SL-01 ( Fig. 5a ). Because the inhibition of the p53–MDM2 interaction with compounds causes accumulation of p53 and MDM2 in cells 12 ( Fig. 5a ), we limited the amount of anti-MDM2 antibody used for the immunoprecipitation, so the antibody was saturated with MDM2 binding and the amount of MDM2 precipitated was controlled at the same level, even though the total MDM2 levels in the lysates were different. Similar as previously reported, nutlin-3 treatment caused significantly less p53 to coimmunoprecipitate with MDM2. Our compound SL-01 displayed a very similar effect in inhibiting the p53–MDM2 interaction ( Fig. 5a ). The structure of SL-01 is shown in Figure 5b . SL-01, a Z-L-Phe chloromethyl ketone, has been reported to be a bovine chymotrypsin A–γ inhibitor. 20 Its inhibitory effect on the p53–MDM2 interaction has never been reported.
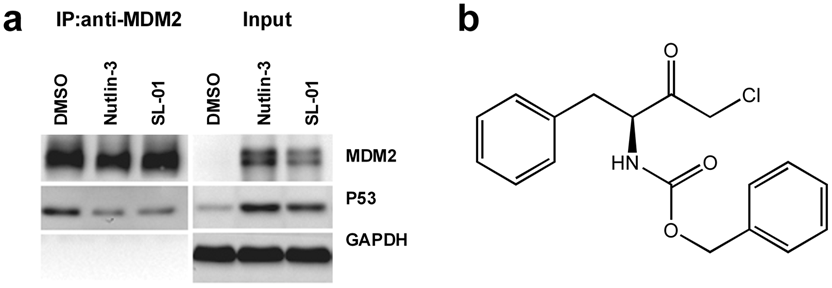
SL-01 blocks the intracellular p53–MDM2 interaction in direct binding assay. (
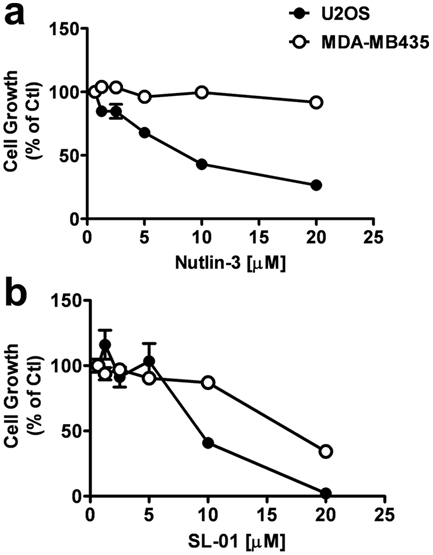
Inhibition of cell growth by compound that blocks p53–MDM2 interaction. U2OS and MDA-MB-435 cells were treated with various concentration of nutlin-3 (
Activation of the p53 pathway causes cell cycle arrest, which leads to cell growth inhibition and/or programmed cell death.12,14 In agreement with previous reports, 12 nutlin-3 potently inhibited cell growth on U2OS (wild-type p53) cells but had no effect on MDA-MB-435 (mutant p53) ( Fig. 6a ). SL-01 also induced a significantly different growth inhibition profile on cancer cell lines according to their p53 status ( Fig. 6b ). Taken together, these data suggest that the compounds identified by the two-hybrid system indeed block the interaction between MDM2 and p53, which leads to the activation of the p53 pathway and induces cell growth arrest in tumor cells with wild-type p53.
We hereby present a two-hybrid assay for HTS of inhibitors of the p53–MDM2 interaction in mammalian cells. With this assay, we screened a library consisting of 3840 compounds and identified six potential inhibitors of the p53–MDM2 interaction. Further analysis revealed that three of these compounds could induce the accumulation of p53 and activated p53-mediated transcription. With a cell growth inhibition assay, we confirmed that SL-01 is a true functional inhibitor of the p53–MDM2 interaction and induced growth arrest in tumor cell lines with wild-type p53. This compound is an ideal starting point for structural modification and warrants further investigation. Thus, the mammalian two-hybrid technology appears to be an efficient cell-based screening strategy for identifying small-molecule inhibitors of the p53–MDM2 interaction. Compared with previous approaches employed in the design and discovery of p53–MDM2 interaction inhibitors,7–11 this assay was performed in a more physiological environment and provided a more efficient way to discover p53–MDM2 inhibitors, although developing a stable cell line is highly desirable to further improve assay stability and throughput. In addition, this assay can also serve as a general strategy for the discovery of protein–protein interaction inhibitors.
Footnotes
Acknowledgements
This project was supported by grants from the National Natural Sciences Foundation of China (90713047), the Ministry of Science and Technology of China (2009ZX09302-001, 2009CB940900), and Shanghai Commission of Science and Technology (08DZ2291300, 09DZ2291200).