
Abstract
We applied a novel profiling approach using receptor binding domain (RBD) ligands to cell surface domains of a panel of nutrient transporters to characterize the impact of a number of tyrosine kinase inhibitor anticancer drugs on human stem cell–derived cardiomyocytes. High-content screening and flow cytometry analysis showed diagnostic changes in nutrient transporter expression correlating with glycolysis and oxidative phosphorylation–based cell metabolism in glucose and galactose media. Cluster analysis of RBD binding signatures of drug-treated cells cultured in glucose medium showed good correlation with sensitization of mitochondrial toxicity in cells undergoing oxidative phosphorylation in galactose medium. These data demonstrate the potential for RBD ligands as profiling tools to improve the clinical predictivity of in vitro cell assays for drug toxicity.
Introduction
There is increasing evidence that nutrient transporters in general, and solute-linked carriers (SLC) 1 in particular, are key players in cell metabolism. These proteins play a pivotal role in cell homeostasis and in metabolic adaptation and regulation induced by, or required for, a wide range of cellular processes. It has been shown that SLC are directly involved in cell differentiation,2,3 transformation, 4 proliferation,5,6 development, 7 and viral infection and pathophysiology.8,9 Analysis of these regulators of nutrient fluxes therefore offers great opportunities to monitor cell metabolism and physiology under normal or perturbed conditions.
Until recently, quantification of nutrient transporter expression has relied on measurement of mRNA expression, as monoclonal antibodies targeting the highly conserved and poorly immunogenic extracellular epitopes of SLC are not available.10,11 As mRNA analysis does not necessarily indicate protein abundance and because nutrient transporter activities depend on their subcellular localization, accurate and detailed analysis of changes in SLC cell surface expression associated with modulation of cell metabolism requires measurement of the functional pool of transporters expressed at the cell surface. 12 Development of novel nonimmunoglobulin SLC ligands, termed receptor binding domains (RBD), derived from envelope glycoproteins evolved across a range of phylogenetically related retroviruses that use SLC to interact with host cells,13–16 provides unique tools to probe this elusive area of cell biology and to further explore and understand the physiological roles of nutrient transporters.17,18
We have used profiling with a panel of seven RBD ligands to characterize the impact of a range of anticancer tyrosine kinase inhibitors on the metabolism of human stem cell–derived cardiomyocytes. Cardiac tissue is metabolically flexible, shifting the balance of substrate utilization to maintain the production of adenosine triphosphate (ATP) necessary to maintain contractile function under normal and stressed conditions. Cardiomyocytes are extremely rich in mitochondria, 19 and the direct or indirect action of anticancer drugs, including kinase inhibitors, on mitochondria has been identified as a key contributing factor in cardiotoxicity in oncology therapeutics 20 and in other drugs. 21 It is therefore important to take account of the variable mitochondrial metabolism of the heart as a “metabolic omnivore” 22 when assessing the potential cardiotoxicity of new drugs. Cells cultured in glucose culture media generate ATP predominantly by glycolysis (the Crabtree effect) and consequently may be partially or wholly resistant to mitochondrial mechanisms of toxicity. 23 Culture of cells in galactose media shifts cellular metabolism to oxidative phosphorylation (OXPHOS), more reflective of cellular metabolism in vivo, and we 24 and others 25 have previously shown that the cardiotoxicity of certain anticancer drugs affecting mitochondrial metabolism and bioenergetics can be increased very significantly in cells using OXPHOS ATP generation.
To fully access the investigative possibilities offered by RBD ligands, it is of prime importance to generate assay protocols dedicated to a wide range of applications. In this study, we have adapted existing flow cytometry protocols 12 to develop a high-content screening assay for profiling nutrient transporter expression in live human stem cell–derived cardiomyocytes in 384-well plates and profiled the metabolic impact of kinase inhibitors on cardiomyocytes cultured in glucose and galactose media.
Materials and Methods
Production of RBDs
IgG-Fc–tagged RBD fusion proteins were produced as follows. H2.RBD.mFc, Ko.RBD.mFc, and RD114.RBD.mFc ligands were derived from the sequences encoding the first 178, 252, and 223 amino acids of HTLV-2, KoRV, and RD114 Env, respectively. The corresponding RBD-encoding sequences were fused to the mouse IgG1-Fc fragment (mFc). A.RBD.rFc, Xeno.RBD.rFc, FeLVC.RBD.rFc, and PERVA.RBD.rFc ligands were derived from the sequences encoding the first 243, 293, 232, and 309 and amino acids of Amphotropic-MLV, Xenotropic-MLV, FeLVC, and PERVA Env, respectively. The corresponding RBD-encoding sequences were fused to the rabbit IgG1-Fc fragment (rFc).
All constructs were inserted into the eukaryotic pCSI expression vector
26
and transfected into 293T cells by calcium phosphate precipitation. Cells were washed 16 h later and incubated for a further 48 h in serum-free Optipro SFM medium (Life Technologies, Carlsbad, CA) supplemented with glutamine and nonessential amino acids. Conditioned media were harvested, filtered through 0.45 µm filters and concentrated 100-fold by centrifugation at 3500 × g on 10 kDa cutoff Centricon Plus-70 units (Millipore, Billerica, MA). Samples were aliquoted and stored at −80 °C. Each preparation was verified for integrity by Western blotting. See
High-Content Screening Analysis of Drug Toxicity
Plates with 384 wells were coated with Matrigel (BD Biosciences, Franklin Lakes, NJ), and Cytiva cardiomyocytes27,28 (GE Healthcare, Little Chalfont, UK) derived from H7 human embryonic stem cells were seeded at 15,000 cells per well in RPMI1640+B27 (Life Technologies). Cells were cultured for 5 d in glucose media followed by 1 d in glucose or galactose media. Test compounds were diluted from DMSO stocks into glucose or galactose media to yield final assay concentrations in the range of 0.05 µM to 100 µM. Plates were incubated for 72 h, and Hoechst 33342 (1 µM) and TOTO-3 (1 µM; Life Technologies) were added for 1 h prior to imaging. Cardiomyocytes were imaged on IN Cell Analyzer 2000 (GE Healthcare) using excitation and emission filters for Hoechst and TOTO-3. Automated image analysis was performed with IN Cell Investigator (GE Healthcare) to measure total cells (Hoechst staining) and nonviable cells (TOTO-3 staining).
High-Content Screening Measurement of RBD Binding to Nutrient Transporters
Cardiomyocytes were cultured and treated with test compound as described for drug toxicity assays with 0.1% DMSO used as a control. Following 72 h compound exposure, media were aspirated, cells were blocked for 20 min at room temperature with phosphate-buffered saline (PBS) containing 5% fetal calf serum (FCS) and 0.1 mg/mL human immunoglobulin G (IgG), and then RBD probes were added in glucose or galactose culture media with 1 µM Hoechst and incubated for 15 min at 37 °C. Cells were washed twice with 5% FCS and 1 µM Hoechst in PBS at room temperature and then incubated for 30 min at 4 °C with Alexa Fluor 647 chicken anti-rabbit IgG (1:400; Life Technologies) or R-PE–labeled goat anti-mouse IgG (1:200; Life Technologies) in 5% FCS and 1 µM Hoechst in PBS. Following antibody labeling, cells were washed twice with 5% FCS in PBS with 1 µM Hoechst and fixed for 30 min at 4 °C in 5% FCS in PBS with 1 µM Hoechst and 0.1% paraformaldehyde. Cardiomyocytes were imaged on IN Cell Analyzer 2200 (GE Healthcare) using excitation and emission filters for Hoechst, R-PE, and Alexa Fluor 647. Automated image analysis of RBD staining intensity was performed with IN Cell Investigator (GE Healthcare). To avoid cytoplasmic membrane segmentation artifacts arising from variable RBD intensities, cardiomyocyte nuclei were segmented using the Hoechst channel and RBD intensity measured in the R-PE/Alexa Fluor 647 channel in a region defined by expansion of the nuclear segmentation region. RBD intensity data are expressed as mean RBD fluorescence intensity/cell. Hierarchical clustering of intensity data was carried out using Spotfire (TIBCO, Palo Alto, CA).
Flow Cytometry Analysis of RBD Binding to Nutrient Transporters
Ninety-six–well plates were coated with Matrigel (BD Biosciences), and cardiomyocytes were seeded at 40,000 cells per well in RPMI1640+B27 (Life Technologies). Cells were cultured for 5 d in glucose media followed by 1 d in glucose or galactose media. Antimycin A, nifedipine, imatinib, sorafenib, and mubritinib were diluted from DMSO stocks into glucose or galactose media to yield 1, 10, or 30 µM final assay concentrations. DMSO 0.1% was used as negative control. After 24 h of drug treatment, cells were prepared for RBD binding to nutrient transporters. Cardiomyocytes were detached using trypsin 0.25% (Life Technologies) for 5 min at 37 °C and transferred into a 96-well V-shaped microplate. RBDs were premixed pairwise (one mFc- and one rFc-fused RBD) in culture medium containing 0.1% sodium azide and 1 mM EDTA. RBDs were added to cardiomyocytes and incubated at 37 °C for 15 min. Cells were washed once with PBS/2% FCS and then incubated with Alexa Fluor 647 goat anti-rabbit IgG (1:800; Life Technologies) and R-PE goat anti-mouse IgG1 (1:200; Life Technologies) antibodies in binding buffer (PBS/2% FCS/0.1% sodium azide/1 mM EDTA) for 30 min at 4 °C. Cells were washed twice with binding buffer and labeled with mouse anti-human CD90-FITC antibody (BD Biosciences; clone 5E10, 1:500) and 1 µg/mL DAPI to restrict RBD binding analysis to CD90-negative cells (mainly composed of cardiomyocytes, as evidenced by CD90/cardiac troponin T labeling; data not shown). After 30 min of incubation at 4 °C, cells were washed and resuspended in binding buffer before flow cytometry analysis.
Fluorescent signals were acquired on a FACSVerse flow cytometer (BD Biosciences) with 405, 488, and 640 nm excitation, and data analysis was performed using Flowjo software (TreeStar Inc., Ashland, OR). Dead cells and CD90-positive cells, representing noncardiomyocytes, were excluded from the analysis. Fluorescence minus one controls were used to establish background levels in RBD channels (R-PE and AF647). Signals were converted into molecules of equivalent soluble fluorochrome (MESF) values using calibration beads (R-PE and AF647 MESF quantum beads; Bangs Laboratories, Fishers, IN) according to the manufacturer’s instructions. Clustering of RBD binding data was performed with dChip software (Harvard, http://www.hsph.harvard.edu/cli/complab/dchip/).
Results
To analyze the effect of cellular metabolism and bioenergetics on compound toxicity in cardiomyocytes, we measured the impact of drug treatment following shifting cellular metabolism from glycolysis in glucose medium to oxidative phosphorylation in galactose medium ( Fig. 1 ). Amiodarone, an antiarrhythmic drug with known mitochondrial toxicity used as a positive toxic control in this study, showed equivalent toxicity in both glucose and galactose media. Nifedipine, a calcium channel blocker used here as a negative control, showed no impact on cell viability in either media at the concentrations tested. Antimycin A, an inhibitor of cytochrome c reductase that interferes with the electron transport chain of oxidative phosphorylation, showed minimal toxicity in glucose medium and extreme toxicity in galactose medium, confirming a metabolic shift from glycolysis to oxidative phosphorylation following culture of cardiomyocytes in galactose medium.
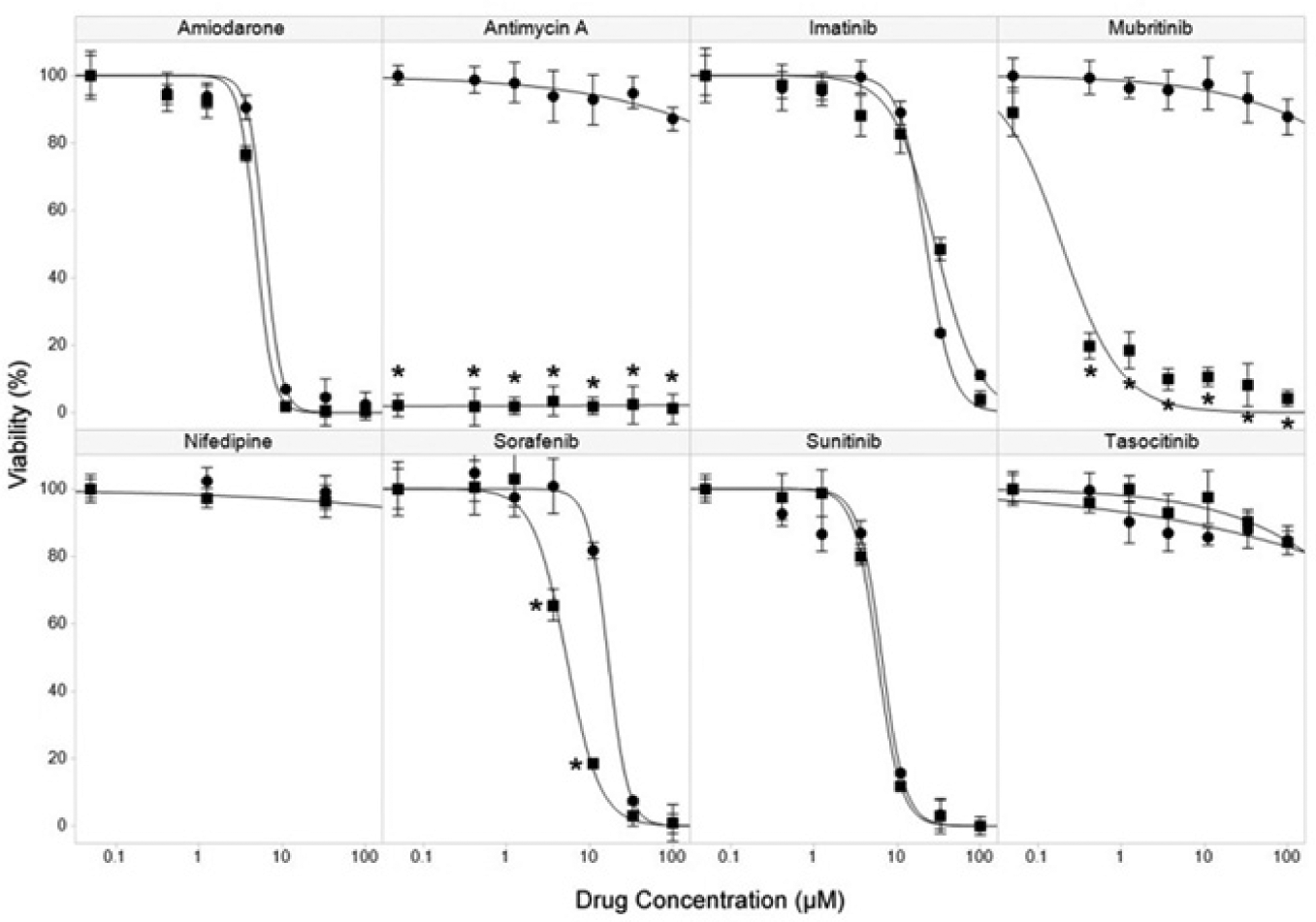
Cardiotoxicity of test drugs. Cell viability was determined by imaging of TOTO-3 staining of nuclei of nonviable cells following incubation of cardiomyocytes with the indicated concentrations of drugs for 72 h in glucose (•) or galactose culture media (■). Data are the mean and standard deviation of three replicates, *p < 0.001. Drug IC50 values (µM) were determined by logistic regression curve fit (drug IC50 glucose/IC50 galactose): amiodarone 6.2/4.8, antimycin A >100/<0.05, imatinib 22.4/28.8, mubritinib >100/5.2, nifedipine >100/>100, sorafenib 16.6/5.2, sunitinib 6.5/5.8, and tasocitinib >100/>100.
Among the five anticancer drugs tested, three (imatinib, sunitinib, and tasocitinib) gave very similar results in both media, with imatinib and sunitinib showing equivalent toxicity and tasocitinib showing minimal toxicity. Both sorafenib and mubritinib showed sensitization of toxicity in galactose medium, with sorafenib showing significantly increased (p < 0.001) toxicity at 3.7 µM and 11 µM, resulting in a greater than threefold reduction in IC50 between glucose and galactose media. In the case of mubritinib, sensitization of toxicity was extremely pronounced, changing from minimal impact on viability in glucose medium to extreme toxicity in galactose medium (p < 0.001 at concentrations >0.05 µM). This dramatic increase in toxicity, associated with changing cardiomyocyte metabolism from glycolysis to oxidative phosphorylation, is a strong indication that mubritinib toxicity is mediated through mitochondria and has significant implications for the predictivity of standard in vitro cell assays carried out in glucose media to reflect the highly adaptive metabolic state of cardiomyocytes in vivo.
To investigate whether monitoring metabolic changes occurring in drug-treated cardiomyocytes could be predictive of metabolism-dependent drug toxicity, we profiled the cell surface expression of a panel of seven key SLC nutrient transporters using RBD ligand binding analyzed by high-content screening and flow cytometry.
Immunofluorescence imaging of RBD binding ( Fig. 2A ) showed visible changes in RBD binding to nutrient transporters with media and drug treatment, including a significant increase in GLUT1 levels in DMSO-treated cells in galactose medium compared with glucose medium ( Fig. 2A , panels A and B) that was confirmed by quantitative image analysis. Increases in cell surface GLUT1 were also observed in glucose medium following exposure to certain drugs, including mubritinib ( Fig. 2A , panels A and C). Analysis of cardiomyocytes treated with the DMSO vehicle and low concentrations of the nontoxic compounds nifedipine and tasocitinib ( Fig. 2B ) showed distinctly different RBD intensity profiles in glucose and galactose media, reflecting changes in nutrient transporter expression under altered metabolic conditions in the absence of drug toxicity. The observed increase in cell surface expression of GLUT1 in galactose medium is consistent with a cellular shift in energy source in the absence of glucose, that is, up-regulation of GLUT1 to attempt to scavenge glucose in response to reduced availability and to transport available galactose. 29
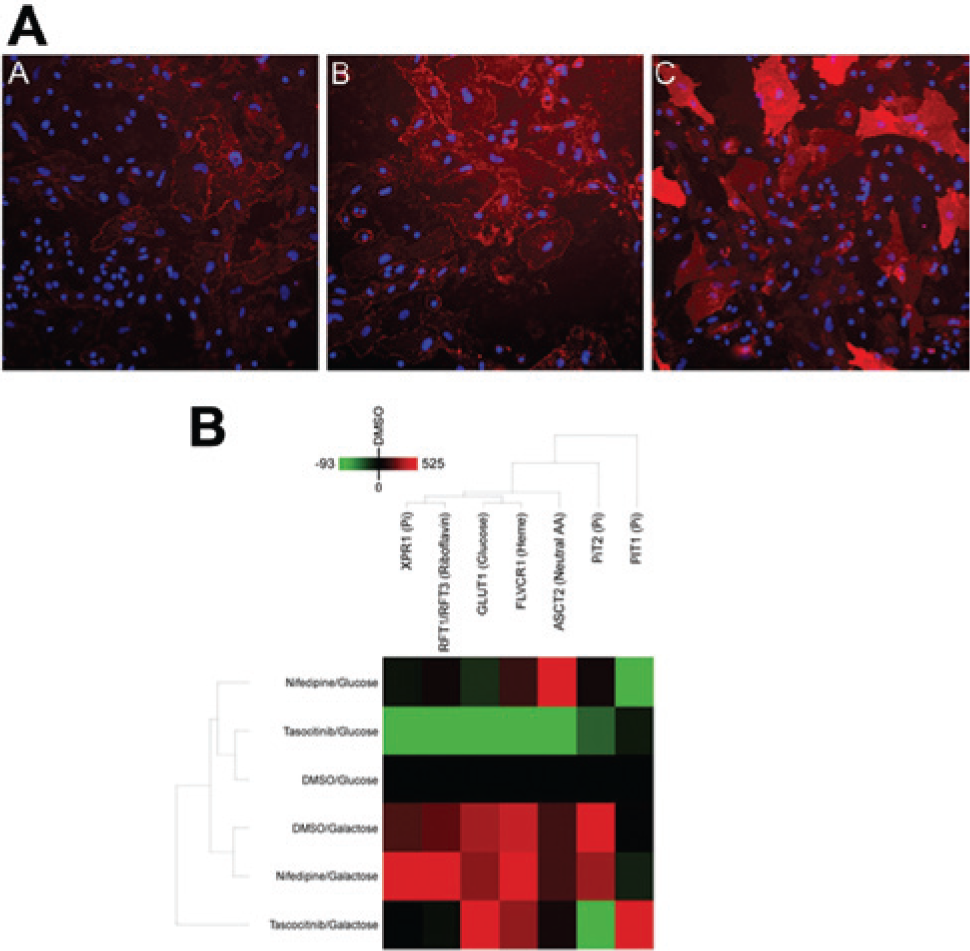
(
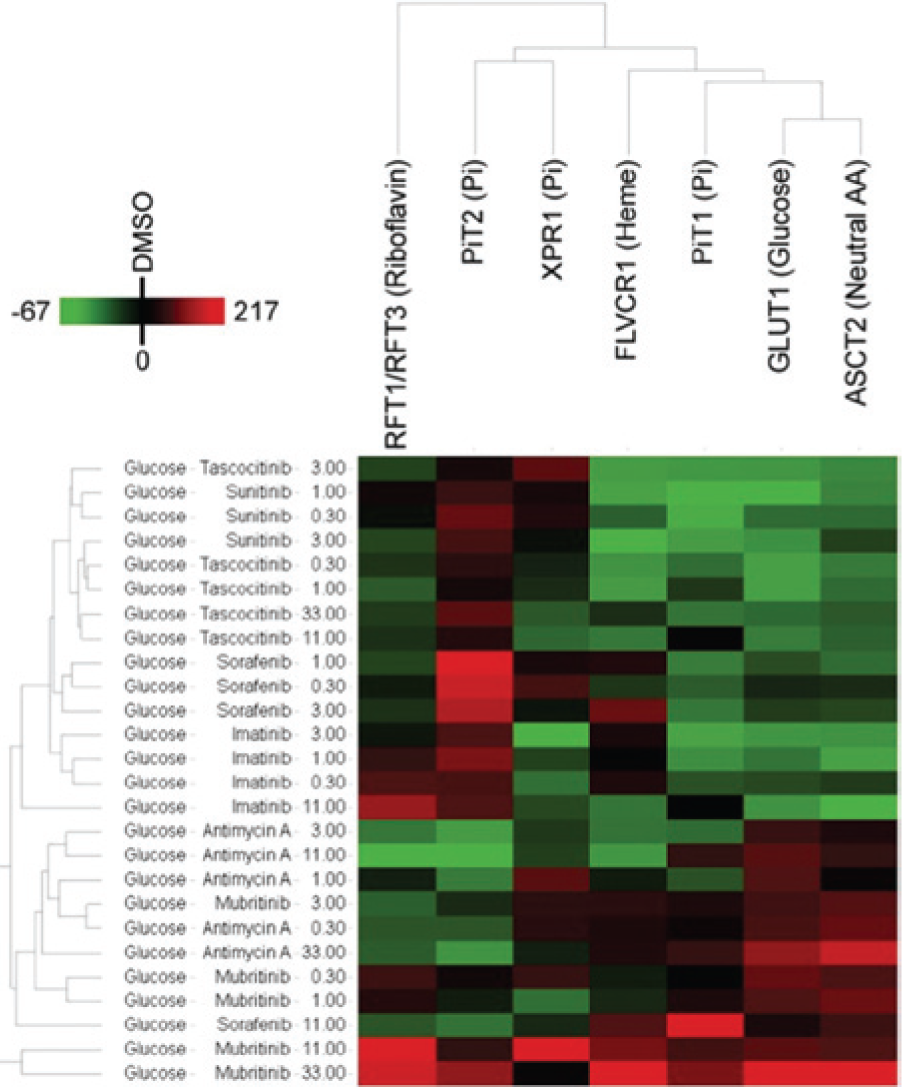
Full analysis of RBD binding data obtained at increasing drug concentrations (

Profiling of effects of drug treatment on receptor binding domain (RBD) binding. Parallel coordinate axis analysis of immunofluorescence imaging data for RBD binding to cardiomyocytes treated with increasing concentrations of drugs for 72 h in glucose and galactose media ( 0.3 µM,  1.0 µM,
1.0 µM,  3.0 µM,
3.0 µM,  11.0 µM,
11.0 µM,  33.0 µM). Cells showing significant toxicity following drug treatment (antimycin A and mubritinib in galactose medium and higher concentrations of amiodarone, imatinib, sorafenib, and sunitinib in glucose and/or galactose media) were not analyzed for RBD binding. RBD intensity data were calculated relative to values in DMSO-treated cells in glucose medium, and parallel coordinate axes are scaled to minimum (0%) and maximal (100%) intensities for each separate RBD. Data are the means of three replicates.
33.0 µM). Cells showing significant toxicity following drug treatment (antimycin A and mubritinib in galactose medium and higher concentrations of amiodarone, imatinib, sorafenib, and sunitinib in glucose and/or galactose media) were not analyzed for RBD binding. RBD intensity data were calculated relative to values in DMSO-treated cells in glucose medium, and parallel coordinate axes are scaled to minimum (0%) and maximal (100%) intensities for each separate RBD. Data are the means of three replicates.
The most significant finding in this analysis was the dose-dependent changes in RBD signatures observed for sorafenib and mubritinib in glucose media ( Fig. 3 ). In both cases, these compounds, which had previously been shown to have sensitization of toxicity in galactose media ( Fig. 1 ), showed profile changes that were not observed with other kinase inhibitors that showed equal toxicity in both media. Sorafenib at 11 µM, a concentration producing significant toxicity in galactose medium but not in glucose medium, showed an increase in GLUT1 and PiT1 expression and a decrease in several other transporters. Mubritinib showed a marked change in RBD profiles with increasing drug concentrations, with significant up-regulation of GLUT1 and the heme transporter FLVCR1 at 11 µM and 33 µM, drug concentrations at which a minimal impact on cell viability was observed ( Fig. 1 ). Increased cell surface GLUT1 expression is consistent with mubritinib’s perturbing mitochondrial function and generation of ATP by OXPHOS, leading to increased cellular demand for glucose to maintain ATP synthesis by glycolysis. Up-regulation of FLVCR1 may be further indicative of loss of mitochondrial integrity, leading to leaching of free heme into the cell cytoplasm and a subsequent increase in expression and/or mobilization of the export transporter to remove the toxic heme from the cell.
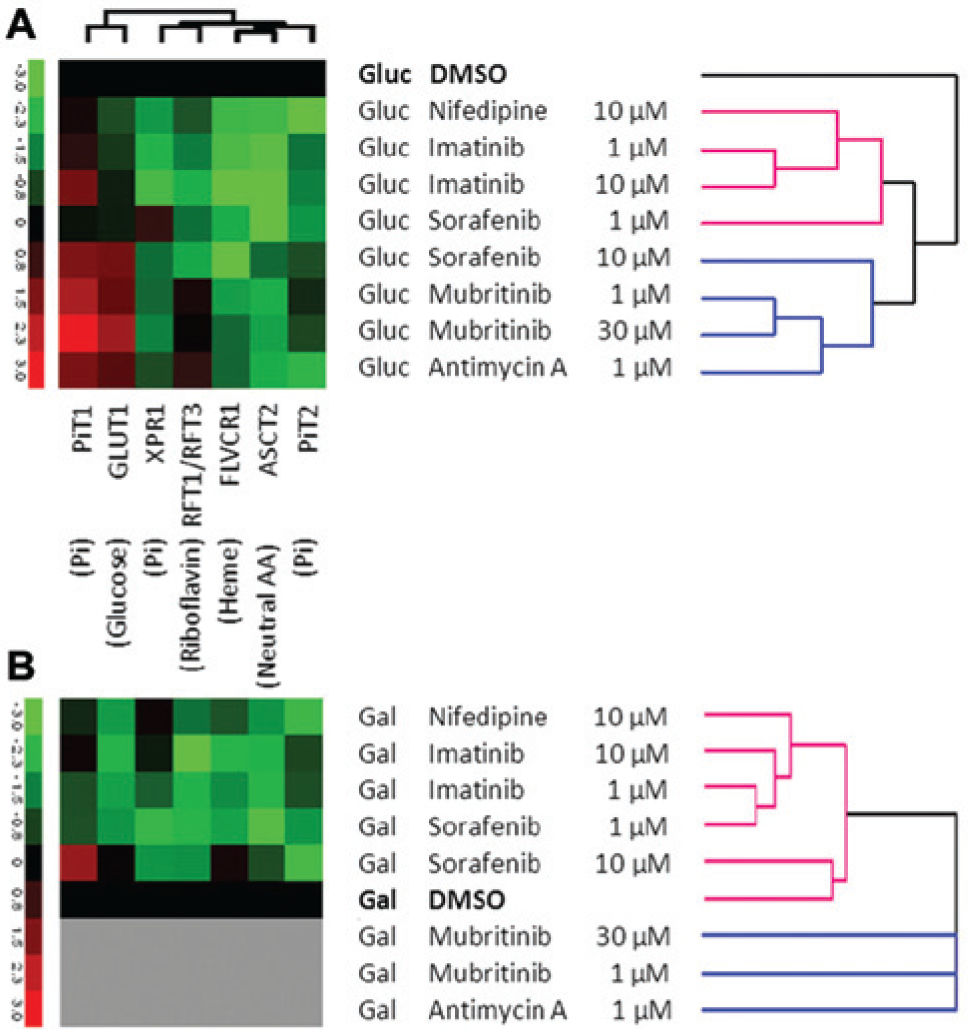
Hierarchical cluster analysis of RBD binding data acquired in glucose media ( Fig. 4 ) confirmed these observations and the predictive potential of RBD profiling to identify compounds with metabolic modulation of toxicity. All concentrations of mubritinib, antimycin A, and 11 µM sorafenib were clustered separately from lower concentrations of sorafenib and from all doses of the compounds not showing metabolic sensitization of toxicity, imatinib, sunitinib, and tasocitinib. These findings show significant potential for RBD binding analysis to predict bioenergetic modulation of toxicity, potentially obviating the need for dual media screening of compounds.
Clustering of receptor binding domain (RBD) imaging data. Immunofluorescence intensity data for RBD binding to cardiomyocytes exposed to drugs at concentrations of 0.03 µM to 33.0 µM in glucose medium for 72 h were analyzed by hierarchical clustering. Drug concentrations causing significant toxicity were excluded from analysis. RBD intensity values were calculated relative to intensity values in DMSO-treated cells in glucose medium. Data are the mean of three replicates.
To determine whether these observations could be detected at a shorter drug treatment time, to determine the utility of using RBD ligands to profile cardiomyocyte nutrient transporter expression using an alternate analysis method, and to confirm our findings in a second laboratory, we carried out flow cytometry measurement of RBD binding to cardiomyocytes treated for only 24 h with a subset of the drug treatments used for high-content screening (
Fig. 5
;
Flow cytometry profiling of receptor binding domain (RBD) binding to cardiomyocyte metabolite transporters. RBD binding was quantified by flow cytometry on cells treated with the indicated drug concentrations for 24 h in (
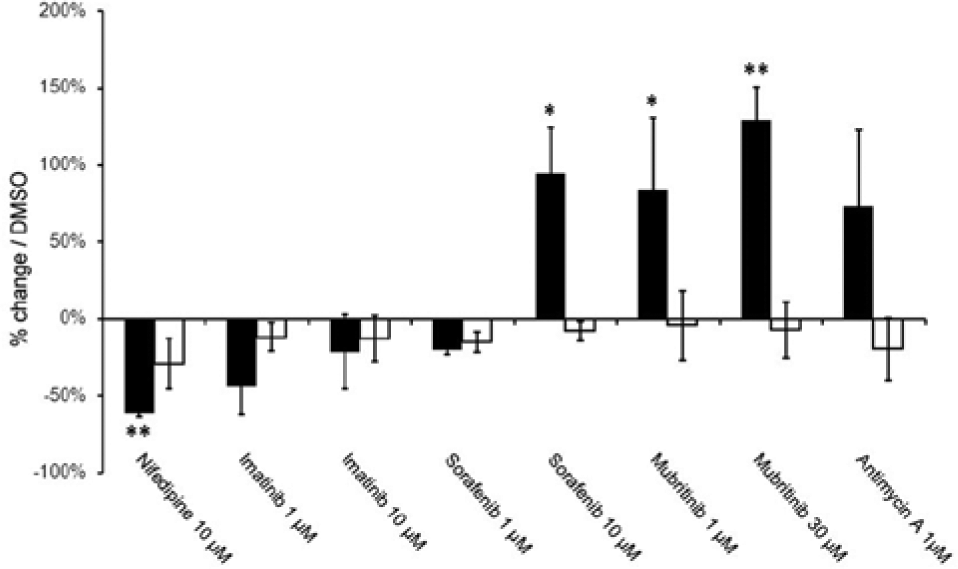
Receptor binding domain (RBD) binding to GLUT1 and PiT2 transporters in drug-treated cardiomyocytes in glucose medium. RBD binding to GLUT1 (■) and Pit2 (□) was measured by flow cytometry in cells exposed to the indicated drug concentrations for 24 h. Mean and standard deviation of three experiments, *p < 0.05, **p < 0.01.
Flow cytometry data confirm the findings from high-content screening that RBD profiling is predictive of metabolic sensitization of toxicity and that clustering of RBD signatures from cells in glycolysis correlates with the degree of toxicity likely to be observed in cells reliant on oxidative phosphorylation for ATP production.
These findings demonstrate that although potential bioenergetic modulation of toxicity of kinase inhibitors and other drugs can be addressed by dual screening in glucose and galactose media, profiling using RBD ligands readily detects subtle differences in cell metabolism triggered by kinase inhibitors in a single screen in glucose medium and provides predictivity of the relative toxicity of the compounds under shifted metabolic conditions. The method also provides insight into mechanisms associated with drug toxicity, for example, the up-regulation of glucose and heme transporters by mubritinib, diagnostic of drug-induced mitochondrial damage that leads to increased reliance on glucose as an energy source for glycolysis and an increasing requirement to export toxic heme released from damaged mitochondria. These results exemplify the potential power of this new RBD-based high-content screening assay to unravel the toxicity of kinase inhibitors in human stem cell–derived cardiomyocytes.
Discussion
Cardiotoxicity is a leading cause of drug failure during development and in the clinic, and there is a strong interest in developing more predictive in vitro assays for use in preclinical assessment. 30 In this study, we have used a new profiling approach to analyze the impact of a number of anti–kinase inhibitors, some of which are still in clinical use and one that failed during drug development, on human stem cell derived–cardiomyocytes. Use of high-content screening and flow cytometry using RBD ligands to profile cell surface nutrient transporter expression has provided evidence that such profiling is predictive of metabolic modulation of drug toxicity, which may be an important contributing factor to functional cardiotoxicity of drugs in vivo. Because drug-induced metabolic changes will arise from a combination of on-target and off-target drug impacts, some of which that may result in toxicity, common metabolic signatures can be shared by different molecules targeting or altering the same pathways, allowing new drug candidates to be profiled and ranked alongside compounds with available clinical data.
We have shown that culture of cardiomyocytes for 24 h in galactose medium produces a rapid shift in cellular metabolism from glycolysis to oxidative phosphorylation, which is accompanied by a change in nutrient transporter expression profiles including significant up-regulation of GLUT1, highlighting the potential for individual RBDs to provide predictive or diagnostic data for defined toxicity mechanisms such as mitochondrial toxicity. This metabolic shift is characteristic of the known metabolic flexibility of cardiac tissue in vivo and allows cell analysis of drug impact on cardiomyocyte integrity in vitro to be potentially more predictive of clinical response than measurements made under standard tissue culture conditions.
RBD profiling of kinase inhibitors showed that mubritinib, which we had previously shown using standard dual media screening to have extreme sensitization of toxicity on metabolic shift to oxidative phosphorylation, caused a dose-dependent change of the RBD binding profile in glucose media, which was not observed with other kinase inhibitors not exhibiting bioenergetics-dependent toxicity. Mubritinib (TAK-165), an irreversible inhibitor of human epidermal growth factor receptor 2 tyrosine kinase, 31 was the subject of a phase 1 clinical safety and tolerability study, but no trial results were reported. 32 Standard preclinical toxicity assays of this compound using cells in glucose media would not have detected bioenergetic sensitization of toxicity, and it is possible that this played a role in the clinical failure of this drug. Development of improved stem cell toxicity models coupled with more sophisticated analytical approaches such as those described here have great potential to speed drug development and reduce costs by providing improved and earlier insight into potential clinical drug toxicity based on more predictive in vitro analyses.
RBD profiling in a physiologically relevant human cell model provides a key enablement over standard methods of assessing drug impact(s) in deciphering subtle dynamic changes in cell metabolism and physiology, which may vary significantly over a range of drug doses. Highlighting metabolic stress that does not translate into overt toxicity, as determined by conventional methods such as measurement of cell viability, provides a test system with improved sensitivity and is potentially more predictive than standard assays, particularly for assessing subacute toxicity at low drug doses.
The data presented here highlight the potential of RBD profiling for early toxicity assessment in drug development. Profiling drug impact(s) with a panel of RBD probes and hierarchical clustering provides a generic method to classify drug effects on cell metabolism acting through a wide range of mechanisms. Consequently, RBD profiling, as a rapid and informative approach to characterizing cellular metabolism via measurement of nutrient transporters as surface biomarkers of cell metabolism, has broad potential across a range of applications, including drug efficacy, functional quality control of cells for cell therapy, disease diagnostics, and so forth.
Footnotes
Acknowledgements
We thank Dr Jean-Luc Battini and Dr Marc Sitbon for continuous and fruitful discussions on nutrient transporters roles.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.