
Abstract
Excess caloric consumption leads to triacylglyceride (TAG) accumulation in tissues that do not typically store fat, such as skeletal muscle. This ectopic accumulation alters cells, contributing to the pathogenesis of metabolic syndrome, a major health problem worldwide. We developed a 1536-well assay to measure intracellular TAG accumulation in differentiating H9c2 myoblasts. For this assay, cells were incubated with oleic acid to stimulate TAG accumulation prior to adding compounds. We used Nile red as a fluorescent dye to quantify TAG content with a microplate reader. The cell nuclei were counterstained with DAPI nuclear stain to assess cell count and filter cytotoxic compounds. In parallel, we developed an image-based assay in H9c2 cells to measure lipid accumulation levels via high-content analysis, exploiting the dual-emission spectra characteristic of Nile red staining of neutral and phospholipids. Using both approaches, we successfully screened ~227,000 compounds from the National Institutes of Health library. The screening data from the plate reader and IC50 values correlated with that from the Opera QEHS cell imager. The 1536-well plate reader assay is a powerful high-throughout screening platform to identify potent inhibitors of TAG accumulation to better understand the molecular pathways involved in lipid metabolism that lead to lipotoxicity.
Keywords
Introduction
An obesity pandemic is driving an unprecedented increase in the prevalence of type 2 diabetes,1–4 creating enormous health problems and increasing costs due to associated burdens of chronic disease and disability related largely to the high incidence of insulin resistance. Rational therapeutic approaches to this pervasive medical problem require comprehensive understanding of the pathogenesis of metabolic dysfunction, including the molecular mechanism by which excess caloric intake and insulin resistance result in ectopic lipid accumulation and obesity. Evidence has emerged that, in the context of chronic caloric excess, increased delivery of fatty acids to skeletal muscle results in excessive intramyocellular lipid accumulation (IMCL), composed largely of triglyceride (TAG). Many studies demonstrate that increased IMCL is tightly linked to the development of insulin resistance,5,6 but the molecular consequences of IMCL that lead to insulin resistance remain unclear. Although lipotoxic intermediates such as ceramide and diacylglycerol inhibit insulin receptor signaling, 7 evidence indicates that these perturbations can also occur downstream of the insulin signaling pathway. 8 Reducing IMCL accumulation represents one important therapeutic approach to improving insulin resistance. We hypothesize that IMCL accumulation can trigger both adaptive and maladaptive (lipotoxic) responses relevant to muscle insulin resistance and that a dynamic balance between these responses determines the evolution of muscle insulin resistance and diabetes. A chemical biology screening campaign may identify probes to study the mechanistic underpinnings of the association between IMCL and insulin resistance and provide new therapeutics to improve insulin resistance.
To interrogate a small-molecule collection and identify highly potent chemical inhibitors of hepatic lipid droplet formation, we used a novel high-content imaging-based phenotypic assay in a 384-well format that was developed to quantify the number and size of lipid droplets formed in a murine hepatocyte cell line (AML 12). 9 Our goal was to apply our expertise in phenotypic high-throughput screening (HTS) using high-content analysis of lipid droplet formation and inhibition to identify specific inhibitors of IMCL in muscle cells. H9c2 cells are a well-characterized rodent-derived myocyte-like cell line relevant to our hypothesis and suitable for HTS. These cells are immortalized lines, derived from rat heart, that exhibit skeletal muscle characteristics upon differentiation, including expressing skeletal muscle-specific genes PGC1α and PGC1β and active TAG synthesis and subsequent storage in lipid droplets. As a follow-on to HTS, and to establish relevance of the H9c2 cell model, we developed a 96-well TAG accumulation assay in differentiated primary human skeletal myotubes and tested candidate compounds in dose response.
Small-molecule inhibition of diacylglycerol acyltransferase (DGAT-1) and stearoyl-CoA desaturase (SCD1) has been shown to decrease IMCL,10 –12 but the compounds exhibit mechanism-based toxicity. Thus, the need is urgent to identify novel pathways associated with striated muscle lipotoxicity. Our goal is to identify molecular probes, other than DGAT-1 inhibitors, of cellular processes that control skeletal myocyte lipid homeostasis. To filter inhibitors of TAG synthesis pathway targeting DGAT-1, we developed and tested compounds in a radiolabeled extraction assay using a human DGAT-1 assay.
We used Nile red dye and high-content image analysis to quantify lipid content in differentiating H9c2 cells to identify compound activities based on phenotypic lipid accumulation-based algorithms. Nile red fluorescence for TAG accumulation was multiplexed with the DAPI fluorescence for cell count as an indirect measure of compound cytotoxicity. To leverage the speed and lower cost of a plate reader assay, we simultaneously developed a nonimaging 1536-well cell-based assay by measuring total fluorescence. HTS screening plates from the National Institutes of Health (NIH) Molecular Libraries Small Molecule Repository (MLSMR) collection were tested and analyzed using imaging and nonimaging platforms. The same screening plates were read sequentially on the EnVision plate reader and the Opera QHES (PerkinElmer, Waltham, MA). The hit sets correlated well between the platforms, as did the IC50 values of compounds that reconfirmed in the nonimaging assay with those determined by high-content imaging.
Methods and Materials
Reagents
H9c2 cells were purchased from ATCC (Manassas, VA). Primary human skeletal myocytes were obtained from the Translational Research Institute (Orlando, FL); Dulbecco’s modified Eagle’s medium (DMEM) was from Fisher Scientific (Mediatech, Manassas, VA); fetal bovine serum (FBS) was from Hyclone (Logan, UT); and L-glutamine, penicillin streptomycin solution, trypsin, epidermal growth factor, α-MEM, Nile red, and DAPI were from Invitrogen (Carlsbad, CA). Triacsin C came from Enzo Life Sciences (Farmingdale, NY); oleic acid–albumin, dexamethasone, and fetuin and bovine serum albumin (BSA) were from Sigma-Aldrich (St. Louis, MO). Paraformaldehyde (EMS, Fort Washington, PA) was made up as a 6% solution and kept at 4 °C. Cell culture flasks and CellBIND 1536-well clear-bottom plates came from Corning (Corning, NY); 96-well clear-bottom plates were from Greiner Bio-One (Monroe, NC). To determine viability, ATPLite luciferase reagent and AlamarBlue were used (PerkinElmer, Waltham, MA, and AbD Serotec, Raleigh, NC, respectively) and Human DGAT-1 recombinant virus was expressed with N-terminal 6His-tag in SF9 insect cells using pFastBacHT vector by Sanford Burnham’s protein core facility. [14C] decanoyl-CoA was from ARC (St. Louis MO), 1,2-dioleoyl glycerol oil was from Avanti (cat. 800811O; Avanti, Alabaster, AL), and Microscint-E was from PerkinElmer. The DGAT-1 inhibitor A922500 used as a control was from Tocris Bioscience (Minneapolis, MN).
Cell Culture and Compound Treatments
H9c2 cells were cultured in T225 tissue culture flasks in DMEM supplemented with 10% (v/v) FBS, 1% antibiotics, and 1 mM L-glutamine (growth medium) at 37 °C and 5% CO2. Cells were typically maintained at less than 70% confluency and subcultured every third day. H9c2 myoblast differentiation was initiated (day 0) in flasks by aspirating and replacing with the growth medium, with FBS reduced to 2% (H9c2 differentiation medium) and incubated for 2 days. Primary human skeletal myocytes from three lean male donors were pooled and expanded in DMEM supplemented with 10% (v/v) FBS, 1 nM dexamethasone, 0.5 mg/mL fetuin, 0.5 mg/mL BSA, and 10 ng/mL human epidermal growth factor. Proliferating primary human cells earlier than passage 6 were differentiated for 7 days in α-MEM containing 2% (v/v) FBS and 0.5 mg/mL fetuin (human primary differentiation medium) with media changes every second day.
For dose-response titration in 1536-well, compounds were prepared as 20-mM, 2.5-mM, and 0.312-mM stock concentrations in 100% DMSO. From each stock concentration, 2-fold serial dilutions were done in 2.5-nL increments by acoustic dispensing using Echo 555 (Labcyte, Sunnyvale, CA) into plates starting from 50 µM (final concentration in the assay). For dose-response titration in 96-well, compounds were serial diluted manually in 100% DMSO from 20-mM stock concentrations and further diluted into human primary differentiation medium and added to plates starting at 30 µM (final concentration in the assay). Final concentration of DMSO did not exceed 0.25% in the dose-response assays.
1536-Well TAG Assay
On day 2, using the straight tips of a Kalypsys (San Diego, CA) dispenser/washer, 4 µL cells were seeded into 1536-well black clear-bottom CellBIND plates at a density of 800 per well and treated with 200 µM oleic acid. Plates were centrifuged at 500 rpm and incubated for 24 h at 37 °C and 5% CO2 with Kalypsys metal lids. On day 3, test compounds (12.5 µM final concentration) and triacsin C (5 µM final concentration) or DMSO were added. The plates were incubated another 24 h at 37 °C and 5% CO2. On day 4, cells were fixed with 3% paraformaldehyde and washed three times with a Kalypsys dispenser/washer. After washing the plates, 4 µL of freshly prepared Nile red solution (0.5 µg/mL in acetone) diluted into phosphate-buffered saline (PBS) (3.25 µM working solution) was added to the 2.5-µL residual volume in the plates (2 µM final concentration) and incubated at room temperature for 0.5 h. The plates were read on an EnVision (PerkinElmer) microplate reader in the bottom-read mode using an FITC dichoric mirror (FITC excitation filter [485 nm] and Texas Red emission filter [555 nm]).
After this reading, 1 µL of DAPI nuclear stain in PBS solution prepared from a 1-mg/mL stock in water was added (133 µM final concentration) and incubated at room temperature for at least 1 h. Plates were read on the EnVision plate reader, and DAPI fluorescence was measured using a Lance/Delphia dichoric mirror (UV excitation filter [340 nm] and cyan fluorescence protein (CFP) emission filter [486 nm]). A blank plate with no cells was used to subtract the background DAPI signal. Cells were counted by comparing the DAPI fluorescence with the DMSO controls.
Images (three fields per well) were acquired on an Opera QEHS (PerkinElmer) at settings 20× 0.45 NA air objective; acquisition camera set to 2-by-2 binning for an image size of 688 × 512 pixels, with each channel acquired sequentially; Exp1Cam4 = DAPI (nuclei) using 365-nm Xenon lamp excitation and 450/50-nm emission filters; Exp2Cam1 = Nile red 515/575 (yellow-gold fluorescence) using 488-nm laser excitation and 540/70-nm emission filters; and Exp3Cam3 = Nile red 552/635 (red fluorescence) using 561-nm laser excitation and 690/70-nm emission filters.
96-Well Human Skeletal Myocyte Assay
Human primary cells were seeded at a density of 8000 per well in 96-well and differentiated for 7 days in differentiation medium (see Materials and Methods). On day 8, myotubes were treated with 100 µM oleic acid–albumin and the test compounds either at decreasing dose range of 30 to 0.032 µM or 1 µM triacsin C. After 24 h, potential cell toxicity was detected by incubation with 1:10 AlamarBlue for 2 h at 37 °C, 5% CO2; fluorescence was detected using a BioTek (Winooski, VT) plate reader with 540-nm excitation and 590/20-nm emission. The cells were fixed with 4% paraformaldehyde for 10 min at room temperature and washed once with PBS. Neutral lipids were stained with 1:20 solution of Nile red in PBS for 10 min at room temperature. Following two washes with PBS, the Nile red fluorescence was read using a BioTek plate reader with 485/20-nm excitation and 590/20-nm emission.
DGAT-1 Enzyme Assay
To assess compounds as potential inhibitors of DGAT-1, human DGAT-1 (10 µg/mL) was incubated with 10 mM [14C]-decanoyl CoA (58 Ci/mol) and 100 µM 1,2-dioleoyl glycerol (DODAG) at room temperature for 20 min in triplicate in 60 µL in a 96-well solvent-resistant plate, as described by Dow et al. 11 Control wells contained no DODAG. Compounds were tested at 10 µM in 2.5% DMSO. Reaction was stopped with HCL and neutralized, and 90 µL microscint E was added and radioactivity from [14C]-TAG counted on a TopCount scintillation counter (PerkinElmer).
Data and Statistical Analysis
Edge effects and systematic dispensing patterns across the plates in the pilot and HTS phases were corrected with Genedata Screener Assay Analyzer 10.0.1 (Genedata, Basel, Switzerland). Percent inhibition of lipid accumulation was calculated based on the positive (triascin C–treated) and negative (oleate only–treated) controls on each plate: % inhibition = 100 * [(compound value – AVEneg)/(AVEpos – AVEneg)]. Dose-response curves were analyzed using the Chemical and Biological Information Systems (ChemInnovation Software, San Diego, CA) or GraphPad Prism (GraphPad Software, La Jolla, CA) by nonlinear least squares analysis. Assay performance was routinely addressed in terms of signal-to-background (S/B) and Z′ factor (Z′).
Images were analyzed using a proprietary algorithm (
Results
1536-Well Plate Reader TAG Assay Development
As a model of muscle TAG accumulation, we predifferentiated H9c2 cells in 2% serum and exposed them to an oleic acid–BSA complex for an additional day prior to control or inhibitor treatment. Limiting the number of days of low serum exposure is vital: the longer the cells are cultured under these conditions, the fewer lipids are absorbed, decreasing S/B acceptable for HTS. To ensure that the H9c2 cells cultured under these conditions remained relevant for studying skeletal myocyte lipid accumulation, quantitative PCR was studied on cells exposed to low serum for 0, 2, and 4 days to determine levels of PGC1α and PGC1β. The cells showed 5- and 2-fold gene induction of PGC1α and PGC1β, respectively, when grown in 2% serum compared with myoblasts grown in 10% serum (data not shown). Gene induction between 2 and 4 days’ differentiation in low serum did not differ significantly. Although H9c2 syncytia was not observed under conditions described in Materials and Methods and visualized by high-content analysis, the experimental HTS-compatible system models differentiating cells containing upregulation of important genetic biomarkers of skeletal muscle.
Triacsin C was added after the first 24 h of oleic acid incubation as a positive control to allow the cells to initiate lipid uptake and synthesis. Triacsin C is a potent inhibitor of long-chain fatty acyl-CoA synthetase, the enzyme that catalyzes the formation of fatty acyl-CoA, and thus inhibits the initial step in incorporating fatty acids into intracellular TAG. To measure the lipid accumulation in cells in a high-throughput manner, we used commercially available, inexpensive Nile red dye, which, in the hydrophobic environment of lipids, is strongly fluorescent. Early studies in cultured aortic smooth muscle cells and macrophages proved Nile red a sensitive stain for detecting both polar and nonpolar lipids. Cytoplasmic lipid droplets can be selectively visualized over phospholipid vesicles using fluorescence microscopy by examining Nile red–treated cells for yellow-gold (528 nm) rather than red fluorescence (>610 nm).13–15
Saturation of the fluorescence signal is evident in differentiating H9c2 cells treated with 150 to 200 µM oleic acid for the 2 days, as detected by Nile red fluorescence, and triacsin C blocks this lipid accumulation ( Fig. 1A ). In the presence of oleate, triacsin C suppressed the signal to that of no oleate control. In its absence, triacsin C suppressed the signal below the no-oleate control. Oleic acid acts as a nutrient source, and cellular response proved to be more reproducible over the course of the experiment if control wells were incubated with oleic acid plus triacsin C rather than with vehicle only. We performed the high-throughput screen using 200 µM oleic acid as the high control and the same concentration of saturating concentrations of oleic acid and 5 µM triacsin C as the low control, which gave a maximum S/B of 2.5-fold and Z′ = 0.6.
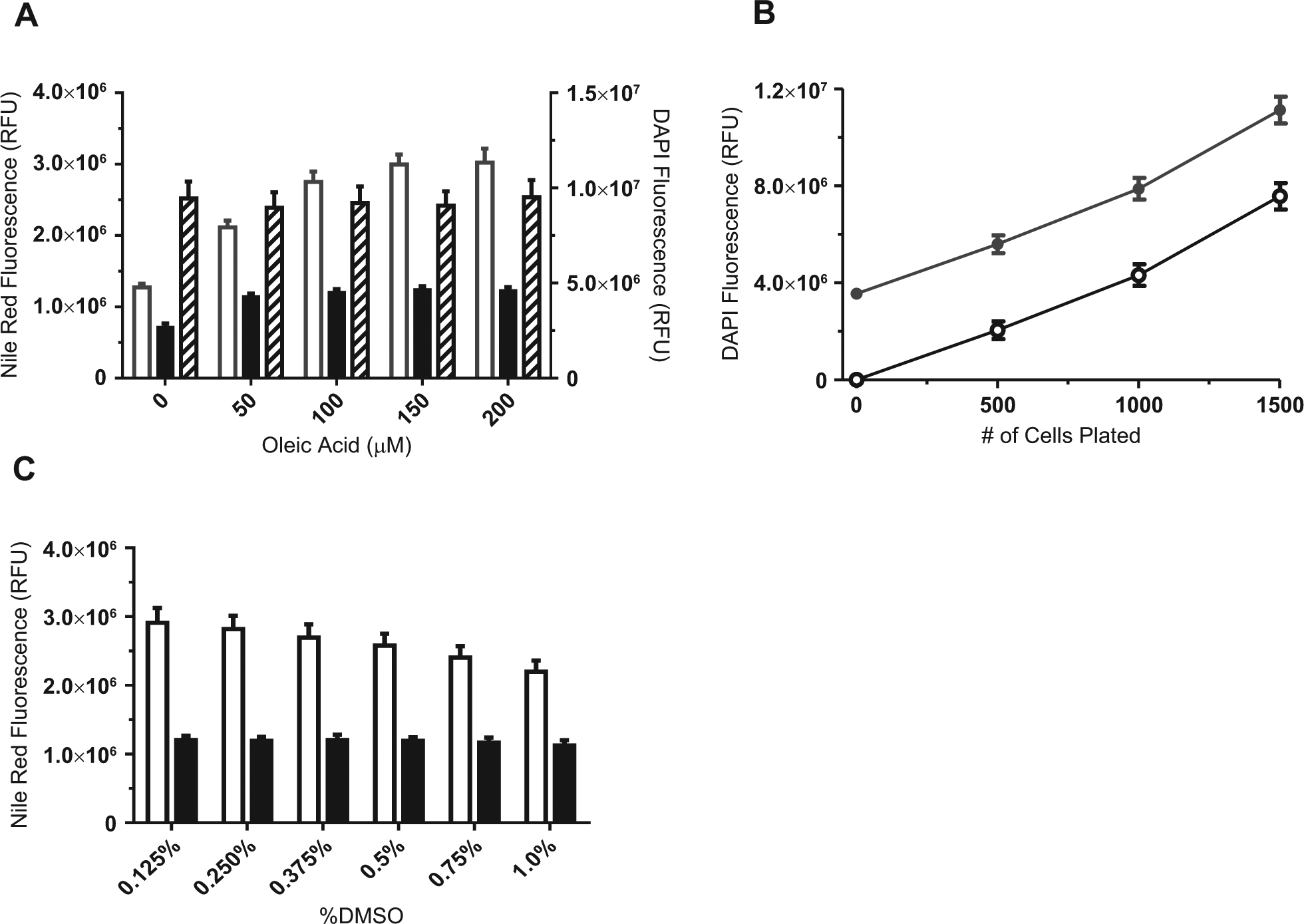
Assay development of 1536-well plate reader assay. (
To assess compound-induced cell toxicity and remove false-positive hits, we developed a simple plate-based method for quantifying cell number. Cells were counterstained with DAPI nuclear dye after Nile red quantification and plates read on an EnVision plate reader. DAPI fluorescence was proportional to cell number ( Fig. 1B ) after subtracting the background fluorescence of the DAPI in blank wells (no cells). Oleic acid treatment did not affect cell count ( Fig. 1 ). This multiplexed approach allowed us to normalize well-to-well compound inhibition with compound cytotoxicity.
Because MLSMR compounds are solubilized in DMSO, which can introduce cellular artifacts and affect S/B of the assay, we tested the effect of various DMSO concentrations in differentiating H9c2 cells. We observed a significant decrease in the S/B at DMSO concentrations >0.2% ( Fig. 1C ). Due to the high sensitivity of this assay to DMSO, a 10-mM compound library was chosen to screen compounds at the desired concentration while maintaining a final concentration of DMSO <0.2% (v/v).
1536-Well High-Content TAG Assay Development
Nile red has properties that make it ideal as a fluorescent tracer dye for high-content imaging. The spectrum is biphasic and dependent on the relative polarity of the stained lipids. Neutral lipids such as TAG and cholesterol esters in lipid droplets exhibit a more gold-yellow fluorescence (shorter wavelength, 515-nm excitation, and 575-nm emission) and can be visualized using a green laser and a filter for fluorescein. More polar lipids, such as phospholipids (useful to detect cell membrane), can be selectively imaged with a red laser and filter suitable for far red dyes (longer wavelength, 552-nm excitation, and 635-nm emission).
Using the strategy in the supplemental material ( Fig. 1 ; Materials and Methods), TAG uptake was quantified from the images for DAPI-stained nuclei, Nile red-red fluorescence phospholipids, and Nile red-yellow-gold TAG fluorescence. Images of Nile red–stained H9c2 cells treated with increasing concentrations of oleic acid, using the same plate from the plate reader experiment in Figure 1A , displayed a concentration-response of TAG accumulation as visualized by the gold-yellow fluorescence ( Fig. 2A ) and quantified per cell as integrated TAG intensity of the cytoplasm ( Fig. 2B ). The maximum S/B in cells treated with 200 µM oleic acid was 3-fold compared with the 5 µM triacsin C control.
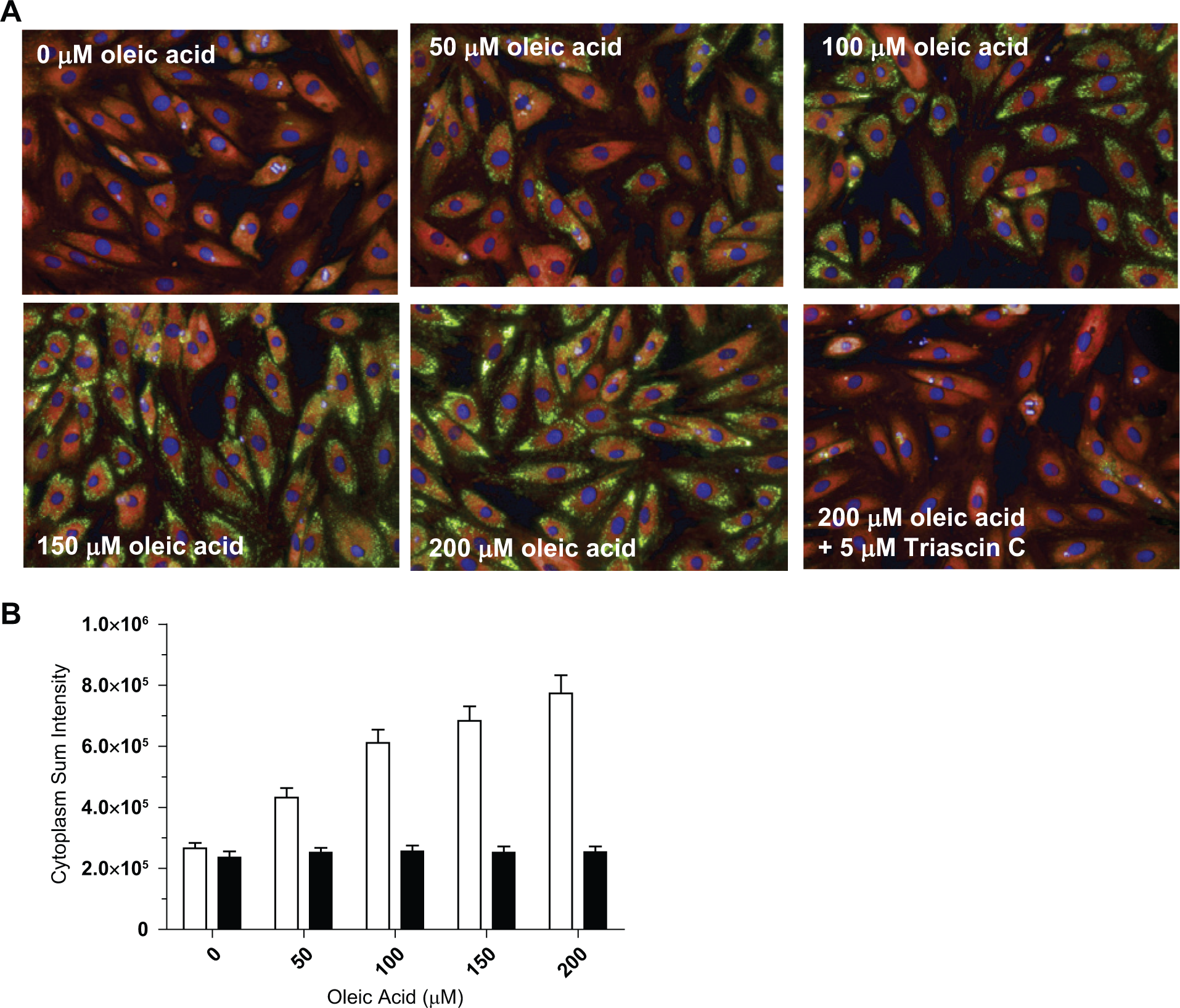
Assay development of 1536-well high-content assay. (
Contrasted with the total florescence signal measured on the EnVision, the cytoplasm sum intensity measurement did not plateau between the 150- and 200-µM oleate treatment plate reader ( Fig. 2B ). Treating the cells with concentrations of oleate above 200 µM disrupted cell adhesion, rendering us unable to fully assess saturation in the high-content assay. The two readouts differ: the plate reader measures the sum intensity of all cells in the well, including nonspecific signals due to background in the cell and surrounding liquid; however, in the high-content images, we set the cytoplasm sum threshold intensity to eliminate nonspecific intensities due to cell background. Only cells that pass viability filters based on DAPI-stained nuclear size, shape, and brightness are measured for TAG fluorescence; thus, the cytoplasm sum intensity readout should incorporate a larger dynamic range and greater sensitivity than the total fluorescence readout. In addition, the high-content imager images a 5-µm slice of the cell, unlike the plate reader, which detects signal from the entire cell. These may explain the difference in apparent oleate saturation.
1536-Well HTS Pilot Screen
A pilot screen of ten 1536-well plates comprising 13,633 compounds compared compound inhibition of TAG accumulation as measured in both the plate reader and the high-content imaging assay. As Figure 3A shows, an average S/B across all plates of 2.5- and 3-fold was obtained for the EnVision plate reader and Opera QEHS, respectively, with average coefficients of variation (CVs) of 4% to 5%. The Z′ was robust and consistent throughout and averaged ~0.6 for both detection modes. Despite achieving only 2.5-fold signal between triacsin C–treated and triacsin C–untreated controls in the plate reader assay measuring overall fluorescence intensity, excellent Z′s, identical to those obtained by high-content imaging and by applying an advanced high-content algorithm to specifically detect neutral lipid fluorescent intensity, were obtained. These data demonstrate that the plate reader assay is a powerful screening tool for detecting IMCL with equivalent robustness to high-content imaging but at a fraction of the resources that screening a high-content assay demands.
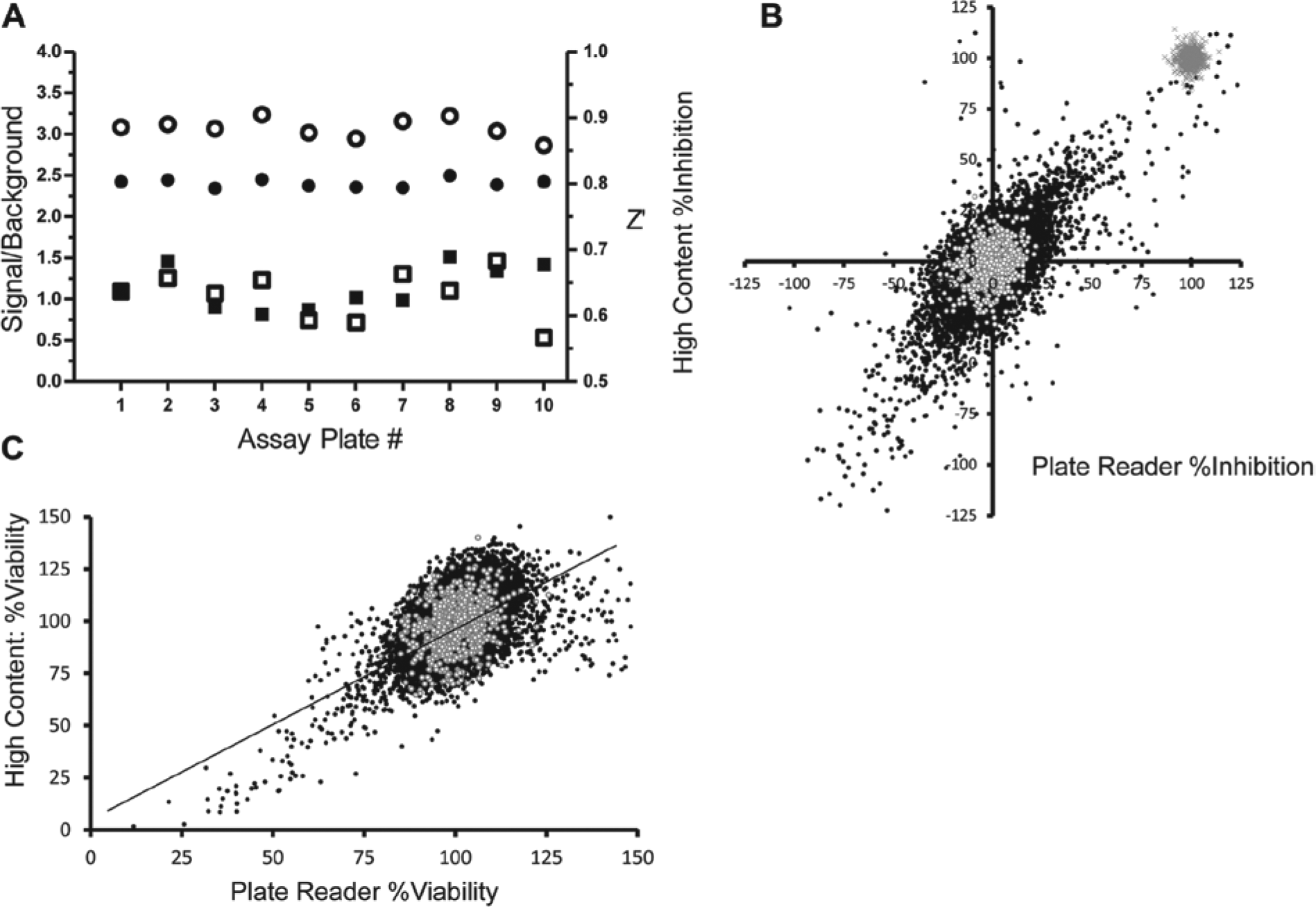
Results from the pilot screen study comparing plate reader versus high content. (
The hit rate was 0.82% for compounds tested at 12.5 µM, taking a cutoff criterion of ≥50% inhibition as normalized from the negative control wells treated with oleate only and the positive control wells treated with oleate plus 5 µM triacsin C. Figure 3B compares the Genedata-corrected percent inhibition for all test compounds in the pilot determined from the plate reader assay plotted on the x-axis and the high-content imaging assay on the y-axis. The hit rate was higher for the EnVision-read plates than for the Opera-read plates (0.82% and 0.64%, respectively). However, DAPI-based viability is inherent in the high-content analysis, and hits appearing as cytotoxic are filtered based on a cutoff of a ≥50% decrease in cell viability. After filtering cytotoxic compounds identified by a ≥50% reduction in DAPI fluorescence in the plate reader assay ( Fig. 3C ), the hit rate decreased to 0.5%. Of the hits determined from the EnVision-read plates, 48% also met the cutoff criteria for a hit on the Opera-read plates.
1536-Well HTS Screen
We screened 227,000 compounds from the MLSMR collection in differentiating H9c2 cardiomyocytes induced with 200 µM oleic acid to accumulate intracellular TAG. The assay performed robustly with a Z′ average over 167 plates of 0.6 and S/B of 2.3. The binned raw data displayed a Gaussian distribution centered over the negative controls (data not shown). Because data collection and analysis was significantly faster using the EnVision plate reader and our pilot study showed that the results from the plate reader assay were as robust as from the high-content imaging assay, we used results from the plate reader assay to filter screening hits. We identified 2303 hits that were reduced to 1502 noncytotoxic hits showing ≤50% inhibition of DAPI fluorescence using ≥50% inhibition of Nile red fluorescence as a cutoff criterion. Similar to our pilot screen results, 45% of these also met the cutoff criteria in the high-content imaging assay. Figure 4A shows a scatter plot of the overlap of percent inhibition for these active compounds in both screening platforms. Correlation analysis of the scatter plot showed good correlation between the platforms (Pearson r = 0.69, p < 0.0001).
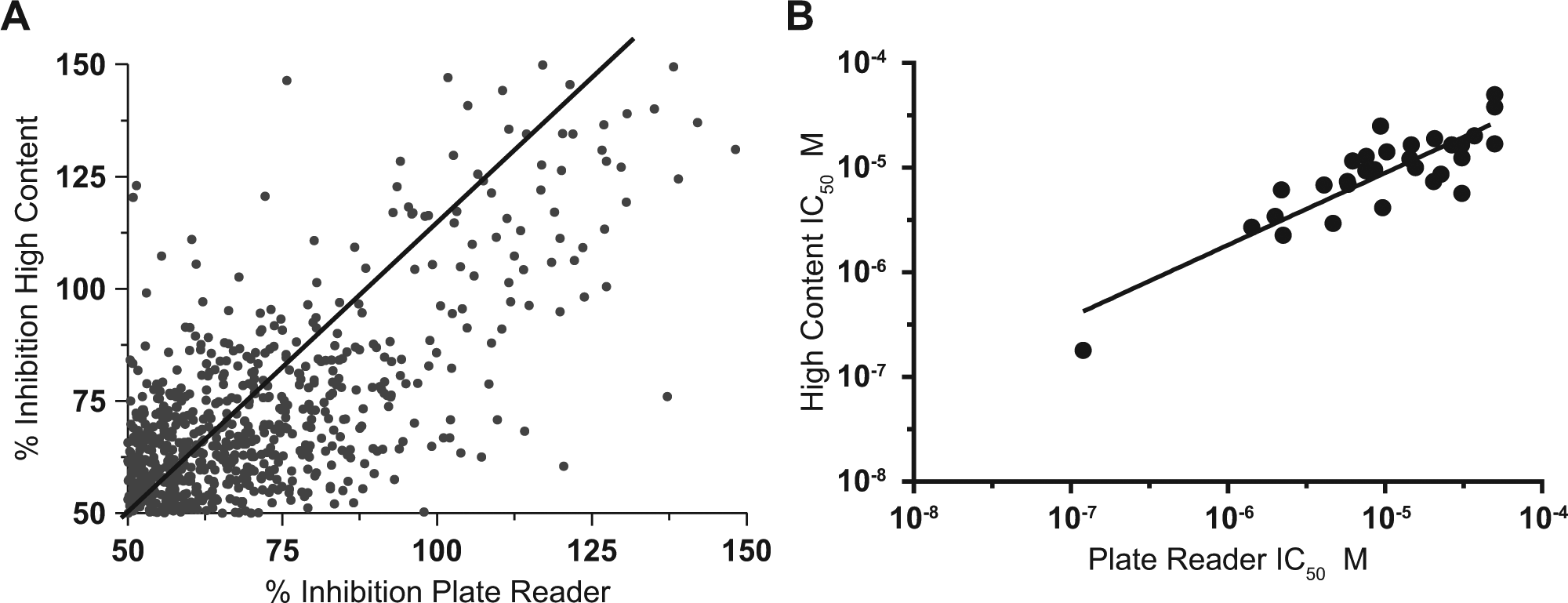
Results from high-throughput screening comparing plate reader versus high content. A cutoff criterion of ≥50% inhibition was applied to both the hit sets from the plate reader and the high-content imaging. (
After eliminating known PAINS 16 (Pan Assay Interference Compounds) and promiscuous compounds, we received 987 hits from the MLSMR as fresh 10-mM stocks and retested the compounds under the same conditions as the HTS to obtain a reconfirmation rate of 71%. We tested 702 compounds in 12-point concentration-response mode; 282 gave calculated IC50 values ≤10 µM inhibition of Nile red fluorescence and IC50 values ≥25 µM (highest concentration tested) inhibition of DAPI fluorescence. We identified 29 structures representing unique, chemically tractable scaffolds that were reordered as dry-powder stocks and retested in the primary assay at 10 concentrations. The pIC50 values for these 29 compounds, determined from the plate reader versus the high-content imaging assay, showed strong correlation (Pearson r = 0.86, p < 0.0001) ( Fig. 4B ).
Validating the Chemical Scaffold Identified by HTS
From this set of confirmed noncytotoxic screening hits, we identified a single lead scaffold. A representative of this scaffold, SBI-942, exhibited IC50 values of 2.5 ± 0.1 and 1.3 ± 0.1 µM in the EnVision plate reader and Opera high-content imager, respectively, with maximal efficacy ( Fig. 5A ). A structural analogue, SBI-819, exhibited IC50 values of 2.2 ± 0.1 and 1.6 ± 0.1 µM in the EnVision and Opera, respectively, with 70% efficacy ( Fig. 5B ). Figure 5A , B also shows the concentration-response data as percent viability for both compounds. As an additional assay for cell viability, we used the luciferase-based adenosine triphosphate (ATP) detection kit, ATPLite. The 29 active compounds were tested in concentration-response after oleate treatment. For SBI-942, loss in cell viability was observed at the two highest concentrations tested; however, the fold selectivity between potency and cytotoxicity was >10-fold. For the remaining hit compounds, including SBI-819, no significant loss in viability was observed up to 50 µM, indicating that the nuclear dye fluorescence intensity readout provided a good filter for eliminating cytotoxic hits.
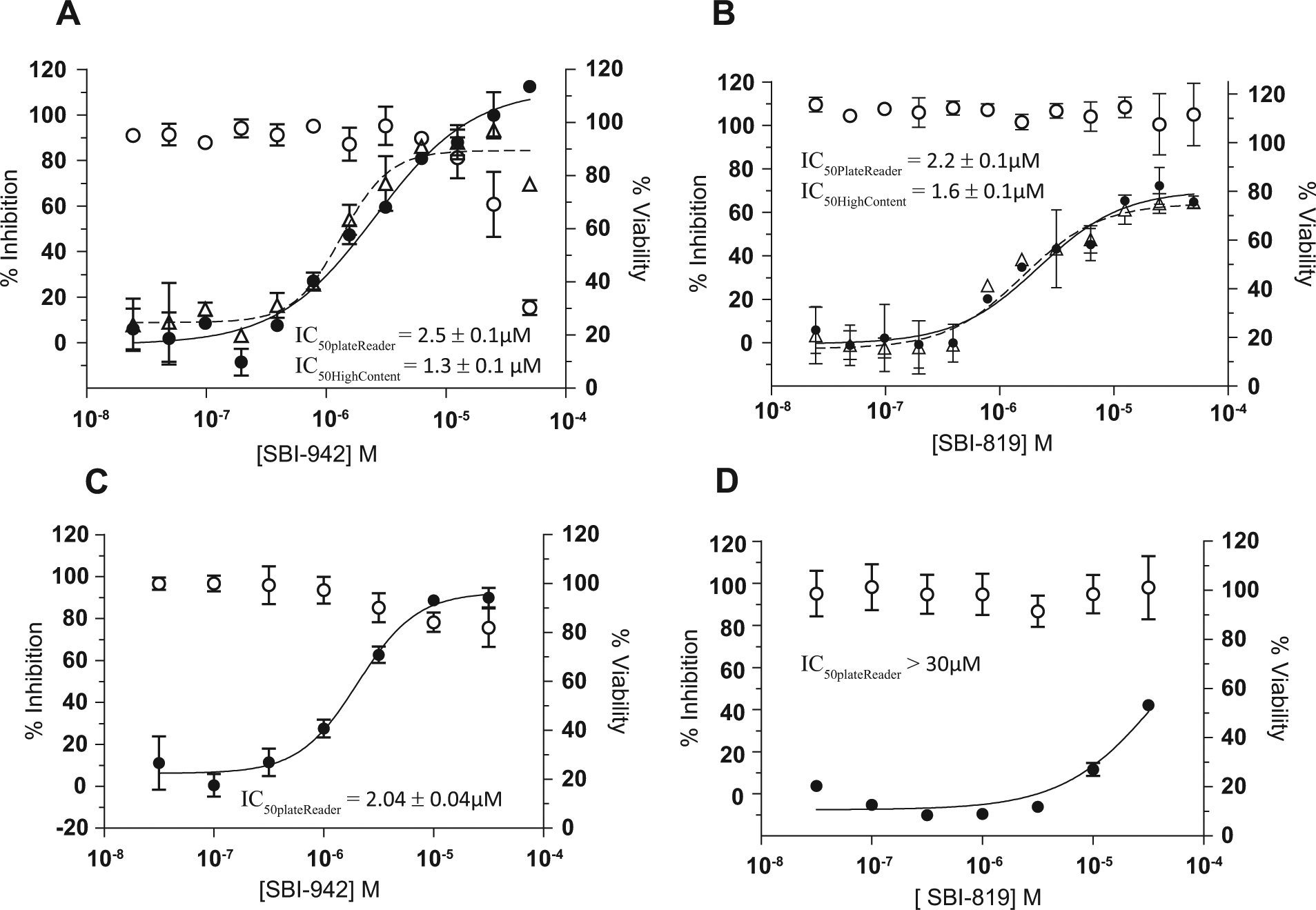
Dose responses of lead compounds in rodent and human TAG accumulation assays. (
To validate human activity of the lead scaffold and establish relevancy of the H9c2 rodent model, we tested SBI-942 and SBI-819 in human skeletal myotubes using a modified version of the H9c2 TAG accumulation assay. We confirmed SBI-942 activity in the human myotube assay (IC50 value of 2.04 ± 0.04) ( Fig. 5C ). However, SBI-819 potency significantly decreased in the human myotube assay, indicating a pharmacological difference within the scaffold ( Fig. 5D ). AlamarBlue was used to assess viability at each dose. As observed in the ATPLite assay in the H9c2 cells, SBI-942 exhibits a viability loss at doses of 10 µM and higher but >10-fold selectivity between primary activity and cytotoxicity. SBI-819 does not affect viability at the concentration range tested, similar to that observed in the ATPLite assay. SBI-942 inhibition of TAG accumulation in H9c2 cells translates to human skeletal myotubes, confirming the compound’s activity in human cells and validating the relevance of the H9c2 cell model for identifying inhibitors of TAG accumulation in skeletal muscle.
SBI-942 was further tested for potential DGAT-1 inhibition using human DGAT-1 enzyme and monitoring accumulation of the [14C]-TAG tracer. A922500, a known, commercially available DGAT-1 inhibitor, decreased [14C]-TAG levels 94% at a 100-nM concentration. SBI-942 at a 10-µM concentration showed no inhibition of DGAT-1, suggesting that the mechanism by which SBI-942 inhibits TAG accumulation differs from the DGAT-1 inhibitor, A922500 ( Fig. 6 ).
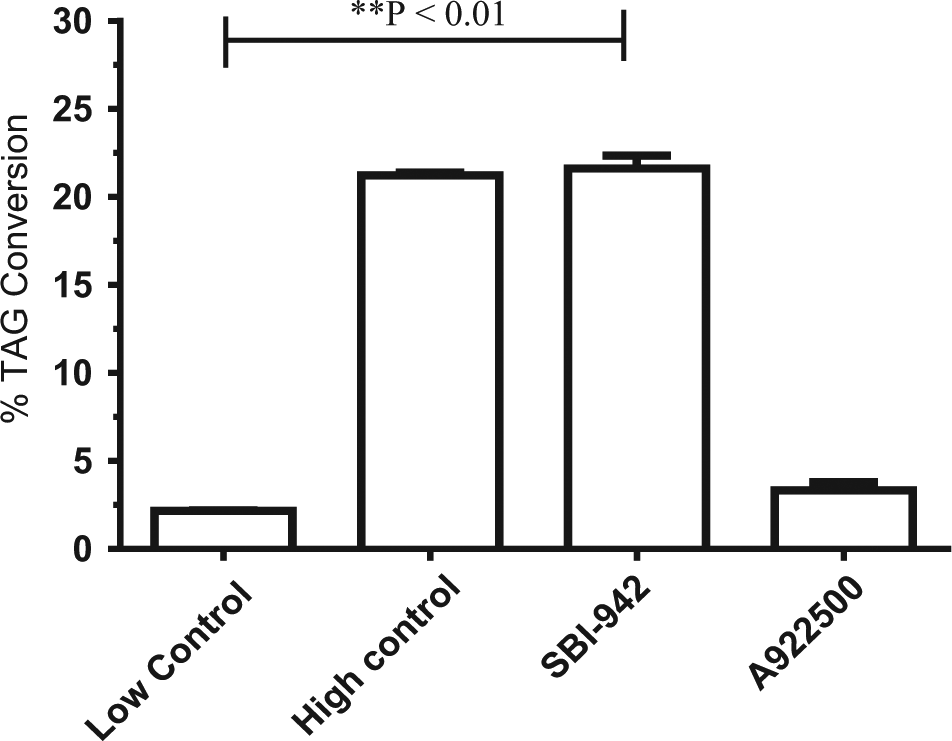
DGAT-1 activity of lead scaffold. SBI-942 was tested at a 10-µM concentration in a DGAT-1 radiolabeled assay as described in Materials and Methods. The commercially available DGAT-1 inhibitor A922500 was used as a positive control at 100 nM. The high control wells contained DGAT-1, [14C]-decanoyl, and 1,2-dioleoyl glycerol. The low control wells contained DGAT-1 and [14C]-decanoyl only. SBI-942 was statistically significant compared with the low control by a two-tailed unpaired t test (p = 0.0013).
Discussion
Phenotypic chemical biology screening is powerful for identifying relevant molecular probes of novel targets and pathways regulating lipid accumulation. Here we report a novel, multiplexed 1536-well, cell-based, nonimaging platform for detecting inhibition of TAG accumulation in differentiating H9c2 cells. The assay leverages the unique spectral qualities of the fluorescent dye Nile red to quantify neutral and esterified lipids in cells. Nile red is virtually nonfluorescent in aqueous solvents such as cell media, but in a nonpolar environment, such as a lipid vesicle, fluorescence is enhanced. The assay was multiplexed by adding DAPI to determine cell count, providing a normalization factor and eliminating cytotoxic compounds. High-content screens for lipid accumulation have been reported in 96- and 384-well.9,17 To our knowledge, however, this is the first report of a 1536-well HTS plate reader and high-content assay that can measure cellular TAG accumulation. We validated the assay by screening the MLSMR collection and correlating the resulting hit set to that obtained by high-content analysis. A single scaffold with superior chemical tractability was identified. Powder stocks of two compounds representative of the core scaffold were assayed for purity by high-performance liquid chromatography (HPLC) and tested in the primary assay at a range of concentrations. SBI-942 and SBI-819 were confirmed as low micromolar potent inhibitors of TAG accumulation in differentiating H9c2 cells. The potencies of these compounds were consistent across the plate reader and high-content detection platforms. In addition, SBI-942, but not SBI-819, exhibited equivalent potency and efficacy in inhibiting TAG accumulation in primary human skeletal myotubes. SBI-942 represents a strong starting point over SBI-819 for medicinal chemistry efforts. Hit-to-lead optimization will focus on prioritizing compounds by confirmed activity in the human primary TAG accumulation assay. This strategy, incorporated into structure-activity relationship studies, will increase success in identifying potent and selective first-in-class probes that interfere with human skeletal TAG formation.
That inhibiting DGAT-1 reduces cellular lipid accumulation overall is widely recognized. Thus, we sought to eliminate compounds acting through this mechanism, as they are unlikely to be useful in probing novel pathways of IMCL. As a secondary assay, we developed a radiolabeled DGAT-1 assay using 14C-Acyl-CoA and purified human DGAT-1 protein to filter compounds that inhibit DGAT-1. Preliminary results demonstrated that SBI-942 does not significantly inhibit DGAT-1 activity at 10 µM, a concentration five times the measured IC50 activity in the primary assay, but that A922500 tested at 100 nM reduced DGAT-1 activity 95%. These findings support using the TAG assay to identify small-molecule probes of lipid accumulation with potential novel mechanisms of action.
A lack of understanding exists of the links between myocyte lipid accumulation and insulin action where intramyocellular TAG accumulation is associated with improved muscle performance and metabolic flexibility. 18 This paradox emphasizes the importance of conducting unbiased studies to unravel the adaptive and maladaptive cellular responses of the skeletal myocyte to increased lipid import. SBI-942 and SBI-819 are important new experimental tools with significant utility for biomedical researchers studying these responses. To this end, we will assess the effects of these probes on insulin-stimulated glucose uptake and glycogen synthesis in primary murine skeletal myotubes. Given the importance of mitochondrial function in linking skeletal myotube lipid accumulation to insulin sensitivity and glucose utilization, we will also determine the effect of these probe compounds on mitochondrial respiratory function and biogenesis. Profiling studies, including transcriptional and metabolomic, will help identify target pathways perturbed after exposing myotubes to the probes. We will also use standard biochemical approaches, including mass spectrometry, to evaluate the direct interaction of the probes to candidate targets. Further medicinal chemistry structure-activity studies and pharmacologic analyses and optimization will assess the suitability of the candidate probes for in vivo studies of insulin sensitivity and glucose tolerance in rodents. These probes will allow us to identify new pathways and corresponding mechanisms that link alterations in myotube lipid metabolism to glucose utilization and insulin sensitivity. Long term, this work could lead to a new class of drugs aimed at preventing insulin resistance and reducing the end-organ complications of type 2 diabetes.
Footnotes
Acknowledgements
We thank Steve Smith, MD, at the Translational Research Institute, Orlando, Florida, for providing the primary human skeletal myocytes. We recognize our compound management, cheminformatics teams, and the protein core facility at Sanford Burnham, La Jolla, California, for their assistance on this project.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health Roadmap grant U54 HG005033 and R03 grant DA026213-01.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.