
Abstract
Zinc is an essential micronutrient that is crucial for many vital cellular functions such as DNA and protein synthesis, metabolism, and intracellular signaling. Therefore, the intracellular zinc concentration is tightly regulated by zinc transporters and zinc-binding proteins. The members of the SCL39 transporter family transport zinc into the cytosol. The SLC39A2 (hZIP2) protein is highly expressed in prostate epithelial cells and was found to be involved in prostate cancer development. Thus far, there is no specific modulator available for the SLC39 transporters. The aim of this study was to develop a screening assay for compound screening targeting hZIP2. Employing the pIRES2-DsRed Express 2 bicistronic vector, we detected human ZIP2 expression at the plasma membrane in transiently transfected HEK293 cells. Using the FLIPR Tetra fluorescence plate reader, we demonstrated that ZIP2 transports Cd2+ with an apparent Km value of 53.96 nM at an extracellular pH of 6.5. The cadmium influx via hZIP2 was inhibited by zinc in a competitive manner. We found that hZIP2 activity can be measured using cadmium in the range of 0.1 to 10 µM with our assay. In summary, for the first time we developed an assay for human ZIP2 that can be adapted to other zinc transporters.
Introduction
Zinc is an essential trace element that is ubiquitously present in the human body. It is a cofactor of more than 300 enzymes and a key structural component of zinc-finger motifs in many proteins.
1
Zinc is crucial element for transcription factors that require zinc to maintain their structure and their function to bind DNA. Zinc deficiency leads to growth retardation, cognitive impairment, testicular atrophy, and immune dysfunction in humans.
2
Even though severe zinc deficiency is rare, mild zinc deficiency is widespread in developing countries. In contrast, high levels of zinc are toxic for cells. Hence, intracellular zinc levels are tightly regulated by zinc transporters and other transport proteins, which are found at the plasma membrane in intracellular compartments. There are two main superfamilies of zinc transporters: the SLC30 (ZnT) zinc transporter solute carrier 30 family and SLC39
Because zinc is essential for the proper functioning of many proteins, crucial cellular processes including cell proliferation, hormone and neurotransmitter secretion, and apoptosis are greatly affected by the status of cellular zinc homeostasis. Mutations in the gene of SLC39A4 (ZIP4) have been shown to be associated with the disease acrodermatitis enteropathica, 6 and malfunction due to genetic alterations of other ZIP proteins has already been implicated in many other diseases.
There are several studies showing the implication of the hZIP2 protein in the development of prostate cancer. Among all soft tissues in the human body, zinc content is the highest in prostate. Zinc levels are exceptionally high in secretory epithelial cells of the peripheral prostate, where they are 3- to 15-fold higher compared with other soft tissues. High zinc levels promote the accumulation and the secretion of citrate into the prostatic fluid as well as the mitochondrial apoptogenesis in these cells. In prostate carcinoma cells, zinc levels have been found to be very low, creating anti-apoptotic conditions with high-energy supplies of ATP favoring proliferation (for a more detailed review, see Franz et al. 7 ). The hZIP2 protein, together with the hZIP3 protein, has been located on the apical side of healthy epithelial cells, indicating a possible role in reabsorption of zinc from the prostatic fluid. 8 In prostate cancer, the expression of hZIP2 is significantly decreased.
The goal of this study was to establish a screening assay to identify specific modulators of hZIP2 that can be used as therapeutic lead compounds (e.g., for prostate cancer prevention and to study the functional role of hZIP2 in normal prostate epithelial cells). In addition, this assay could be further adapted for use with other SLC39 proteins.
Methods
Cell Culture and Cloning
HEK293 cells were cultured in complete Dulbecco’s modified Eagle medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 10 mM HEPES, 100 µM minimal essential medium nonessential amino acids, and 1 mM sodium pyruvate (Gibco, Carlsbad, CA) at 37 °C in a cell culture incubator. Cells were subcultivated when confluency reached 90%.
Molecular Cloning
The entire coding region of human ZIP2 (hZIP2) cDNA was amplified by PCR reaction using two primers, the 5′-primer hZIP2 5′-TAGCTCGAGATATGGAGCAACTAC-TAGGAATAAAAC-3′ with an Xho1 restriction site and the 3′-primer hZIP2: 5′-GATGAATTCTCAGGCCCACAAG-GCAATAAAGGC-3′ with an EcoRI restriction site, which were used to clone hZIP2 cDNA into the pIRES2-DsRed-Express2 vector.
Transfection and establishment of Stable hZIP2-Expressing HEK293 Cell Clone
For all experiments, cells were transfected 24 h after plating. Transfection was carried out along the manufacturer’s protocol of lipofectamine 2000 (Invitrogen) reagent, except that only 50% of the recommended amount of DNA and lipofectamine 2000 was used. After transfection, medium was changed 4 h later to antibiotic-free DMEM. The transfection efficiency was estimated to be 60% to 70% using fluorescence microscopy. To establish hZIP2-expressing clones, transfected cells were cultured in 100 µg/mL G418-containing cell culture medium. After the death of the majority of the nontransfected cells, cells were plated in a 96-well cell culture plate at 1 cell per well density. Cells showing red fluorescence were selected and tested functionally.
Real-Time PCR
HEK293 cells were seeded at a density of 3 × 105 cells/well into a six-well plate coated with 100 µg/mL poly-D-lysine. After 24 h, cells were transfected with hZIP2-DsRed, the empty DsRed vector, or left untransfected. Cells were harvested at 6 h, 12 h, 24 h, 48 h, and 72 h after transfection, and mRNA was extracted using TRIzol (Life Technologies, Carlsbad, CA) following the manufacturer’s protocol. The concentration of purified mRNA was determined using an Eppendorf BioPhotometer device. Reverse transcription was performed using the TaqMan reverse transcription reagents (Applied Biosystems, Foster City, CA). The qPCR experiments were carried out with a ViiA7 real-time PCR platform using prevalidated TaqMan gene expression assays (hZIP2: Hs01113547_g1, hGAPDH: Hs02758991_g1). Data were analyzed using the ΔCT method normalizing hZIP2 expression to the housekeeping gene hGAPDH.
Immunofluorescence
HEK293 cells were seeded at a density of 2.5 × 104 cells/well onto sterile No. 0 glass coverslips coated with 100 µg/mL poly-D-lysine placed into a six-well plate. After 24 h, cells were transfected with hZIP2 containing pIRES2 DsRed-Express2 vector. The next day, cells were washed thoroughly with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde at 37 °C for 20 min. The cells were not permeabilized in order to stain only the hZIP2 proteins expressed in the plasma membrane using the hZIP2 antibody raised against an extracellular epitope. Thereafter, samples were washed three times with PBS. After blocking the cells with PBS containing 0.5% bovine serum albumin (Sigma, St. Louis, MO) and 10% goat serum (Jackson ImmunoResearch, West Grove, PA) for 1 h, cells were stained with anti-hZIP2 (1:200, Abcam, ab99071) antibody at room temperature for 1 h. The cells were then washed three times with PBS followed by incubation with an Alexa488-conjugated goat-anti-rabbit secondary antibody (1:5000; Invitrogen). After washing the cells four times with PBS, the samples were mounted with CitiFluor AF2 (EMS). Fluorescence images were obtained using a Nikon C1 confocal laser scanning microscopy system.
Cell Biotinylation and Western Blotting
For cell surface biotinylation experiments, HEK293 cells were seeded into poly-D-lysine–coated 60 mm dishes at a density of 5 × 105 cells/well. On the following day, cells were transfected with the appropriate expression vector with/without the full length of hZIP2 cDNA. The cell surface biotinylation was performed at 4 °C. After rinsing the cells with PBS-Ca-Mg (PBS containing 0.1 mM CaCl2 and 1 mM MgCl2), the surface was biotinylated by incubating cells with 1.5 mg/mL sulfo-NHS-SS-biotin in biotinylation buffer (10 mM triethanolamine [pH 7.4], 1 mM MgCl2, 2 mM CaCl2, and 150 mM NaCl) for 60 min with horizontal shaking at 4 °C. Samples were washed with quenching buffer (PBS containing 1 mM MgCl2, 0.1 mM CaCl2, and 100 mM glycine) at 4 °C for 20 min before rinsing them three times with PBS-Ca-Mg. After removing any remaining liquid, cells were lysed in lysis buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 10 mM MgCl, 1% NP 40) for 30 min and lysates were cleared by centrifugation. Protein concentrations were adjusted to 1 mg/mL 9 . Cell lysates were incubated with streptavidin agarose beads at 4 °C overnight. Samples were centrifuged at 4 °C at 8000g for 1 min, and the supernatant containing the cytosolic proteins was stored. Beads were washed sequentially with solutions A (50 mM Tris·HCl [pH 7.4], 100 mM NaCl, and 5 mM EDTA) three times, B (50 mM Tris·HCl [pH 7.4] and 500 mM NaCl) twice, and C (50 mM Tris·HCl [pH 7.4]) once. Biotinylated surface proteins were released by adding 2× Lämmli buffer and boiling them at 95 °C. Proteins from the total lysate or the cytosolic fraction were diluted with 4× Lämmli buffer.
Samples were run on a 12% SDS gel with 30 µg protein loaded into each lane. Using the semi-dry transfer method, samples were transferred onto a polyvinylidene fluoride membrane. Membranes were blocked with PBS containing 5% milk and 0.05% Tween 20 for 1 h. Afterward, samples were incubated in blocking buffer containing the used primary antibody (hZIP2; Abcam, ab99071) at 4 °C overnight followed by three washes with PBS containing 0.1% Tween 20. An HRP-conjugated goat-anti-rabbit antibody (1:20,000; Promega, Madison, WI) was used as secondary antibody. After three consecutive washes with PBS, the enhanced chemiluminescence method was used for detection. As a loading control, the membranes were stripped of antibodies by incubating at 50 °C for 30 min in a stripping buffer (62.5 mM Tris-HCl pH 6.7, 100 mM 2-β mercaptoethanol, 2% [w/v] SDS) followed by washing the blot with PBS–0.1% Tween 20. Finally, the blot was reprobed with rabbit-anti-β actin (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA) primary antibody.
Functional Ion Influx Measurements Using the FLIPR Tetra
Cells were plated in 96-well, clear-bottom, black-well plates coated with poly-D-lysine at a density of 1.5 × 104 cells per well in 100 µL volume. The next day, cells were transfected with hZIP2-pIRES2 DsRed-Express2 or control vector. Twenty-four hours later, cells were loaded with calcium-5 fluorescence dye in 100 µL modified Krebs buffer (117 mM NaCl, 4.8 mM KCl, 1 mM MgCl2, 10 mM D-glucose, 10 mM HEPES) at 37 °C for 1 h. Fluorescence ion measurements were carried out using a FLIPR Tetra high-throughput fluorescence microplate reader at 37 °C. Cells were excited using a 470 to 495 nm LED module, and the emitted fluorescence signal was filtered with a 515 to 575 nm emission filter. In each experiment, a stable baseline was established for 50 s followed by the experimental protocol. Sampling rates were 2 Hz around the time of the administration of the substrate and 1 Hz for the rest of the experiment. Following the FLIPR Tetra guide, the buffer addition controls were assigned as negative control groups, whereas positive control groups were assigned to wells that give the maximum response to cadmium. Experiments were done with three to five repeats per group at least twice. To measure the zinc transporter activity, the change of the increase of the fluorescence intensity in response to cadmium administration was taken. The difference of the maximum and the minimum of the normalized fluorescence signal intensity during the first 25 s after the addition of the substrate was measured.
Statistics
Results are presented as mean ± SEM. Data were compared with the Student t test. p < 0.05 was considered significant. To validate our assay, the Z-factor was calculated as described by Zhang et al. 10
Results
hZIP2 Expression after Transient Transfection into HEK293 Cells
First, we examined the temporal pattern of the expression of the mRNA and protein of hZIP2 when it was overexpressed in HEK293. We observed an increased level of the mRNA of hZIP2 at 6 h and a decreased level after hitting its maximum at 24 h ( Fig. 1A ). The predicted molecular weight of hZIP2 is 33 kDa. Three bands were visible at this height of the Western blot, which might correspond to different glycosylation states of hZIP2. A band consisting of hZIP2 dimers was visible at 66 kDa. The hZIP2 protein started to be expressed at 12 h and reached its maximum at 24 h. In addition, the used anti-hZIP2 antibody was found to be specific because no band was observed in hZIP1-expressing HEK293 cells ( Fig. 1B ).
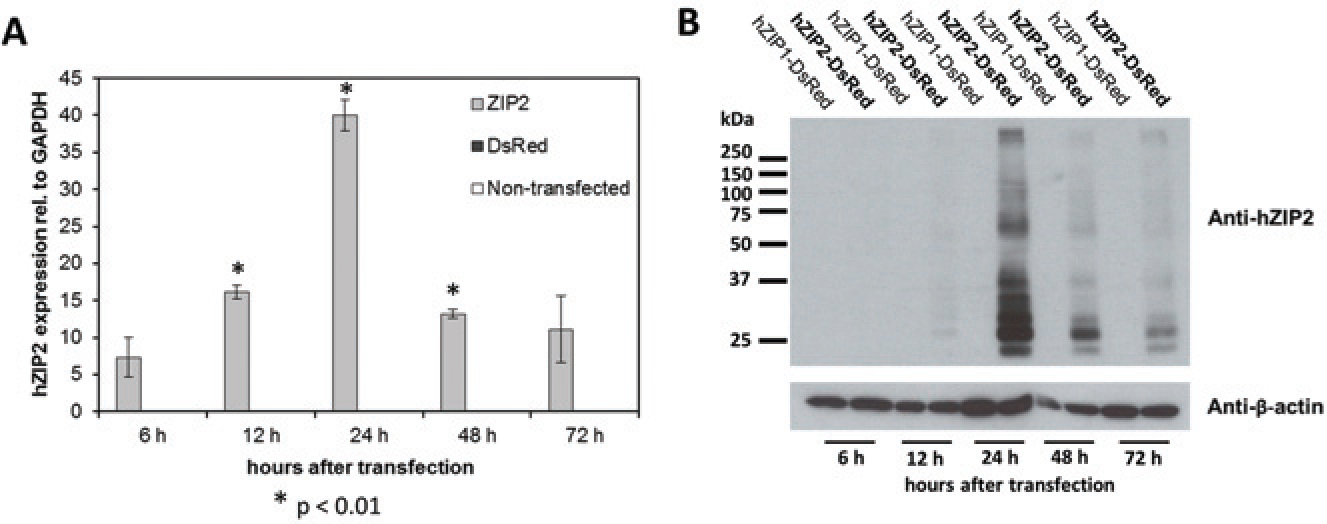
mRNA and protein expression of human ZIP2 (hZIP2). HEK293 cells were transiently transfected with pIRES2-DsRedExpress 2-hZIP2 construct. mRNA (
To examine whether the overexpressed proteins are targeted to the plasma membrane, we first performed surface biotinylation experiments. These show clear expression of hZIP2 at the plasma membrane ( Fig. 2A ). A similar expression pattern was observed as seen in Figure 1B . The antibody did not recognize hZIP1. We also applied confocal laser scanning microscopy to confirm our findings of hZIP2 surface expression obtained with surface biotinylation. The topology of the protein predicts that both N- and C-termini are located on the extracellular side of the plasma membrane. Because the epitope recognized by the used anti-ZIP2 antibody is located at the N-terminus (aa 71–120), fluorescence staining obtained with no permeabilization indicates protein expression at the plasma membrane. Using this technique, we observed a green ring along the cell membrane in hZIP2-expressing HEK293 cells (showing red fluorescence due to DsRed Express2 expression; Fig. 2B ). When the cellular membranes were permeabilized, we obtained an additional, strong, intracellular fluorescent signal that corresponds to protein expressed in the endoplasmic reticulum and Golgi (data not shown).
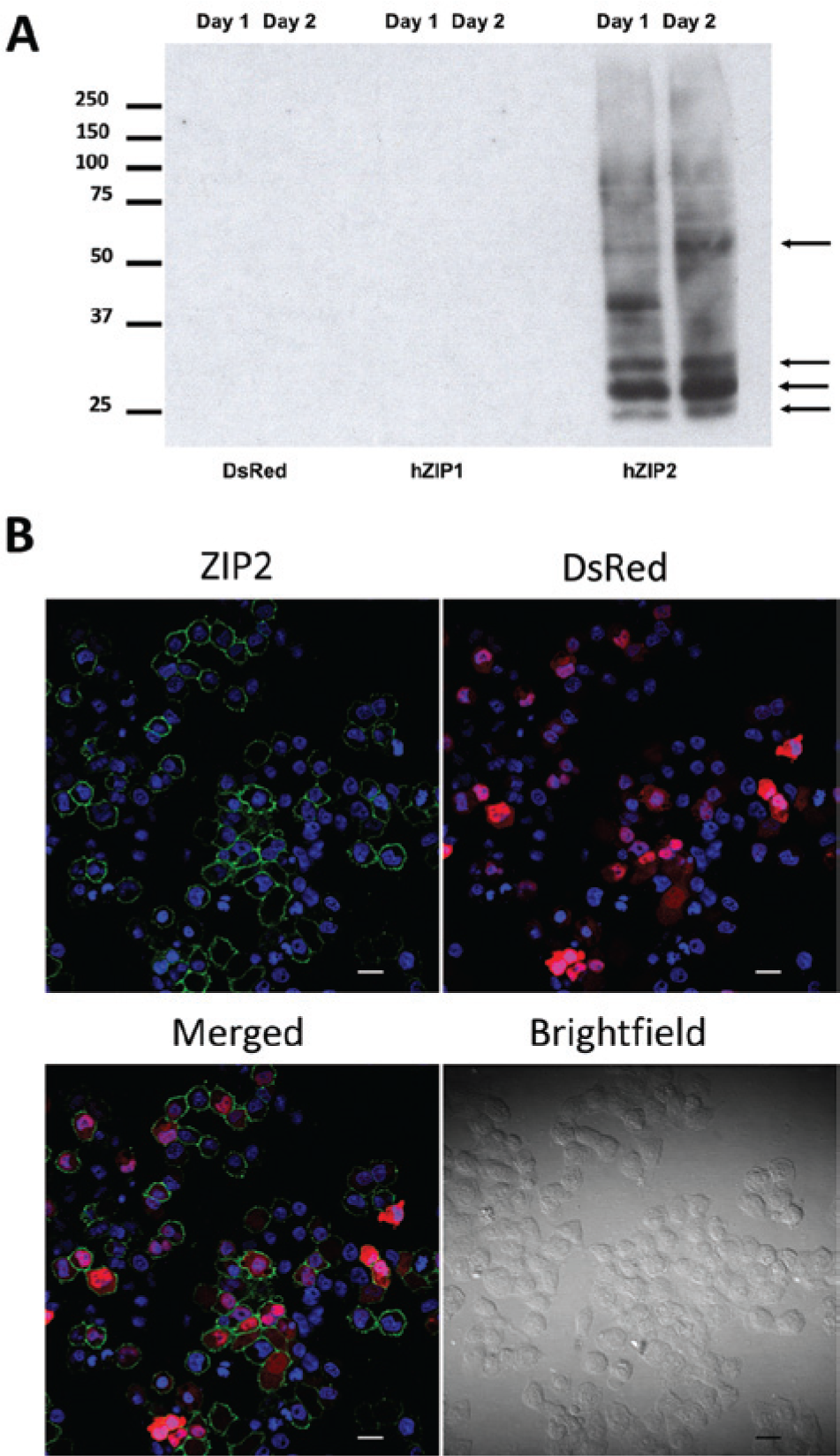
Expression of human ZIP2 (hZIP2) at the plasma membrane. Surface biotinylation experiments revealed that hZIP2 is expressed in the membrane fractions of hZIP2-expressing HEK293 cells (
hZIP2 Transport Activity at Different Extracellular pHs
Cadmium was reported to be transported by hZIP2.11,12 We also published previously that cadmium, as a substrate for ion channels such as TRPV5 and TRPV6, provides a powerful assay.13–15 When using a fluorescence-based cadmium influx assay at an extracellular pH of 7.4, we observed cadmium influx in HEK293 cells transfected with empty (DsRed Express2) vector only at the higher µM Cd concentrations ( Fig. 3A ). In contrast, hZIP2-expressing cells take up cadmium at low µM concentrations, but the increase in uptake was small ( Fig. 3B ). Because hZIP2 is expressed at the luminal surface in prostate epithelial cells and the pH of the prostatic fluid is ~6.2, we tested the activity at lower pH values. We observed that reduction of the pH of the extracellular buffer from pH 7.4 to pH 7.0 or 6.5 significantly increased the cadmium uptake in the hZIP2-expressing HEK293 cells but not in the DsRed Express2–expressing HEK293 cells ( Figs. 4A , B ). The highest hZIP2 activity was at pH 6.5. hZIP2 transports cadmium with an apparent Km of 30.4 nM or 53.96 nM at pHs 7.4 and 6.5, respectively.
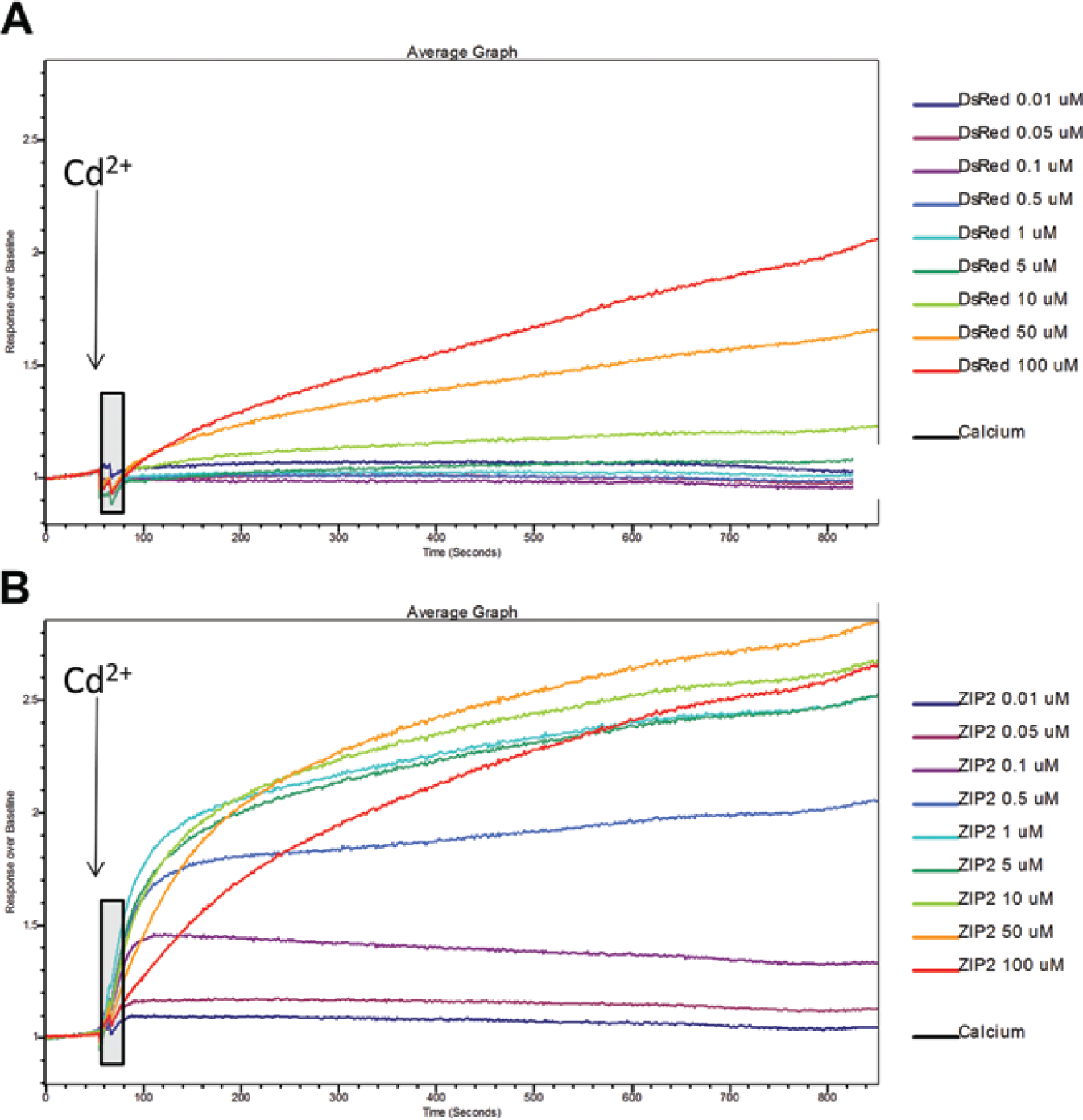
Cadmium transport via human ZIP2 (hZIP2). Representative, averaged calcium-5 tracings obtained with a FLIPR Tetra fluorescence, high-throughput screening microplate reader show cadmium influx in DsRed Express2– (
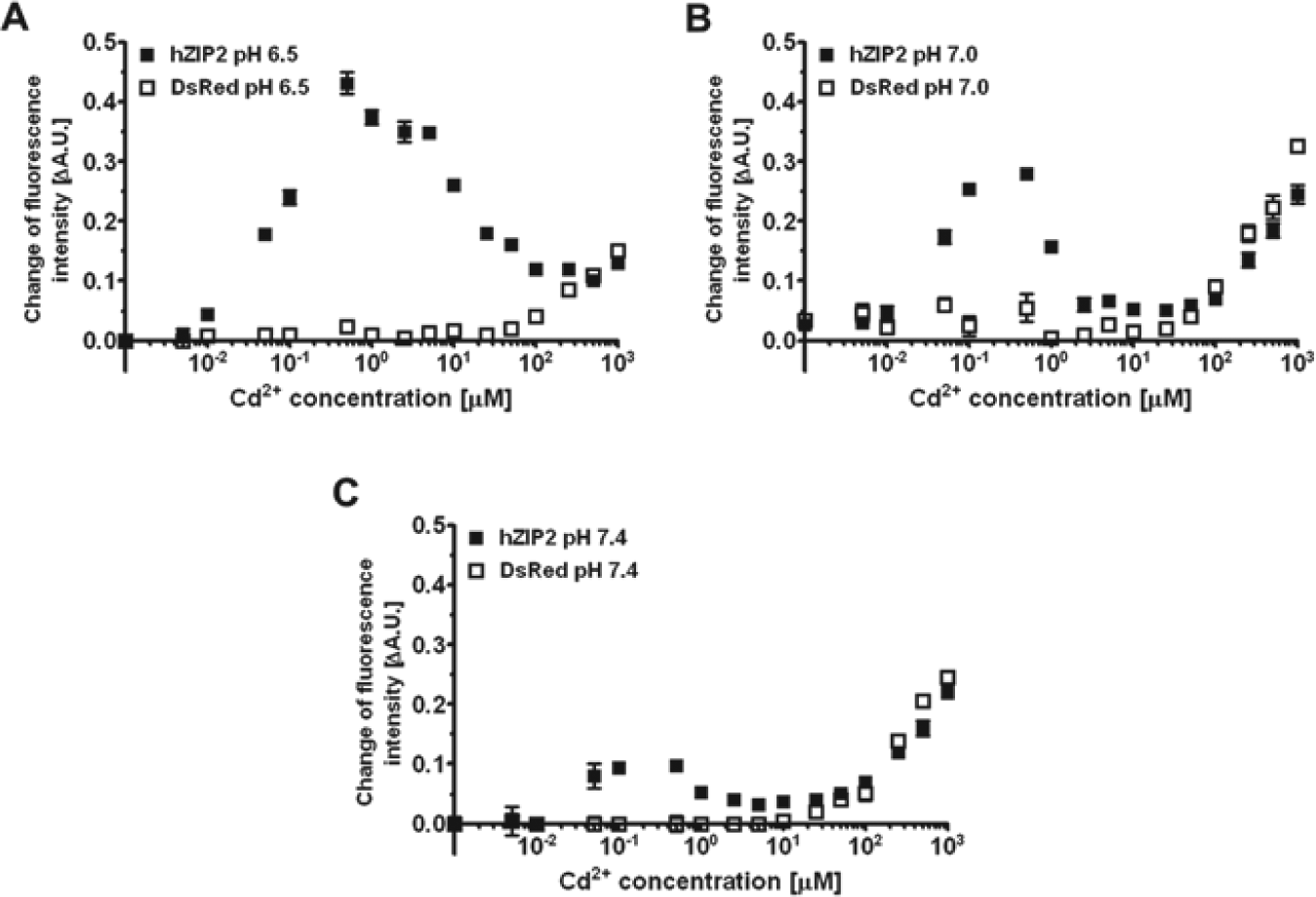
Cadmium transport activity of human ZIP2 (hZIP2) at different extracellular pHs. Changes of fluorescence intensity of calcium-5 in response to different cadmium concentrations were measured in transiently transfected DsRed Express2–expressing (open squares) and hZIP2–expressing (closed squares) HEK293 cells at extracellular pH 6.5, 7.0, and 7.4 (
Inhibition of hZIP2-Mediated Cadmium Uptake by Zinc
To confirm that the observed increased cadmium influx is indeed mediated by hZIP2, we tested the effect of zinc on the cadmium influx via hZIP2. The fluorescence of the calcium-5 dye was not affected by zinc. In other words, it was not sensitive to zinc (data not shown). Using 0.1 µM cadmium as substrate, we found that zinc inhibited hZIP2-mediated cadmium influx with an apparent Ki of 173.9 nM ( Fig. 5 ). The dose-response curve of zinc was shifted rightward with an increase in cadmium concentration from 0.1 to 10 µM ( Fig. 5 ), indicating competitive inhibition and suggesting that zinc is also transported by hZIP2.
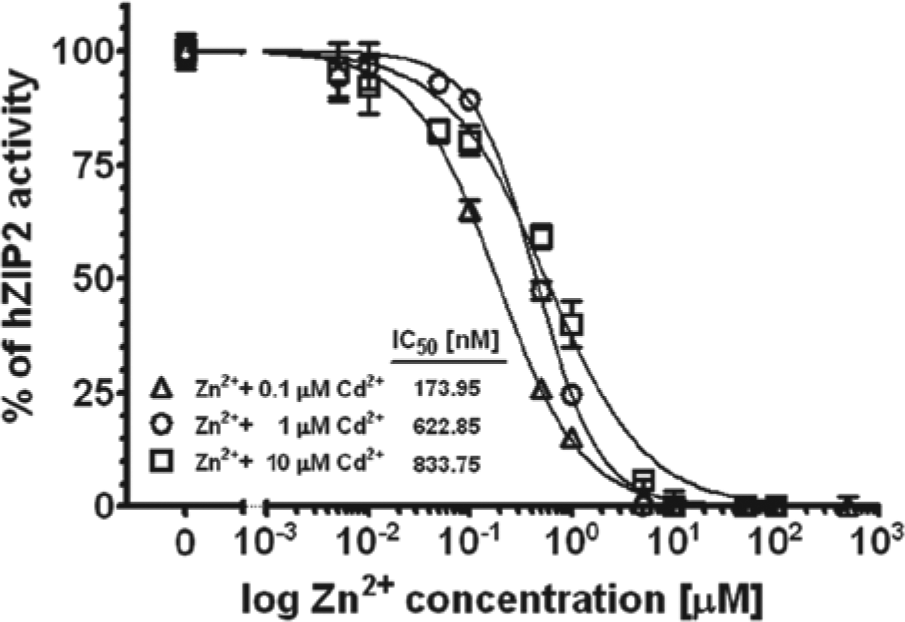
Inhibition of human ZIP2 (hZIP)–mediated Cd2+ transport by zinc. Zinc was applied in the presence of 0.1 µM, 1 µM, or 10 µM of Cd2+. Measurements were performed at an extracellular pH 6.5. Dose-response curves were established and IC50 values were calculated (n = 2).
Screening Assay for hZIP2 Activity
To obtain a large dynamic range for the screening assays, we established a high-throughput screening (HTS) assay using a stably hZIP2-expressing HEK29 clone, measuring transport at pH 6.5. We plated 30,000 hZIP2-expressing clonal cells and native HEK293 cells into a black 96-well plate ( Fig. 6 ). To avoid the “edge effect” in the microtiter plates, no cells were plated into the outer wells. Twenty-four hours later, the wells were subconfluent and ready for the assay. Administration of 10 µM cadmium induced a robust change in fluorescence intensity of calcium-5 in the hZIP2-expressing clone, whereas minimal increase was observed in native HEK293 cells ( Fig. 6A ). The average of the calculated Z-scores from three different experiments was 0.57 ± 0.08. Because the EC50 value for cadmium was 53.96 nM at pH 6.5, we decreased the concentration of the applied cadmium to 1 and 0.1 µM. As was expected, the maximum response was decreased compared with previous data using 10 µM cadmium; however, the obtained Z-scores were still greater than 0.5 (0.74 and 0.54, respectively; Fig. 6B , C ).
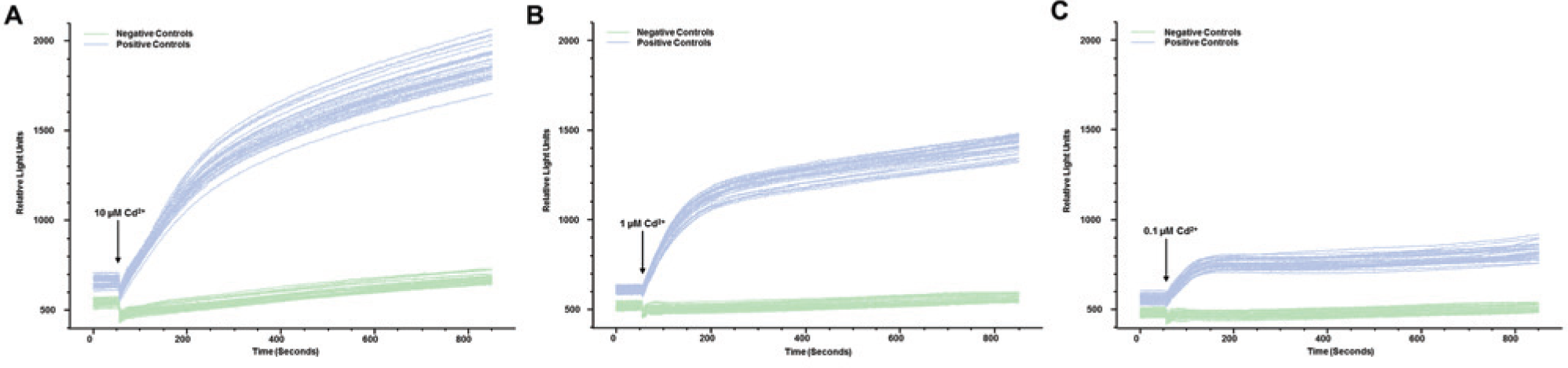
Screening assay validation using a human ZIP2 (hZIP2)–expressing HEK293 clone. Representative tracings show the effect of the administration of 10 µM, 1 µM, or 0.1 µM cadmium on the fluorescence intensity of the calcium-5 fluorescence dye in hZIP2-expressing clone (positive ctrl) and nontransfected HEK293 cells (negative control) using the FLIPR Tetra high-throughput screening fluorescence microplate reader (
Discussion
The ZIP (ZRT1, IRT1-like protein, SLC39A) transporter superfamily contains many members in eukaryotic organisms including plants, fungi, nematodes, and vertebrates. The founding members of this superfamily, ZRT1, ZRT2, and IRT1, were cloned from Saccharomyces cerevisiae and Arabidopsis thaliana more than a decade ago, respectively.16,17 The human ortholog of ZIP2 was identified and cloned some years later. 11 The mRNA of mZIP2 is expressed at a low level as compared with mZIP1 and only in certain tissues, with the highest levels detected in the skin, liver, ovary, and visceral yolk sac. 12 hZIP2 protein was also found to be expressed in high amounts in prostate, where it was localized to the apical membrane of prostate epithelial cells. 8 Development of a functional, screening assay for hZIP2 provides a great tool to examine the function and regulation of Zip2 as well as to develop specific and efficient inhibitors/activators against this protein for therapeutic purposes.
hZIP2 has been characterized in detail using only radioactive 65Zn uptake that revealed zinc as a substrate for this transporter together with Cd2+, Cu2+, Co2+, and Mn2+.11,12 However, the exact transport mechanism has not yet been elucidated.
In this study, first we cloned hZIP2 into a DsRed-Express2–containing, bicistronic expression vector. The transient transfection of the hZIP2-containing bicistronic construct resulted in a large overexpression of the hZIP2 protein in HEK293 cells. Surface biotinylation experiments revealed that hZIP2 is expressed at the plasma membrane in high amounts. These data show that our expression system is suitable for the screening for modulators of hZIP2 expressed in the plasma membrane.
Previously, we reported that cadmium influx by the epithelial calcium channel TRPV6 can be measured using the calcium-5 fluorescence dye using the FLIPR Tetra microplate reader.13–15 Because the calcium-5 fluorescence dye is completely insensitive to zinc, we selected cadmium as a suitable divalent cation to measure hZIP2 activity using FLIPR Tetra. Given that hZIP2 is a transporter, we kept the temperature at 37 °C throughout our entire assay (because transporters are more temperature sensitive than channels). We performed our experiments in modified Krebs solution under bicarbonate-free conditions at pH 7.4 using different cadmium concentrations as a starting point. Our solution contained chloride as a sole anion to prevent any precipitation with cadmium.
Under these conditions, we observed cadmium influx at higher µM Cd concentrations when using the vector-only transfected control cells. This influx is presumably due to the permeability of calcium channels. In contrast, cadmium uptake was significantly higher, down to the nM and µM Cd range in hZIP2-expressing cells compared with vector-only transfected cells. At the high µM Cd range, no difference could be observed between transfected and control cells, suggesting that hZIP2 activity is inhibited by a high concentration of extracellular cadmium.
Because the differences in the cadmium uptake rates between the hZIP-expressing and control cells were relatively small, we tried to find the right conditions to increase the signal-noise ratio by increasing the hZIP2 activity. Accordingly, we performed experiments at different extracellular pH values. Our data revealed that lowering the extracellular pH dramatically enhanced cadmium-induced changes in the fluorescence signal in hZIP2-expressing cells. The data are supported by the observation that the pH of the prostatic fluid is acidic (6.1–6.6) due to the secretion of the high amount of citrate, which would favor zinc reabsorption via ZIP2 at the apical surface of the prostate epithelial cells under physiological conditions.18–20
As was reported previously for other ZIP transporters (ZIP8 and ZIP14), the cadmium uptake via hZIP2 was decreased by copper and zinc through competitive inhibition experiments, suggesting that these divalent cations are transported by hZIP2 as well.21,22 To further support that our assay indeed measures hZIP2 activity, we tested the effect of extracellular zinc on the cadmium influx via hZIP2. We found that zinc inhibits hZIP2-mediated cadmium uptake with an apparent IC50 of 53.96 nM at 10 µM extracellular cadmium concentration. The IC50 value decreased when extracellular cadmium concentration was reduced, whereas the maximal inhibition by zinc remained 100%, suggesting competitive inhibition. These data confirm that our assay measures specifically hZIP2 activity.
Based on our obtained data, we validated the screening assay at an extracellular pH of 6.5 using 0.1, 1, and 10 µM extracellular cadmium concentrations. Under these conditions, we found a negligible endogenous cadmium influx and a large increase in the fluorescence intensity of calcium-5 in response to cadmium administration in hZIP2-expressing cells. As expected, increasing the concentration of extracellular cadmium resulted in enhanced response of the calcium-5 fluorescence signal. The obtained Z-factor was greater than 0.5 in each case (0.54, 0.74, and 0.57, n = 3, respectively), which shows that our assay is a reliable and useful for HTS, looking for specific hZIP2 modulators. As previously observed for TRPV5 and TRPV6 HTS, the outer wells of the 96 microplates were not suitable for measurement because of significant deviations of the fluorescence signal when compared with the inner wells.
In summary, we established a highly reliable, nonradioactive fluorescence-based assay that is suitable for measuring the activity of the human ZIP2 overexpressed in HEK23 cells using the FLIPR Tetra fluorescence microplate reader. The developed fluorescence-based screening assay can be employed for compound screening to identify specific inhibitors or activators of ZIP2. This assay can be adapted to other membrane transport systems that mediate cadmium flux through the plasma membrane. Identified modulators of hZIP2 can be useful tools to study the role of hZIP2 in normal prostate epithelial cells. In addition, these compounds might be of therapeutic relevance for the prevention of prostate cancer.
Footnotes
Acknowledgements
The authors would like to thank Maria Feher for her excellent cell culture assistance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.