
Abstract
The target of this study, the PfM18 aspartyl aminopeptidase (PfM18AAP), is the only AAP present in the genome of the malaria parasite Plasmodium falciparum. PfM18AAP is a metallo-exopeptidase that exclusively cleaves N-terminal acidic amino acids glutamate and aspartate. It is expressed in parasite cytoplasm and may function in concert with other aminopeptidases in protein degradation, of, for example, hemoglobin. Previous antisense knockdown experiments identified a lethal phenotype associated with PfM18AAP, suggesting that it is a valid target for new antimalaria therapies. To identify inhibitors of PfM18AAP function, a fluorescence enzymatic assay was developed using recombinant PfM18AAP enzyme and a fluorogenic peptide substrate (H-Glu-NHMec). This was screened against the Molecular Libraries Probe Production Centers Network collection of ~292,000 compounds (the Molecular Libraries Small Molecule Repository). A cathepsin L1 (CTSL1) enzyme-based assay was developed and used as a counterscreen to identify compounds with nonspecific activity. Enzymology and phenotypic assays were used to determine mechanism of action and efficacy of selective and potent compounds identified from high-throughput screening. Two structurally related compounds, CID 6852389 and CID 23724194, yielded micromolar potency and were inactive in CTSL1 titration experiments (IC50 >59.6 µM). As measured by the Ki assay, both compounds demonstrated micromolar noncompetitive inhibition in the PfM18AAP enzyme assay. Both CID 6852389 and CID 23724194 demonstrated potency in malaria growth assays (IC50 4 µM and 1.3 µM, respectively).
Introduction
According to the World Health Organization (WHO), the parasites responsible for causing malaria—Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, and Plasmodium malariae—infect up to 500 million people per year and result in an estimated 650,000 fatalities. 1 Current antimalarial regimens are not fully effective due to emerging drug resistance and lack of their ability to prevent transmission. 2 Approximately 40% of the world’s populations live in areas where the risk of malaria transmission is high, typically the tropic zones located closer to the equator. The symptoms of malaria have a rapid onset with recurrent cycles of fever and chills, muscle aches, headaches, vomiting, and jaundice, but may be extended to 8 to 10 months after the initial infected mosquito bite occurs. 3
The life cycle of P. falciparum, the most prevalent and severest etiologic agent of malaria, is complex. It is transmitted by the female Anopheles mosquito to its human host, where the parasites first enter the liver cells before emerging and invading erythrocytes. It is during this stage that P. falciparum uses protein-protein interactions and enzymatically driven processes to allow entry, growth, and escape from the human erythrocyte. One such enzyme is PfM18AAP, a ~67-kDa metallo-aminoipeptidase that oligomerizes and is the sole aspartyl aminopeptidase (AAP) present in the malaria parasite. 4 PfM18AAP is thought to play an important role in the process of protein degradation and, working together with other exo-aminopeptidases, is believed to digest host hemoglobin, an important source of amino acids for the parasite.5,6 PfM18AAP may also interact with the erythrocyte membrane protein, spectrin, presumably to regulate the integrity of the infected erythrocyte membrane skeleton during parasite growth, allowing for host cell expansion and parasite exit. 7 Antisense RNA knockdown experiments have shown that inhibition of intraerythrocytic PfM18AAP debilitates the malaria parasite, making it a promising drug target. 6 Inhibitors of PfM18AAP will thus provide new tools to further our understanding of this enzyme’s function and increase the potential to develop therapies against P. falciparum transmission.
Currently, there are no known small-molecule inhibitors of PfM18AAP. Previous reports have identified the phosphinic and phosphonic acid analogues of glutamate and aspartate, GluP and AspP, as modest amino acid–derived inhibitors of PfM18AAP in vitro. However, these amino acid derivatives do not reduce malaria growth in culture when tested at concentrations up to 100 µM. 8 Thus, the objective of this research program was to identify compounds that inhibit the activity of PfM18AAP. Described here is our approach to discovering such inhibitors by high-throughput screening (HTS) of compound libraries, which yielded several series of potent compounds that demonstrate selectivity to the enzyme. Two of these compounds are noncompetitive inhibitors of PfM18AAP and also inhibit the growth of P. falciparum.
Materials and Methods
Recombinant Enzymes and Assay Reagents
Functionally active purified recombinant PfM18AAP was prepared at the University of Technology Sydney as previously described. 8 Characterization studies with this enzyme have demonstrated that it has comparable properties to that of native PfM18AAP taken from cytosolic extracts. 8 Recombinant cathepsin L was prepared in the same laboratory as previously described. 9 All other reagents were purchased or made from standard laboratory supplies as listed: H-Glu-NHMec substrate (part I-1180; Bachem, Torrance, CA), Z-Leu-Arg-MCA substrate (part MCA-3210-v; Peptides International, Louisville, KY), 1536-well plates (part 789176; Greiner Bio-One, Monroe, NC), Tris (part 0497; Amresco, Solon, OH), CoCl2.6H2O (part D3247; Univar, Redmond, WA), dithiothreitol (DTT) (part 15508-013; Invitrogen, Carlsbad, CA), ZnCl2 (part 208086; Sigma, St. Louis, MO), Z-Phe-Ala-diazomethylketone (part N-1040; Bachem, Torrance, CA), and bovine serum albumin (BSA) (part 126609; Calbiochem, Billerica, MA). PfM18AAP assay buffer was prepared as 50 mM Tris-HCl (pH 7.5), 4 mM CoCl2, and 0.1% BSA (w/v). PfM18AAP substrate buffer was prepared as 50 mM Tris-HCl (pH 8.8). CTSL1 assay buffer was prepared as 25 mM Tris-HCl (pH 7.5), 1 mM DTT, and 0.1% BSA (w/v).
Compounds
The Molecular Libraries Small Molecule Repository (MLSMR) library was provided by the National Institutes of Health’s Molecular Libraries Initiative. Details regarding compound selection for this library can be found online (http://mli.nih.gov/mli/compound-repository/mlsmr-compounds/). Briefly, the MLSMR library is a highly diversified collection of small molecules (more than 50% of compounds exhibit molecular weights between 350 and 410 g/mol) comprising both synthetic and natural products, from either commercial or academic sources, that can be grouped into the three following categories: specialty sets of know bioactive compounds such as drugs and toxins, focused libraries aimed at specific target classes, and diversity sets covering a large area of chemical space. Solid samples (powders) of CID 6852389 (SID 11532952/87693049) and CID 23724194 (SID 47200698/92117383) were purchased from Sigma (cat. D043) and Vitas-M Laboratory (Moscow, Russia; cat. STK091533), respectively.
PfM18AAP Screening Assay
This enzymatic assay uses a fluorogenic peptide substrate (H-Glu-NHMec), which is incubated with purified recombinant PfM18AAP in the presence of test compounds. Cleavage of the substrate by PfM18AAP liberates the 7-amino-4-methylcoumarin fluorogenic leaving group (NHMec) from the peptide, leading to increased fluorescence (
Fig. 1
). Enzymatic inhibitors block PfM18AAP-mediated cleavage of H-Glu-NHMec and liberation of the NHMec leaving group from the substrate, resulting in decreased fluorescence as measured at 340 nm excitation and 450 nm emission. To reduce the number of compounds that optically interfere with the measurement, initial (T0) and 90-min (T90) measurements of plate fluorescence were taken after addition of substrate. Test compounds were assayed in singlicate at a final nominal concentration of 7 µM. A stepwise assay protocol is presented in
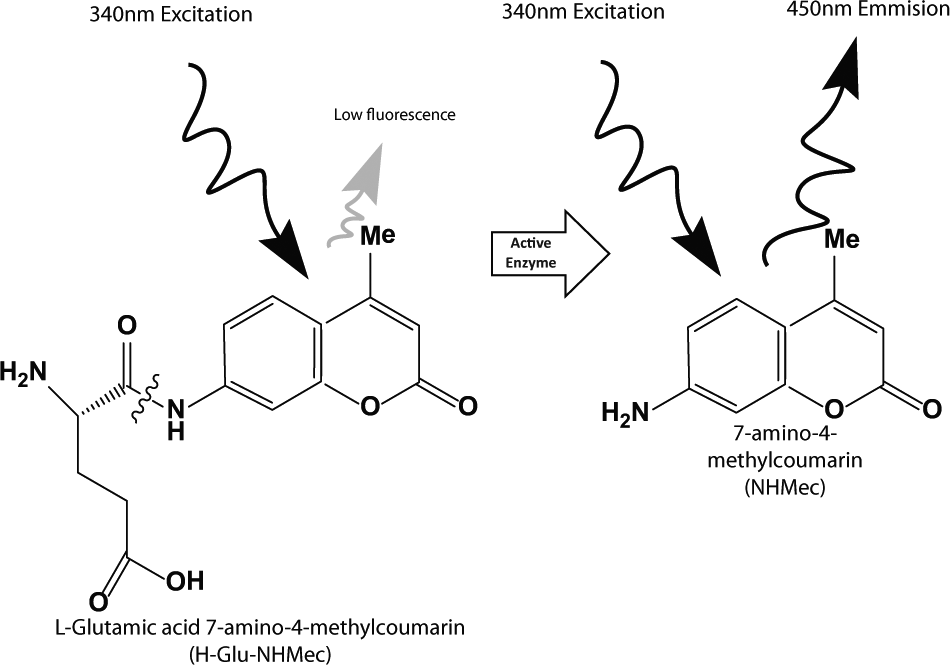
PfM18AAP enzymatic assay principle. PfM18AAP activity is determined by measuring the release of the 7-amino-4-methylcoumarin fluorogenic group (NHMec) from a peptide substrate (H-Glu-NHMec). Represented by the wavy line, cleavage of H-Glu-NHMec’s glutamic acid group by PfM18AAP liberates NHMec from the peptide, leading to increased well fluorescence. As designed, the screening assay identifies compounds that inhibit PfM18AAP-mediated cleavage, resulting in decreased well fluorescence.
PfM18AAP Ultra-High-Throughout Screening Campaign Summary and Results.
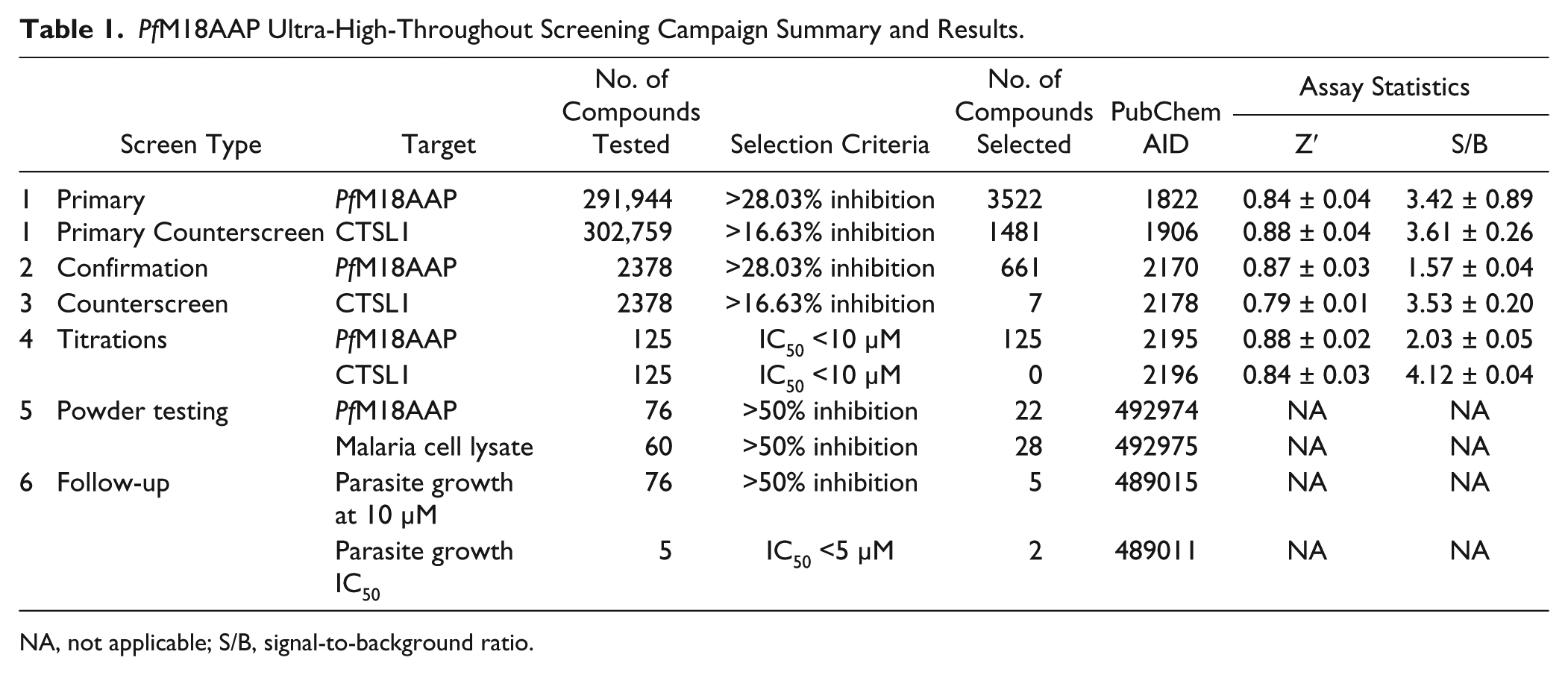
NA, not applicable; S/B, signal-to-background ratio.
Cathepsin L1 (CTSL1) Screening Assay
CTSL1 is a functionally active recombinant form of a cysteine endopeptidase derived from the worm parasite Fasciola hepatica. Similar in principle to the PfM18AAP assay, this enzymatic assay uses a fluorogenic peptide substrate (Z-Leu-Arg-NHMec) that is incubated with purified recombinant CTSL1 in the presence of test compound. This assay serves as a useful counterscreen because it also measures the level of the released coumarin moiety to monitor enzyme activity, albeit using a completely different enzyme. Hence, compounds that inhibit both assays are either nonspecifically inhibiting the enzymes or possibly behaving as fluorescence-quenching artifacts. Either outcome is undesirable and leads to a definitive path to remove unwanted compounds. Final assay conditions are shown in
PfM18AAP HTS Campaign
Details relative to each step of the PfM18AAP and CTSL1 screening assays are shown Table 1 . Briefly, the first stage involved singlicate screening against PfM18AAP (the primary screen) and CTSL1 (the primary counterscreen) at a final compound test concentration of 7 µM or 6 µM, respectively (see PubChem AIDs 1822 and 1906 for protocol details). The final DMSO concentration was 0.7% (v/v) for the PfM18AAP assay or 0.6% for the CTSL1 assay. Well fluorescence was measured with a ViewLux plate reader (PerkinElmer, Waltham, MA), and the percent inhibition of each test compound was calculated on a per-plate basis as further described below. The numerical cutoff used to qualify active (“hit”) compounds was calculated as the average percentage inhibition of all compounds tested plus three times their standard deviation. 10 The confirmation and counterscreens were run on selected hits in the same conditions as the primary screens, except that plates were assessed in triplicate and results for each compound were reported as the average percentage inhibition of the three measurements, plus or minus the associated standard deviation (PubChem AIDs 2170 and 2178). For titration experiments, assay protocols were identical to those described above, with the exception that compounds were prepared in 10-point, 1:3 serial dilutions starting at a nominal test concentration of 74 µM and assessed in triplicate (PubChem AIDs 2195 and 2196). To confirm activity of the best inhibitors, compounds were purchased as powder samples and tested in various secondary assays, including Ki determination and two other assays, including a malaria cell lysate assay and a P. falciparum parasite growth assay (see below).
Screening Data Acquisition, Normalization, Representation, and Analysis
All screening assays were run on a Kalypsys (San Diego, CA)/GNF robotic platform in 1536-well microtiter plates. Fluorescence was measured by ViewLux plate reader (PerkinElmer). Raw data were uploaded into an institutional HTS database (Accelrys, San Ramon, CA) for further processing. As a first calculation, T0 was subtracted from T90 for each individual well (“delta RFU”). Activity of each well was normalized on a per-plate basis using the following equation:

where “test_compound” is defined as wells containing test compound, “negative_control” is defined as the median of the wells containing test compounds, and “positive_control” is defined as the median of the wells containing ZnCl2 or Z-Phe-Ala-diazomethylketone for PfM18AAP or CTSL1, respectively (n = 24 wells).
Each assay plate underwent a quality control check; a Z′ value greater than 0.5 was required for acceptance of data. 11 For titration experiments, triplicate percent inhibition values were plotted against compound concentration. A four-parameter equation describing a sigmoidal concentration-response curve was then fitted with an adjustable baseline using Assay Explorer software (Accelrys). Concentration-response curves and IC50 values presented in this study were generated by Prism (GraphPad Software, San Diego, CA).
Malaria Cell Lysate Assay
This assay used soluble freeze-thaw extracts of parasites and the substrate H-Glu-NHMec as previously published. Briefly, in vitro infected human red blood cells were harvested and washed with phosphate-buffered saline (PBS), and parasites were then released using saponin. The released parasites were subjected to three rounds of freeze-thaw, and extracts were harvested as supernatants. 8 Activity of test compounds versus PfM18AAP extracts was assessed by incubating the compounds with the extracts in the presence of CoCl2 and H-Asp NHMec. Vehicle and background controls were included in the assay. Test compounds that inhibited extract activity decreased the cleavage of the substrate, which was measured as a decrease in fluorescence proportional to the inhibitory activity of the test compound.
Parasite Growth Assay
The whole-cell malaria growth assay was used to identify compounds that inhibit P. falciparum growth in red blood cells (i.e., P. falciparum’s asexual erythrocytic stage as previously published). 12 Briefly, compounds were incubated with P. falciparum–infected red blood cells (RBCs) in hypoxanthine-free media. 3H-hypoxanthine was added to treated cultures. After 48 h, 3H incorporation was determined. Vehicle and background (RBC) controls (DMSO ≤1%) were included on each plate. As designed, compounds that inhibit the growth of P. falciparum in RBCs decreased the level of 3H incorporated.
Ki Determination and Mode of Inhibition
CID 6852389 and CID 23724194 powder samples were subjected to Ki determination versus purified recombinant PfM18AAP. Reaction buffer consisted of 50 mM Tris-HCl (pH 7.5) + 0.1 mM CoCl2. Reactions were run in a 100-µL assay volume for 1 h with 60-s measurement intervals. The substrate Glu-NHMec was tested in the range of 0.5, 1, 10, 100, 250, and 500 µM while the inhibitor concentrations were tested in the range of 1, 5, 10, and 50 µM. Enzyme and inhibitors were incubated 30 min with reaction buffer before substrate addition. GraphPad Prism software was used for graphical (Lineweaver-Burk) and quantitative (nonlinear regression) determination of inhibition mode and Ki.
Cheminformatics
A Maximum Common Substructure hierarchical clustering (ChemAxon LibraryMCS 5.10.1, Cambridge, MA) was used to identify the scaffolds of active compound families from the 125 active compounds found from the PfM18AAP HTS.13–16 In addition to clustering analysis, physical properties of the clustered compounds were calculated (ChemAxon Instant JChem 5.9.4). Calculated results included molecular mass, topological polar surface area, chiral atoms, H-bond acceptors/donors, ring count, and rotatable bonds.
Results
Implementation of the PfM18AAP and CTSL1 Screening Assays
To help identify potent and selective inhibitors, we implemented a screening strategy that uses a counterscreen to triage nonselective hits and assay artifacts at each HTS stage; in this case, recombinant CTSL1, a cysteine peptidase derived from parasitic helminth F. hepatica, was selected as the counterscreen for PfM18AAP compound hits. The PfM18AAP and CTSL1 screening assays were both designed to measure the inhibition of recombinant enzyme by monitoring the fluorescence of a methylcoumarin-labeled peptide substrate (H-Glu-NHMec and Z-Leu-Arg-NHMec, respectively). The measured fluorescence signal increases as enzyme cleaves the substrate and releases aminomethylcoumarin, whereas enzymatic inhibitors leave the substrate intact, resulting in low amounts of measured fluorescence. The assay principle is illustrated for the PfM18AAP screening assay in Figure 1 ; the CTSL1 counterscreen assay was similarly formatted with an appropriate fluorogenic substrate (Z-Leu-Arg-NHMec). As shown in Figure 2A , the positive control inhibitor for the PfM18AAP assay, Zn(II), yielded an IC50 of 662 ± 314 nM (n = 33 plates). Concentration-response analysis of the CTSL1 inhibitor, Z-Phe-Ala diazomethylketone, resulted in an average IC50 of 15.45 ± 2.66 nM (n = 22 plates).
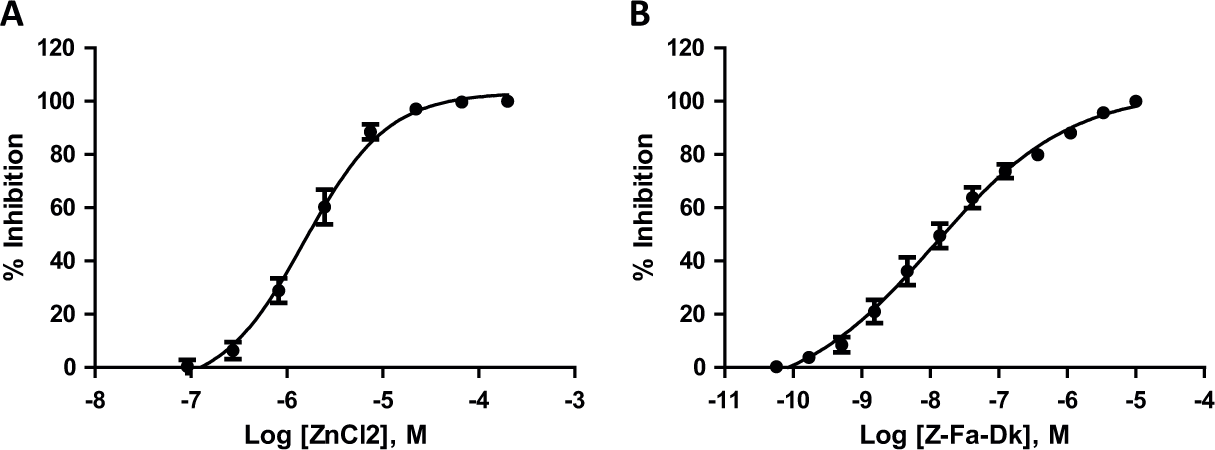
Performance of positive controls in the PfM18AAP and CTSL1 screening assays. (
The PfM18AAP primary assay and CTSL1 counterscreen assay were implemented at a final volume of 5 µL/well in 1536-well plates. The assays were screened against the entire available MLSMR collection; 277,728 unique compounds were tested in both assays. All MLSMR compounds were screened at 7 µM for PfM18AAP and 6 µM for CTSL1. For the PfM18AAP screening assay, an IC100 of Zn(II) was used as a positive control for enzyme inhibition. The median of the wells containing test compounds was used as a negative control (i.e., IC0). Using these controls ( Fig. 3 ), the PfM18AAP assay demonstrated robust screening statistics. It had an average signal-to-background ratio (S/B) of 3.42 ± 0.89 and a Z′ of 0.84 ± 0.04 (n = 241 plates). The CTSL1 assay was similarly robust; using Z-Phe-Ala diazomethylketone and the median of the wells containing test compounds as positive and negative controls, respectively, yielded an S/B of 3.61 ± 0.26 and a Z′ of 0.88 ± 0.04 over 247 plates.
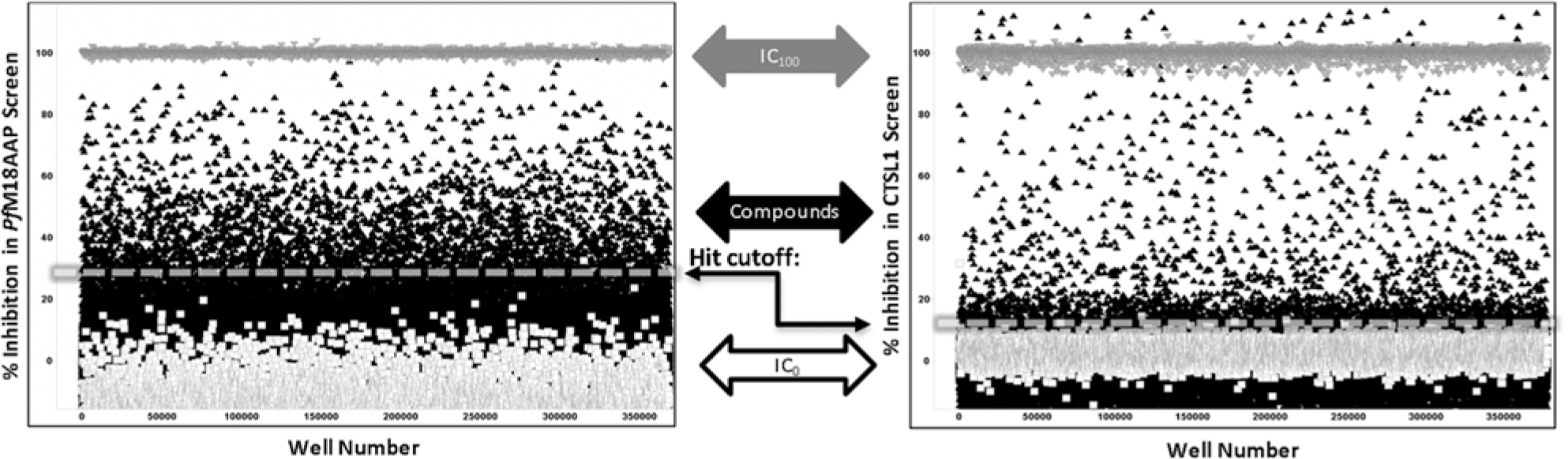
PfM18AAP (left panel) and CTSL1 (right panel) primary high-throughput screening (HTS) assay performance. Positive control wells are shown as inverted gray triangles (IC100 of ZnCl2 or Z-Phe-Ala-diazomethylketone). Results of compound wells (black triangles) and negative control wells (white squares) are also graphed. Calculated hit cutoffs are indicated via dashed lines. Due to the high degree of compound activity found in the PfM18AAP assay, data for both HTS campaigns were normalized to the median of the compound wells (IC0) and the median of the respective IC100, hence the noticeable shift below 0% inhibition for PfM18AAP assay.
Selection of Hits
The excellent screening statistics dictated our ability to select active compounds from singlicate data. 11 Due to an update in the MLSMR collection between the two screening campaigns, PfM18AAP was tested against 291,944 compounds and CTSL1 was tested against 302,759 compounds; this resulted in 277,728 compounds being tested in both assays. For these compounds, the CTSL1 assay results were used as a counterscreen for the PfM18AAP screening assay. That is, any compound found active against PfM18AAP but not active in the CTSL1 assay was prioritized for follow-up in the next round of testing ( Table 1 ). A graphical representation of these selection criteria is shown in Figure 4 . As graphed in the correlation plot, compounds to the right of the PfM18AAP inhibition cutoff (vertical dashed line) were considered active compounds in the PfM18AAP screen (“hits”); hits falling below the CTSL1 inhibition cutoff (horizontal dashed line) were considered selective inhibitors of PfM18AAP.
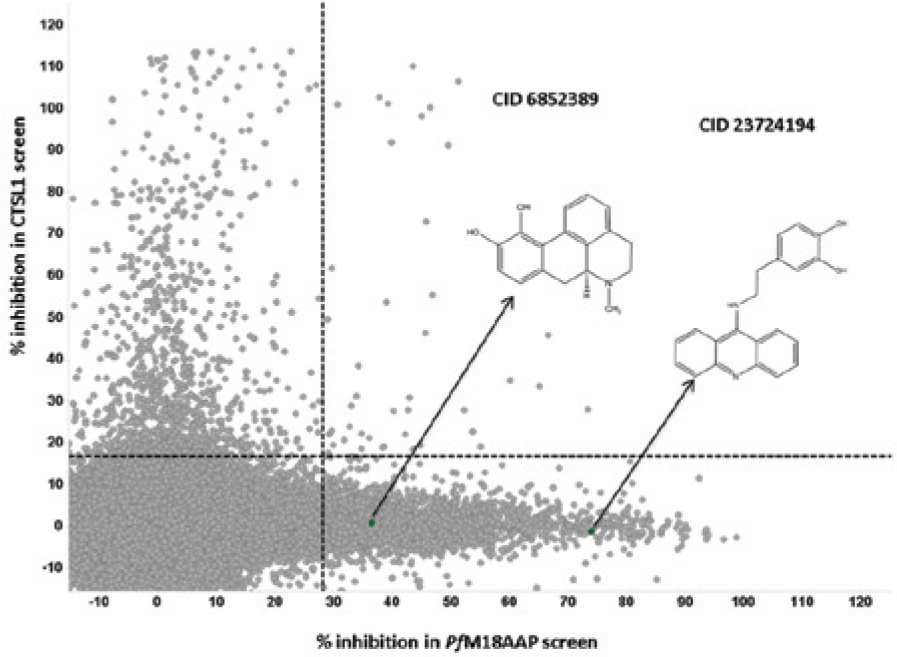
Comparison of PfM18AAP and CTSL1 primary screening results. The results of 277,728 compounds (gray circles) are shown. The horizontal dashed line represents the CTSL1 activity cutoff while the vertical dashed line represents the PfM18AAP activity cutoff. Green circles represent the two “hit” compounds that were identified as part of the primary high-throughput screen. Arrows indicate the screening results for CID 6852389 and CID 23724194.
To further enrich the data set for downstream follow-up studies, fresh aliquots of 2378 hit compounds demonstrating selective inhibition of PfM18AAP were tested in triplicate against both assays. To confirm activity and selectivity, the primary HTS hit activity cutoffs were reapplied to each set of screening results. This yielded 125 compounds that confirmed selective activity against PfM18AAP (i.e., their measured %inhibition was above the PfM18AAP primary HTS activity cutoff and less than the CTSL1 counterscreen HTS activity cutoff). Fresh aliquots of these compounds were obtained for further characterization in titration (IC50) assays. All compounds yielded IC50 values of <10 µM in the PfM18AAP titration assays; none of these demonstrated potency (defined as IC50 <10 µM) against CTSL1.
Cheminformatics Results
Maximum Common Substructure clustering of the 125 compounds analyzed resulted in 30 cluster groups being identified. However, 21 of these clusters had 3 or less members, in which 15 were singletons. In contrast, the four largest clusters (clusters 1, 3, 6, and 9) encompassed ~53% of the 125 compounds evaluated. As shown in
Figure 5
, these four clusters contain catechol, triazole, and benzamide moieties, which are found in compounds with known pharmacology.17–20 The largest grouping of compounds was in cluster 9, containing 33 compounds (~26%) with a methylated catechol moiety. This cluster of compounds has distinguishing properties that include a moderate level of hydrophobicity (ALogP ~2.2) as well as greater enrichment in H-bond acceptors/donors number with respect to the other clusters. Cluster 3 is the second largest grouping with 14 compounds represented by a 1,2,4 triazol-4-yl urea scaffold. These compounds have distinguishing properties that include greater hydrophilicity (ALogP ~0.36), smaller mass, and a lack of chiral centers. Cluster 6 is the third largest grouping with 10 compounds represented by a 1,2,4 triazol-4-yl acetamide scaffold, similar to cluster 3 in its structural and physical properties. Cluster 1 is the fourth largest grouping with nine benzamides. The physical properties generally straddle the values found within the other three clusters. A summary of physical parameters for each cluster is presented in
Figure 5
. Clustering results and calculated physical properties for all compounds are shown in
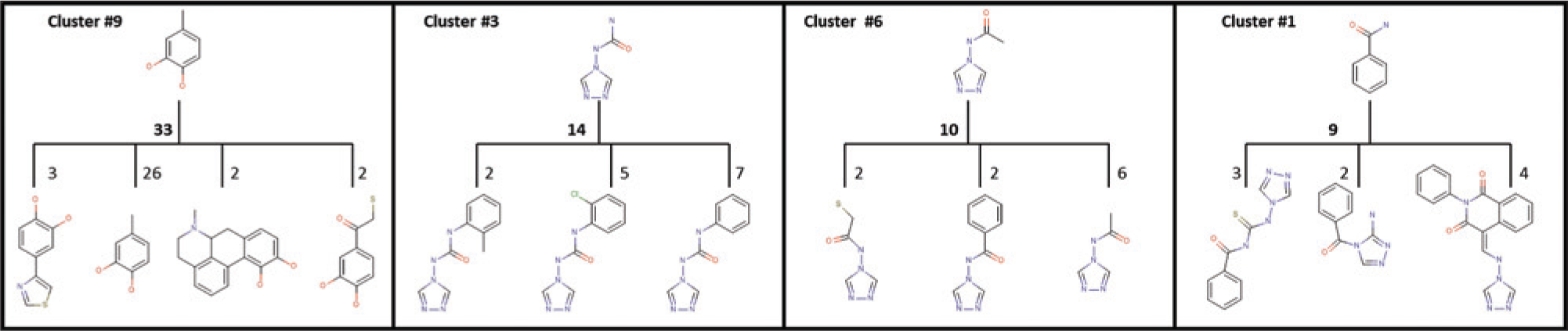
Clustering results of potent PfM18AAP hits. Presented are the most common substructure and respective second-tier derivatives for the four most populous clusters. Cluster 9 (33 compounds total) is represented by a 1,2 catechol moiety; cluster 3 (14 total) is represented by a 1,2,4 triazol-4-yl-urea moiety; cluster 6 (10 total) is represented by a (1,2,4 triazol-4-yl) acetamide moiety; and cluster 1 (9 total) is represented by a benzamide moiety. CID 6852389 and CID 23724194 both belong to cluster 9. Cluster numbers are assigned by the clustering algorithm; all results are provided in the supplemental section.
Mechanistic Assay Results
From the 125 compounds that demonstrated potent and selective inhibition of PfM18AAP, a subset of compounds most tractable for structure-activity relationship (SAR) studies was purchased or resynthesized as powder samples. These powder samples were retested to confirm inhibition of purified recombinant PfM18AAP, as well as in malaria lysates, and also tested for their ability to inhibit P. falciparum parasite growth via 3H hypoxanthine incorporation. Two efficacious compounds were identified from this effort: CID 6852389, (S)-(+)-apomorphine hydrochloride hydrate, and CID 23724194, the hydrochloride salt of 4-[2-(acridin-9-ylamino)ethyl]benzene-1,2-diol. CID 6852389 was found active in the P. falciparum lysate assay (84% efficacy at ≤5 µM; CID 23724194 was unavailable for testing). In the 3H hypoxanthine incorporation assay, CID 6852389 and CID 23724194 yielded 87% inhibition and 96% inhibition at 10-µM test concentrations, respectively.
All compounds that inhibited parasite growth by >50% were titrated and retested as concentration-response curves in the same experiment. CID 6852389 and CID 23724194 had the highest potency in these assays, with IC50 values of 4 µM and 1.3 µM, respectively. CID 6852389 and CID 23724194 were also assayed enzymatically to assess their mode of inhibition. Results of nonlinear regression analysis for Ki values were 3.39 ± 0.36 µM for CID 6852389 and 1.35 ± 0.15 µM for CID 23724194. Mode of inhibition was determined by software-based comparison of fits method using competitive, noncompetitive, uncompetitive, and mixed models of inhibition. The preferred fit was the noncompetitive model of inhibition in all cases. In addition, the mode of inhibition was confirmed by steady-state velocity plots, semilog scale plots, and Lineweaver-Burke plots. As determined by these methods, both behave as noncompetitive inhibitors of PfM18AAP (
Results from previous primary screening assays were used to better understand possible off-target activities and liabilities of CID 6852389 and CID 23724194. When tested in similarly formatted enzymatic inhibition assays run at the Scripps Research Institute Molecular Screening Center (SRIMSC), these compounds were devoid of activity (cf. PubChem AIDs 1859, 1931, 651959). Similarly, where tested, they were also inactive in mammalian cytotoxicity (cf. PubChem AIDs 1486, 1825) and bacterial viability (cf. PubChem AIDs 449731, 651606) screens, as well >100 other primary screens run at the screening center. Remarkably, both compounds were found to be inactive in all cell-based screens tested at the SRIMSC, except for two serotonin receptor screens (cf. PubChem AIDs 612 and 504916). In summary, in silico evaluation of the activity of CID 6852389 and CID 23724194 in a variety of cell-based and biochemical primary screening assays, including screens that used similar detection methodologies to the PfM18AAP primary assay, indicated that the activity of these two compounds was specific to PfM18AAP.
Discussion
Described here is our effort to identify small-molecule inhibitors of PfM18AAP via an HTS approach. As formatted, both the PfM18AAP primary screen and the CTSL1 counterscreen were highly amenable to automated screening in 1536-well microtiter plates, with a throughput of ~20,000 compounds tested per hour. As indicated by Z′ and positive control IC50 values, excellent assay windows and consistent pharmacology were demonstrated throughout the HTS effort, which enabled facile identification of selective PfM18AAP inhibitors. This screening approach identified 125 hit compounds with potent, selective inhibition of PfM18AAP. Two of these compounds, CID 6852389 and CID 23724194, were found to be noncompetitive inhibitors of PfM18AAP, with low-micromolar Ki values.
As described previously,21–24 we applied a “parallel” screening approach for the PfM18AAP HTS, where the CTSL1 counterscreen was used to triage hits at each stage of the PfM18AAP campaign ( Table 1 ). This was implemented since both the PfM18AAP and CTSL1 assays shared similar assay protocols, including the use of the same coumarin-based fluorophore for measurement of activity. CTSL1, a cysteine metalloproteinase, would be expected to be affected by divalent metal ions such as zinc or chelators of such. Divalent metal ions were not added as part of the reaction mix during the counterscreen assay, so identifying chelators is less likely, but similar in principle to PfM18AAP, compounds that may coordinate metal atoms would be expected to affect the activity of CTSL1 and may be identified via this counterscreen. To prioritize viable PfM18AAP inhibitors, we triaged compounds that were active in both assays with the assumption that they optically interfered with the measurement of fluorescence in microtiter plates or were promiscuous inhibitors. The validity of this assumption is partially supported by the results of our in silico analysis of CID 6852389 and CID 23724194; these compounds were inactive not only in the CTSL1 counterscreen but also in all other similarly formatted coumarin-based enzymatic assays run in our screening laboratory.
At the stage of potency assays, recapitulating this parallel approach enabled facile identification of selective PfM18AAP inhibitors in the HTS data sets, allowing us to prioritize compounds for labor-intensive mechanistic assays. Following the HTS stage (i.e., step 4 in Table 1 ), 125 compounds remained that appeared to be selective inhibitors. At this stage, a chemical triage was applied and 76 compounds were procured for testing in the lysate and parasite growth assay. Not all compounds were available in sufficient quantity for testing in both assays, in which case preference for usage was given to the parasite growth assay. Compounds that were active in both the parasite growth assay and the lysate assay (if available), were progressed for IC50 determination in the parasite growth assay. Of those five compounds, 6852389 and CID 23724194 were found to be the most potent.
Although the CTSL1 results were used to identify selective inhibitors for the PfM18AAP HTS effort, it is important to note several MLSMR compounds also selectively inhibited CTSL1 (see Fig. 4 ). Alternatively, it is reasonable to assume that our criteria for prioritizing viable leads in the HTS effort may have triaged genuine PfM18AAP inhibitors. For this reason, all results for the PfM18AAP and CTSL1 screening assays have been placed in a publicly available database, PubChem (https://pubchem.ncbi.nlm.nih.gov/), which enables the reexamination and identification of compounds with apparent activity in either target’s screening assay.
Cheminformatic analysis of the 125 hit compounds provides insight on the possible pharmacology of the hits identified from the HTS effort. As represented by the cluster 9, catechols and catecholamines are well-known stimulants.
17
Derivatives of a 1,2,4 triazol-4-yl urea, found in cluster 3, have been shown to possess anti-inflammatory and antibacterial activities.
18
Cluster 6 and its 1,2,4 triazole derivatives have been associated with a wider range of pharmacology, including anti-inflammatory, antibacterial, antifungal, anticancer, analgesic, and antidepressant.18,19 Finally, cluster 1 and its benzamide scaffold have derivatives with antagonistic activities against various receptors such as dopamine D2, 5-HT2, and 5-HT3.
20
Both CID 6852389 and CID 23724194 are nominally members of the “catechol” cluster 9. While catechols, in general, have been described as pan assay interference compounds (PAINS),
25
there is contradicting evidence that this is the case here (e.g., lack of activity versus >100 assays tested in our laboratory). Initial analysis of the catechols shown in
The PfM18AAP inhibitors detailed here, CID 6852389 and CID 23724194, yield potent, reproducible results across laboratories in both biochemical and whole-cell studies. In addition, these heterocyclic compounds contain a basic nitrogen atom, a physicochemical property that is suspected to encourage lysosomotropism. 27 They are the basis of continuing efforts to identify efficacious small-molecule probes of PfM18AAP, which will be the subject of future reports.
Footnotes
Acknowledgements
We thank Pierre Baillargeon and Lina DeLuca (Lead Identification, Scripps Florida) for compound management.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health’s Roadmap Initiative through grants R03MH084103 (DG, CB, KT, JPD), U54HG005031 awarded to Professor Jeffrey Aubé (PG, PP, JLW, DAW, FJS), and U54MH084512 (TS, VFV, PC, LS, PH). JPD was also supported by the National Institute for Health Research (NIHR, Australia) and Canada Institute for Health Research (CIHR) Tier 1 Canada Research Chair.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.