
Abstract
Neurodegenerative diseases affect millions of people worldwide, and the incidences increase as the population ages. Disease-modifying therapy that prevents or slows disease progression is still lacking, making neurodegenerative diseases an area of high unmet medical need. Target-based drug discovery for disease-modifying agents has been ongoing for many years, without much success due to incomplete understanding of the molecular mechanisms underlying neurodegeneration. Phenotypic screening, starting with a disease-relevant phenotype to screen for compounds that change the outcome of biological pathways rather than activities at certain specific targets, offers an alternative approach to find small molecules or targets that modulate the key characteristics of neurodegeneration. Phenotypic screens that focus on amelioration of disease-specific toxins, protection of neurons from degeneration, or promotion of neuroregeneration could be potential fertile grounds for discovering therapeutic agents for neurodegenerative diseases. In this review, we will summarize the progress of compound screening using these phenotypic-based strategies for this area, with a highlight on unique considerations for disease models, assays, and screening methodologies. We will further provide our perspectives on how best to use phenotypic screening to develop drug leads for neurodegenerative diseases.
Neurodegenerative disease (ND) comprises a collection of conditions with progressive loss of the function and eventually the structure of neurons in the brain, spinal cord, or peripheral nervous system, manifesting a large variety of clinical symptoms depending on the population of neurons affected.1,2 The etiology of neurodegeneration is quite heterogeneous. The onset of diseases could be familial (Mendelian inheritance 3 ), sporadic, or combined.4,5 The most common NDs are Alzheimer’s disease (AD) and Parkinson’s disease (PD), affecting millions of people worldwide. Treatments are available for some NDs, with the majority of them controlling only disease symptoms. These symptomatic control therapies usually work well in the early phase of the disease but gradually lose efficacy when the disease progresses with significant loss of neurons (e.g., donepezil controls dementia only over a short period of time in AD patients 6 ) or even generate significant clinical side effects (e.g., levodopa causes motor fluctuation and dyskinesia in moderate to advanced PD patients 7 ). As a result, the focus of drug discovery for ND has been shifted to the identification of disease-modifying molecules that delay or halt disease progression and offer prolonged or sustained clinical benefits to the patients.8,9
During the past decades, target-based screens and phenotypic screens have been broadly used for drug discovery. 10 Both screening strategies have contributed 45 first-in-class new small-molecule entities (NME) approved by the U.S. Food and Drug Administration (FDA) between 1999 and 2008. 11 Before target-based high throughput screening (HTS) was widely adopted, compound screens based on phenotypic changes, often in animal models or sometimes from observations in humans, were used predominantly for discovering new drugs. 12 Beginning in the 1980s, advances in molecular biology and genomics allowed more efficient mining of gene targets implicated in diseases and triggered the rise of the target-based drug discovery approach. 13 In target-based screening, target identification and validation through expression, gene targeting, and animal disease model studies are used to establish a hypothesis that modulation of the activity of a specific protein target will have beneficial therapeutic effects. Screening of chemical libraries of small molecules is then used to identify compounds that bind/interact with high affinity and specificity to the target. 14 The hits from these screens are used as starting points for both pharmacological target validation and lead discovery/optimization. The strengths of the target-based approach include the ability to apply knowledge of the target to investigate a specific hypothesis, the ability to screen small molecules in high-throughput formats, the ability to facilitate lead optimization with a clear structure-activity relationship (SAR) to enable improvement in compound potency and physicochemical properties, and the ability to predict potential target-associated safety liabilities and toxicity.
However, the increased knowledge of molecular targets has not translated into improved efficacy in discovering disease-modifying therapeutic agents for ND. It is even challenged whether an overreliance on the target-based approach for drug discovery has resulted in reduced success in discovering first-in-class medicines. As an example, a number of disease-specific aggregated proteins were identified as promising targets for neurodegeneration. In the recent clinical trials of the therapeutic antibody bapineuzumab against Aβ, the antibody did not improve cognitive function in AD patients, although it did reduce the amount of amyloid plaque in the brain.15,16 Why bapineuzumab failed to show an effect in the trials is a question with many possible answers. Insufficient target engagement, improper timing of intervention, or even the irrelevance of Aβ as a therapeutic target may have contributed to the failure. 17 Unfortunately, for most NDs, the pathogenesis is complicated and the underlying mechanism is not clear, which resulted in a general lack of well-validated targets for disease modification. Furthermore, modulating a single gene target may not always tune the cellular network for a beneficial outcome. As such, the target-based approach may not provide sufficient therapeutic impact especially for NDs with complex mechanisms. 11
Phenotypic screening addresses these challenges by starting with a disease-driving phenotype to screen for molecules that change the outcome of biological pathways rather than activity at a specific target and offers a complementary approach to find compounds or targets that modulate the key phenotypes of neurodegeneration. The strength of the phenotypic approach is that the assays do not require a full understanding of the molecular mechanisms of the disease, and activity in such assays might be translated more directly into therapeutic impact than in target-based assays. Therefore, with phenotypic screening, there are hopes to discover new therapeutic targets, new disease biology, and, at the same time, drugs for ND.
During the past 20 years, phenotypic screening has contributed to most of the first-in-class small-molecule drugs approved by the FDA. Among all NMEs approved during 1999 to 2008, 28 NMEs were identified through the phenotypic screening approach, whereas the target-based approach contributed the discovery of 17 NMEs. 11 In the central nervous system (CNS) area, phenotypic screening has contributed 7 of 9 first-in-class NMEs.
One successful case is from the epilepsy field, where phenotypic screening has contributed to the boom of antiepilepsy drugs since the 1980s. Owing to the National Institutes of Health–sponsored Anticonvulsant Drug Screening Program, thousands of new chemical entities from academic and pharmaceutical companies have been systematically screened in various rodent models for activity against seizure. This extensive collaboration finally resulted in the discovery and development of vigabatrin, zonisamide, xocarbazepine, lamotrigine, felbamate, gabapentin, topiramate, tiagabine, levetiracetam, pregabalin, lacosamide, and rulfinamide. 18
The previous successes of phenotypic screens in CNS diseases are either built on serendipitous findings or brute force efforts in animal tests using models with good translatability. However, these screens were usually carried out with very limited throughput. With the accumulated knowledge on the biological pathways involved in the neurodegeneration process and the great advancement of screening technologies,19,20 phenotypic screens with higher throughput have been developed using cellular, organotypic, or small animal models (e.g., Caenorhabditis elegans, Drosophila, and Zebrafish). These screens provide the opportunity to identify molecules that could modulate the biological pathways driving disease progression in ND. In this review, we will focus on reviewing the most recent phenotypic screens, the majority of which used cellular models of neurodegeneration. Aspects on compound library selection, the current state-of-art screening technologies, and the follow-up strategies after primary phenotypic screens will be discussed. We also offer our perspectives on challenges associated with phenotypic screening for ND.
Screening against Phenotypes That Recapitulate the Key Pathology of NDs
Phenotypic assays are designed based on disease-relevant phenotypes. In ND, the driver phenotype is progressive dysfunction or death of neurons, which eventually cause the symptoms in the patients. For example, PD is characterized by progressive degeneration of dopaminergic neurons in the substantia nigra. 1 The loss of dopaminergic neurons results in diminished dopaminergic stimulation in the striatum, which eventually causes motor dysfunction. Therefore, agents that ameliorate degeneration of neurons are actively searched for to modify disease progression and provide clinical benefit.
To identify these disease-modifying agents, three phenotypic screening strategies have been considered. The first approach, focusing on toxin removal or toxin reduction ( Fig. 1 ), aims to eliminate the toxins or stresses that cause neurodegeneration. The assumption is that by reducing the toxin/stress levels in the cells or in the environment, the affected neurons will be able to self-recover and regain functions, whereas the unaffected neurons will remain intact. Therefore, reduction of toxin levels will be the primary endpoint for phenotypic-based screens. Neuroprotection, the second approach, is designed to look for molecules or mechanisms that directly protect the neurons against disease-relevant insults. Cell viability will be the primary endpoint for these screens. This approach works only at the stage at which the dysfunctioning neurons can still be rescued. Thus, the key is to apply this approach early in disease before neuronal damage becomes irreversible. Finally, complementary to neuroprotection, the third approach focuses on regeneration (i.e., promoting adult neurogenesis for new functional neurons to be incorporated into the circuit and compensate for the loss of old neurons). An extension of such a strategy would be to seek regeneration of neuronal process such as axons with newly formed functional synapses. Obviously, these three strategies are connected to each other, and molecules/targets affecting multiple mechanisms will be of particular interest to offer more comprehensive intervention against neurodegeneration. In this section, we will summarize and discuss the progress of phenotypic screens based on these strategies with specific examples. We choose to focus on scientific literature and not to include examples from patent publications, so that only peer-reviewed studies are presented.
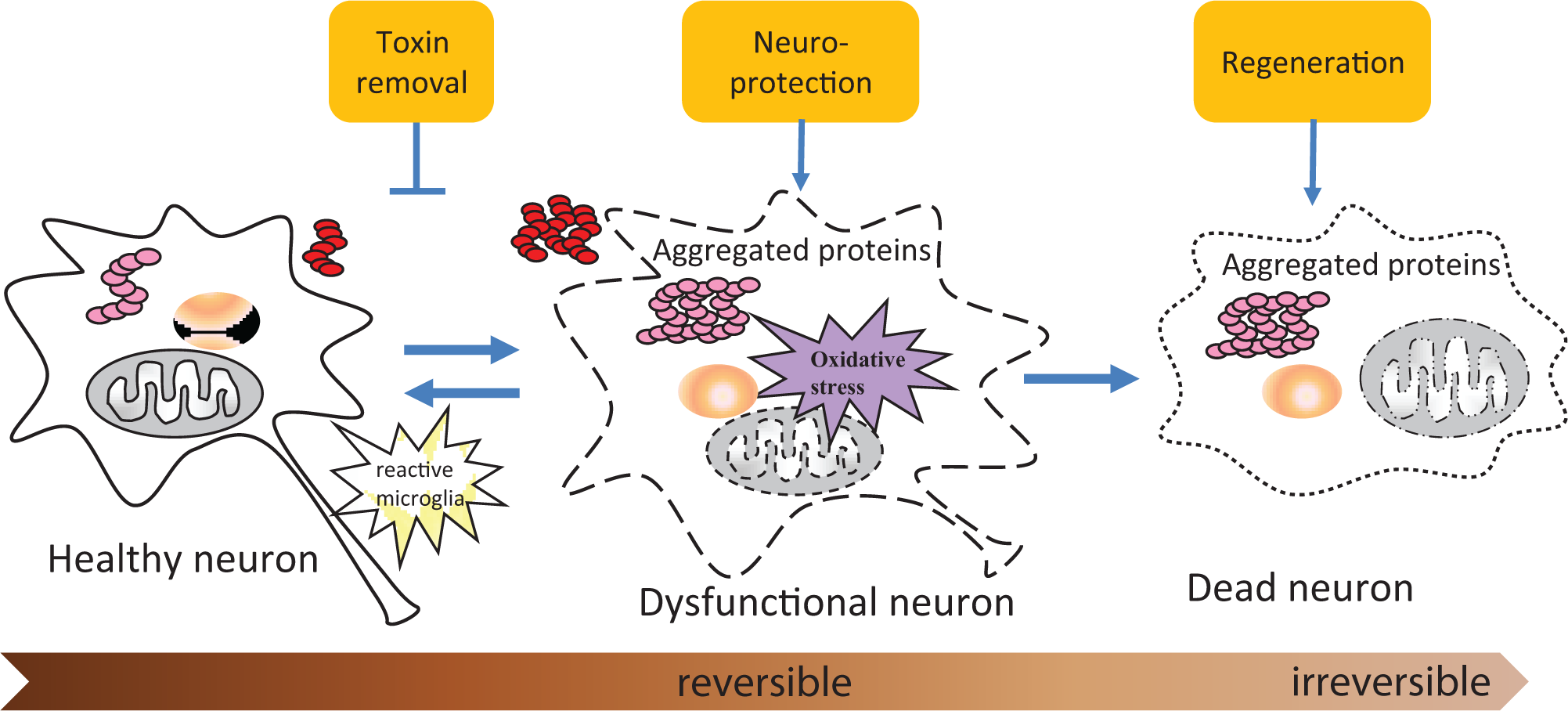
Three phenotypic strategies of drug screening for neurodegenerative diseases. The first approach mainly focuses on removing or reducing the toxic species levels (pathological aggregates, oxidative stress, etc.) that may cause neurodegeneration. The second neuroprotection approach does not directly modulate toxin levels. Instead, it is trying to protect dysfunctional neurons against the detrimental effect of existing toxins. Cell viability or survival is the primary endpoint for the screens for neuroprotectant. The third regeneration approach promotes adult neurogenesis for new neurons to replace old dead neurons in the diseased condition. Axonal regeneration and remyelination are also included as regenerative approaches.
Toxin Removal
Decades of research have focused on uncovering the cause of neuronal dysfunction/death in ND, yet the responsible toxic species in the diseases are still not firmly established. Human genetics, disease pathologies, and advancement of molecular biology have suggested several candidates that may cause neurodegeneration in the patients. Based on these candidate toxicants, phenotypic assays have been designed and chemical screens have been carried out for compounds that could reduce the level of toxins ( Table 1 ).
Nonexhaustive list of phenotypic screens for toxin removal.
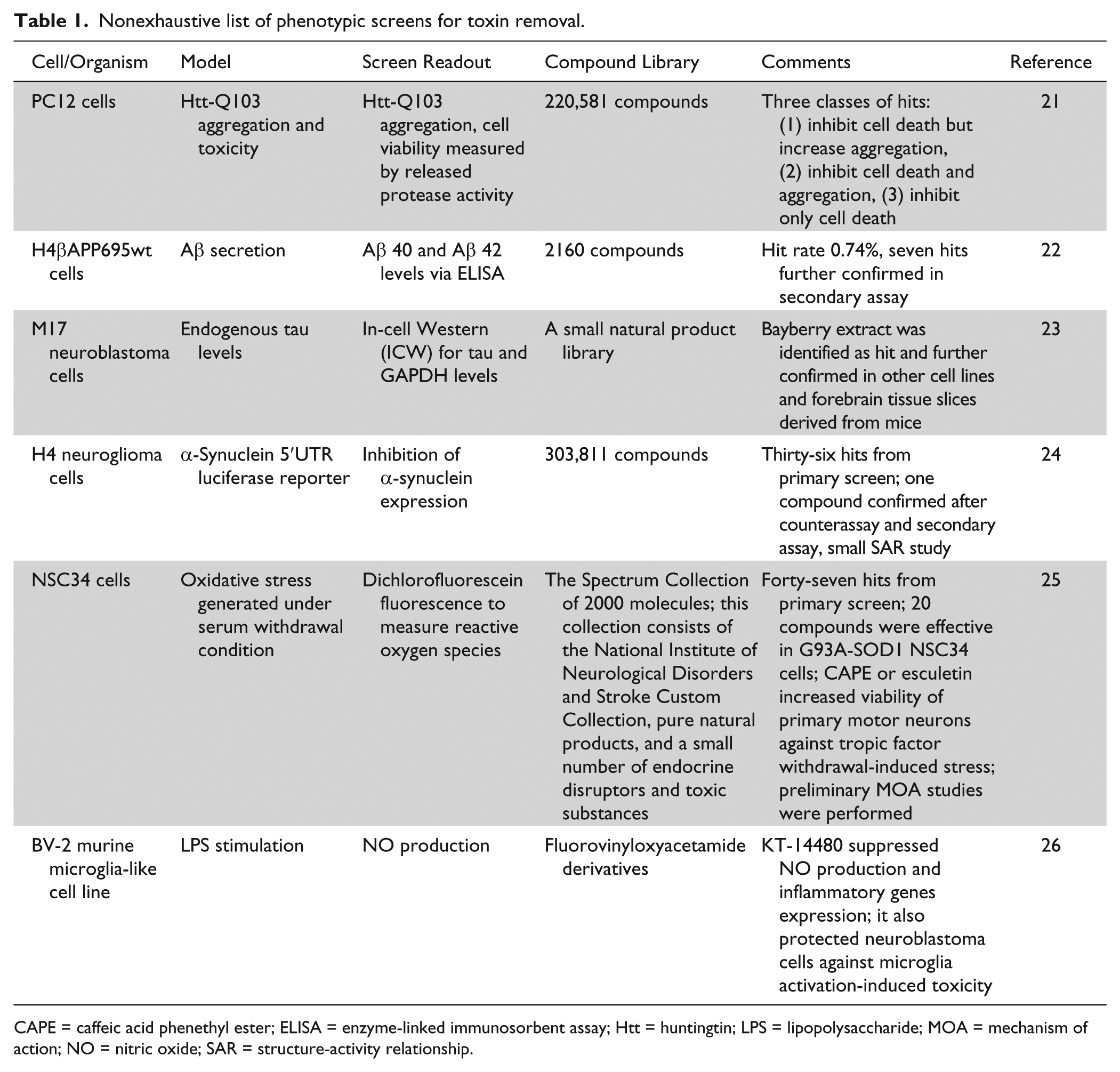
CAPE = caffeic acid phenethyl ester; ELISA = enzyme-linked immunosorbent assay; Htt = huntingtin; LPS = lipopolysaccharide; MOA = mechanism of action; NO = nitric oxide; SAR = structure-activity relationship.
Human genetics has provided significant insight into understanding disease etiology, especially in the familial NDs. Huntington’s disease, for example, is known to be caused by autosomal dominant mutation of the gene Huntingtin (Htt). Characterized by the expansion of CAG triplet repeats (polyglutamine residues, PolyQ) in exon 1 of the Htt gene, 3 the mutant Htt exerts a selective toxicity for striatal medium spiny neurons, although the mechanism is still unclear. 27
Htt with long PolyQ expansion is prone to aggregation, and intranuclear inclusions of mutant Htt in the neurons are pathological hallmarks for Huntington’s disease. 28 Based on this phenotype, a high-throughput assay was designed to quantify intracellular Htt aggregates in PC12 cells expressing Htt-Q103. 21 A library collection of 220,581 compounds was successfully screened with good assay quality. Intriguingly, hits exerted differential regulation of Htt aggregation and cell viability. Some of the hits reduced Htt aggregate levels and rescued Htt-Q103–induced cytotoxcity, whereas others improved cell viability but further enhanced Htt aggregation. This questioned whether Htt aggregates are the cause of neuronal death, which is heatedly debated within the community. 29
Understanding the mechanism of sporadic NDs is much more complicated, as there is no dominant genetic factor contributing to the onset of the diseases. By studying the affected tissues from patients, pathological hallmarks for some sporadic NDs have been identified. For example, AD displays prominent pathological hallmarks of extracellular amyloid plaques composed of Aβ peptides and intracellular neurofibrillary tangles composed of hyperphosphorylated tau. 30 With the assumption that these disease hallmarks are associated with disease progression,31,32 phenotypic screens have been carried out looking for compounds that reduce Aβ or tau levels. In one study, a human cell line secreting Aβ was used for screening. 22 Secreted Aβ40 and Aβ42 peptides were measured by enzyme-linked immunosorbent assay (ELISA). Among 2160 compounds screened, 7 were confirmed to reduce Aβ levels. In another study, M17 neuroblastoma cells expressing high endogenous tau were used to screen natural products that reduce tau protein levels. 23 A Bayberry extract was identified as a potent tau reducer, a finding further confirmed in mouse fresh forebrain tissue slices.
Similarly, PD is characterized by a key pathology of intracellular Lewy bodies composed of α-synuclein. 33 Although the function of α-synuclein in the neurons has not been fully elucidated, several lines of evidence have suggested that α-synuclein accumulation induces dysfunction of neurons.34,35 Recently, a high-throughput phenotypic screening of 303,811 compounds for inhibition of α-synuclein translation in engineered H4 cells was conducted. 24 One compound was confirmed to reduce α-synuclein translation with more than 200-fold selectivity over the prion protein. Interestingly, a SAR study around this compound suggested certain structural features for maintaining the efficacy. The effect of this compound will be further evaluated in primary dopaminergic neurons and in the animal models for PD.
Other than pathological hallmarks, intracellular stressors have also been proposed to cause neuronal dysfunction. Oxidative stress is one of the metabolic loads of neurons with long axons and rapid neural transmitter release. Excessive oxidative stress disrupts proteins, DNA, lipids, and intracellular organelles, which ultimately leads to cell death. 36 In ND, oxidative damages such as oxidized nucleosides and oxidized proteins have been observed in the dying neurons as an early event in the process of neurodegeneration. 37 To identify compounds with antioxidative properties, a phenotypic screen measuring intracellular oxidative stress levels was conducted in NSC34 motor neuron cells under the serum withdrawal condition. 25 Hits were subsequently tested in NSC34 cells overexpressing mutant SOD1 (G93A), a cell-based model for amyotrophic lateral sclerosis. Interestingly, some but not all hit compounds reduced oxidative stress induced by mutant SOD1 and improved cell viability. These compounds were further shown to protect primary motor neurons against trophic factor withdrawal-induced stress.
In addition, extrinsic factors also contribute to neuronal death. In the diseased condition, neuroinflammation characterized by glia cell activation, peripheral lymphocyte infiltration, and increased proinflammatory cytokine levels plays a detrimental role on neurons. 38 Using a microglia-like cell line BV2, fluorovinyloxyacetamide derivatives with anti-inflammatory properties were screened against lipopolysaccharide (LPS)–induced nitric oxide production. 26 The hit compound also reduced LPS-induced production of tumor necrosis factor–α and interleukin-1β and protected neuroblastoma cells against microglia activation-induced toxicity.
Neuroprotection
When the patients of NDs manifest clinical symptoms, there has already been chronic accumulation of toxins in the nervous system, and a significant amount of neurons have already started to degenerate. At this stage, toxin removal may be insufficient or too late to ameliorate the situation. Thus, neuroprotection contributes another important strategy to preserve the remaining functional neurons in a maximal way and slow disease progression.
To establish the assay for screening neuroprotectants, one should recapitulate the key pathologies in ND. The most common models used for screening include aggregated protein-induced cytotoxicity (such as Aβ in AD, α-synuclein in PD), oxidative stress–induced cytotoxicity (including mitochondrial dysfunction), and inflammation-induced cytotoxicity ( Table 2 ). The endpoint in these assays is primarily protection of cell death by test compounds rather than modulating the level of toxic species.
Nonexhaustive list of phenotypic screens for neuroprotection.
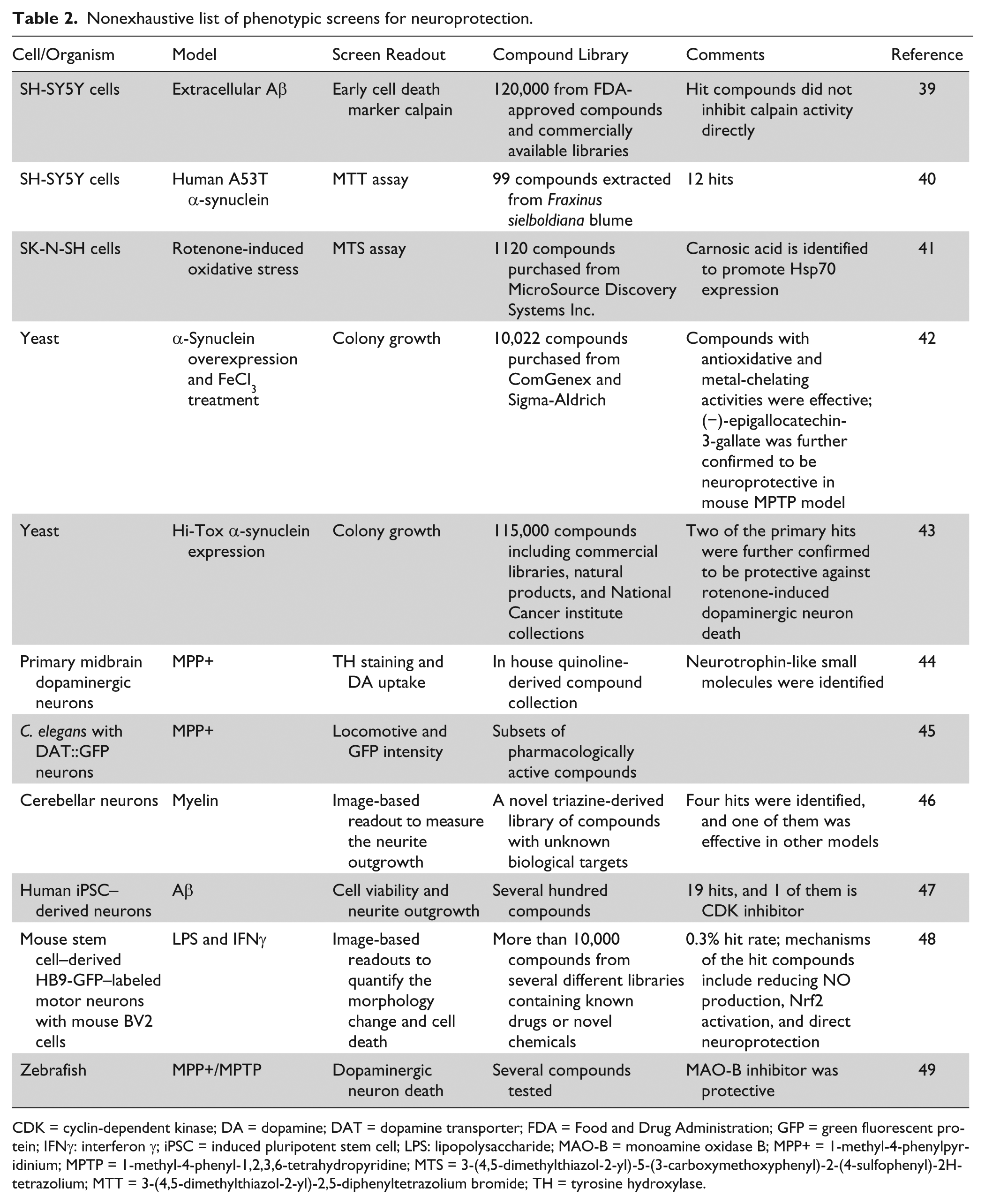
CDK = cyclin-dependent kinase; DA = dopamine; DAT = dopamine transporter; FDA = Food and Drug Administration; GFP = green fluorescent protein; IFNγ: interferon γ; iPSC = induced pluripotent stem cell; LPS: lipopolysaccharide; MAO-B = monoamine oxidase B; MPP+ = 1-methyl-4-phenylpyridinium; MPTP = 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MTS = 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; TH = tyrosine hydroxylase.
In one cellular model for AD, extracellular Aβ was used as an insult to induce cell death in the human neuroblastoma-derived cell line SH-SY5Y. A total of 120,000 compounds (including compounds approved by the FDA, compounds from a purified natural products library, and compounds purchased from diverse libraries) were screened using calpain production as an early readout for cell death. All hits identified blocked calpain production and cell death with no direct inhibition of calpain enzyme activity. 39 In another assay for PD, SH-SY5Y cells were transfected with mutant A53T human α-synuclein and treated with 1-methyl-4-phenylpyridinium (MPP+) and exogenous dopamine to induce cytotoxicity as measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Annexin V, propidium iodide staining, and DNA ladder were further performed to confirm cell apoptosis. Twelve protective compounds were found from Fraxinus sielboldiana blume of the Oleaceae family, which is widely distributed in East Asia. 40
Another model system for evaluating the toxicity of aggregated protein is yeast, in which toxicity can be conveniently observed via the change of cell growth measured by the colony size. In one such study, with α-synuclein overexpression and FeCl3 treatment, 10,022 compounds purchased from ComGenex (Budapest, Hungary) and Sigma Aldrich (St. Louis, MO) were screened. Compounds with antioxidative or metal-chelating activities (such as quercetin and (−)-epigallocatechin-3-gallate) were found to effectively protect yeast cells against α-synuclein and FeCl3-induced toxicity. The protective effect of (−)-epigallocatechin-3-gallate was further confirmed in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model. 42 In another study, a yeast model with a high expression level of α-synuclein was used for screening. Overexpression of α-synuclein generated significant toxic effects on mitochondrial function. Using cell growth, mitochondrial function recovery, and α-synuclein aggregation load as the readouts, 115,000 compounds (including commercial libraries, natural products, and National Cancer Institute collections) were screened, and the hits were subsequently confirmed to reduce α-synuclein–induced toxicity in nematode and primary rat dopaminergic neurons. Two of the hit compounds also demonstrated a neuroprotective effect against rotenone-induced dopaminergic neuron death. 43
In addition to protein aggregates, other commonly used insults are oxidative stress or mitochondrial dysfunction. Yoon et al. 41 reported a screen conducted in the SK-N-SH cell line using rotenone as the insult to induce cell death. A library of 1120 compounds purchased from MicroSource Discovery Systems Inc. (Gaylordsville, CT) was screened, and carnosic acid was shown to be neuroprotective via promoting the expression of Hsp70. 41
Besides neuronal survival, promoting neurite growth against insults has also been considered as a viable surrogate measure in phenotypic screening for assessing neural functions. In one study, mouse cerebellar granule neurons were used to screen compounds that promote neurite outgrowth in the presence of inhibitory myelin substrates. Four hits from a newly synthesized compound library were identified that prevented the inhibitory effect of myelin. One of the hits was subsequently confirmed in a variety of neurite outgrowth assays (various neuron types, with various inhibitory agents) and demonstrated efficacy in in vivo assays with sensory neuron dorsal column transection and optical nerve crush. 46 In another recent report, human induced pluripotent stem cell (iPSC)–derived neurons were treated with extracellular Aβ42, and cell viability was measured as the primary endpoint. Neurite outgrowth was also quantified to confirm the screening result. Several hundred compounds were screened, and 19 hits were identified, one of which was a Cdk2 inhibitor. 47
Neuroinflammation, as discussed in the “Toxin Removal” section of the review, plays a significant role in propagation of the injury to neurons. Reducing inflammation-induced neuronal death is therefore considered as another means of obtaining neuroprotection. As an example, a mixed cell culture, composed of mouse stem cell–derived HB9+ green fluorescent protein (GFP)–labeled motor neurons, mouse stem cell–derived astrocytes, and mouse BV2 microglia-like cells, was treated with LPS and interferon γ. This treatment induced BV2 cells to produce massive levels of nitric oxide (NO), which then killed the motor neurons in the culture. High-content image analysis based on GFP expression in motor neurons was used to quantify neuronal morphological change and survival. More than 10,000 compounds, derived from several different libraries containing known drugs or novel chemical entities structurally enriched to target various target groups, were screened. The hit compounds display diverse mechanisms to reduce neuronal death, including inhibition of NO production by microglia, activation of Nrf2 pathways in both microglia and astrocytes, and direct protection against NO-induced toxicity in motor neurons. 48
In addition, neurotrophic factors are protective molecules to support neuronal survival, and neurotrophic factor deprivation is considered one of the underlying mechanisms for neurodegeneration.50,51 Thus, small molecules with neurotrophin-like activity would be good neuroprotective agents for ND. One such successful phenotypic screen was performed using primary midbrain dopaminergic neurons. Quinoline-derived compounds were screened for hits supporting neuronal survival against spontaneous degeneration. By measuring tyrosine hydroxylase immunoactivity and dopamine uptake, several hit compounds were identified, and they were shown to protect cells from MPP+-induced toxicity and promoted neurite outgrowth. 44 In addition to neurotrophin-like molecules, compounds that induce the expression of neurotrophic factors to enhance neuronal survival will be of particular interest. Information on such a kind of phenotypic screen has not been publically disclosed.
As cell-based assays may not be sufficient to represent all the aspects of disease, ex vivo and in vivo animal models have been attempted for screening, albeit with very limited throughput and capacity. For example, compared with cell-based models, organotypic culture offers a much more real-life–like environment for studying the interaction of different cell types, including neurons, astrocytes, microglia, oligodendrocytes, and other cells. Several compounds have been reported to have effects in promoting remyelination, such as RXR agonist 9-cis retinoid acid, in the brain and spinal cord ex vivo cultures. 52 Organotypic culture now serves mostly as a system for secondary assays to confirm hits from primary assays.
For small sets of compounds, animal models may be another choice for phenotypic screening in which compounds can be tested for effects on functional recovery and behavioral outcome. As expected, the throughput of animal screens is low, and only a few successful cases have been reported. For example, the transgenic C. elegans with GFP-labeled dopaminergic neurons were treated with MPP+ to reduce locomotor activity and dopaminergic neuron–specific GFP intensity. Several active compounds were shown to be protective to the neurons in this model. 45 Zebrafish, a small vertebrate, is also used as a model for Parkinson’s disease. Both MPTP and MPP+ can specifically induce dopaminergic neuron death, and a monoamine oxidase B inhibitor was shown to protect the neurons from death. 49
Regeneration
Promoting regeneration as a way to repair damaged neural tissues is an inspirational goal for the treatment of ND. Two major approaches for regeneration have been studied. The first approach focuses on transplantation of stem cells into the brain to repair damaged neural networks,53,54 whereas the second emphasizes promoting neurogenesis (i.e., differentiation of resident neural stem cells in adult neural stem cell niches) into new neurons, which may migrate to the site of damage and be incorporated into the neural network. 55 Phenotypic assays based on the latter approach have been designed and applied to chemical screening. Besides neurogenesis, remyelination is another important approach to promote neuronal regeneration. Table 3 lists the most recent phenotypic screens for promoting neurogenesis and remyelination. Neural progenitor cells (NPCs) from rodent brains represent one readily available source of cells for neurogenesis screens. The newly differentiated neurons can be measured by quantifying the level of neuron-specific class III β tubulin (Tuj1). One such screen using mouse embryonic NPC, based on cultured neurosphere and Tuj1 ELISA, identified phosphoserine from a library of hundreds of bioactive molecules as an enhancer for neural differentiation and maintenance. 56 Using the same source of NPC, Kim et al. 57 designed an Acumen-based imaging assay to quantify the percentage of Tuj1+ neurons after differentiation. The compounds screened had annotated biological target and pathway. From this screen, GSK3β inhibitors were identified as potent inducers of neurogenesis. In another study, Warashina et al. 58 cultured primary NPC isolated from adult rat hippocampus and screened a combinatorial heterocyclic library of 50,000 compounds for hits that could increase the percentage of Tuj1+ cells after differentiation. A series of 4-aminothiazole compounds were identified, 58 and a lead molecule KHS101 was later discovered after further chemical optimization. This compound was confirmed to increase neurogenesis in rat brain. 59
Nonexhaustive list of phenotypic screens for regeneration.
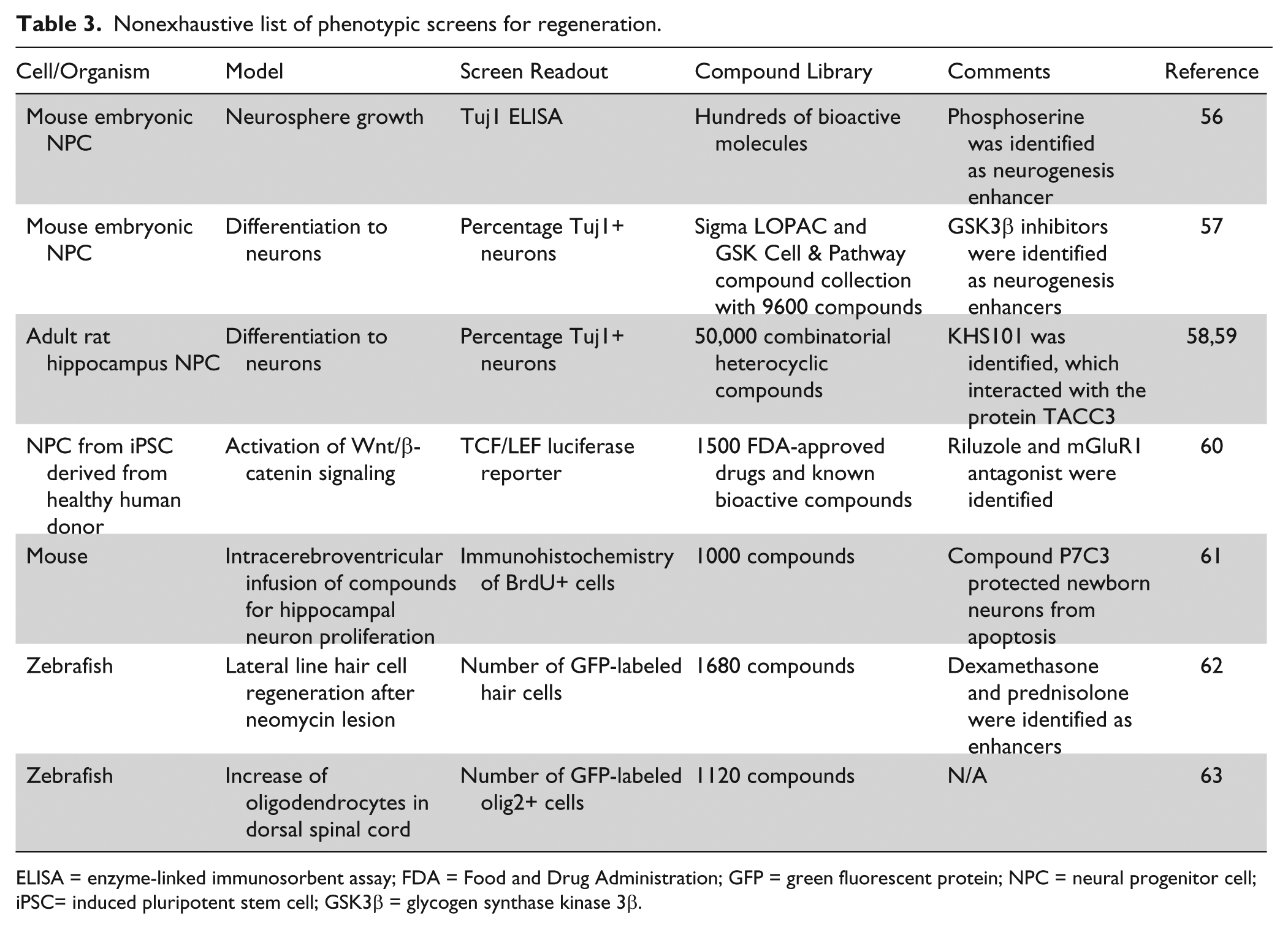
ELISA = enzyme-linked immunosorbent assay; FDA = Food and Drug Administration; GFP = green fluorescent protein; NPC = neural progenitor cell; iPSC= induced pluripotent stem cell; GSK3β = glycogen synthase kinase 3β.
The advancement of iPSC technology has made it possible to obtain NPC cell lines from iPSC reprogrammed from human fibroblasts. In one study, an NPC line was generated from a healthy human adult male. It was further engineered to include a TCF/LEF luciferase reporter for screening of compounds specifically regulating Wnt/β-catenin signaling. This system was validated in 384-well format, and an FDA-approved drug library of 1500 compounds was screened with the identification of several activators for the Wnt/β-catenin pathway. 60 As activation of the Wnt/β-catenin pathway plays an important role in neurogenesis, the screen demonstrated the feasibility of establishing a pathway-specific phenotypic assay for neural regeneration.
In addition to cellular assays, phenotypic screens on neural regeneration have also been carried out in in vivo systems. Pieper et al. 61 screened 1000 compounds in mice using the number of BrdU-positive cells in hippocampus as the readout for in vivo neurogenesis. The compounds were pooled into groups of 10, with each group being injected into two mice, followed by deconvolution of single compound from positive pools. An aminopropyl carbazole compound, designated P7C3, was able to promote neurogenesis by protecting newborn neurons from apoptosis. A study in aged rats also confirmed P7C3’s effects to enhance neurogenesis and preserve cognitive capacity.
Another animal model suitable for phenotypic screening is zebrafish, due to its high regenerative potential, the ease to observe neurons, and the feasibility of establishing a medium-throughput assay in 96-well format. For instance, hair cells exist in small numbers in the inner ear of mammals and are difficult to study. In zebrafish, however, they are located in the lateral line and can be easily observed and quantified. In a recent report, a screen for hair cell regeneration after neomycin-induced cell death was carried out in zebrafish with GFP-labeled hair cells. 62 Among 1680 compounds tested, 2 glucocorticoid compounds were identified as potent enhancers to promote regeneration of hair cells.
In addition to promoting neurogenesis, enhancing remyelination of demyelinated axons represents another approach for neural regeneration, especially for some NDs with the demyelination pathology. Oligodendrocytes are responsible for myelination of axons, and therefore promoting differentiation of oligodendrocyte progenitor cells to functional mature oligodendrocytes can potentially enhance axon remyelination in the diseased condition. In a zebrafish model with GFP-labeled oligodendrocyes, 1120 compounds were screened. Several hits that increased oligodendrocyte counts in dorsal spinal cord were identified. Unfortunately, these hits were not confirmed in the secondary quantitative PCR (qPCR) assay measuring myelin basic protein mRNA levels. 63
The aforementioned screens are by no means meant to be exhaustive, but they do provide good examples to demonstrate the rationale and design of phenotypic assays for ND. It remains to be seen if any of the hits discussed will be developed further and tested in the patients.
Technical Considerations for Phenotypic Screening for NDs
With advanced technologies, many screening models and assay formats have been adopted to facilitate phenotypic screening. In this section, we will provide a systematic review of the technical considerations for phenotypic screening for neurodegeneration, such as the selection of compound libraries, the selection of disease-relevant models, and the application of new technologies for neuronal specific readouts, in the hope of enhancing the chance of success in this challenging area.
Considerations on the Selection of a Compound Library
In the screens discussed in the previous section, various types of compound libraries were chosen for phenotypic screening, such as the marketed drug sets,39,48 annotated compound library with target information,57,64 natural product library,23,25 and a library containing derivatives of known bioactive molecules.26,44 General principles for selection of a compound library, such as enrichment of compounds with good physicochemical properties and structure diversity, are shared issues between target-based screens and phenotypic screens. Despite these, there are additional considerations specifically associated with the unique properties of phenotypic screening.
Phenotypic screening generally has lower throughput, especially when the screens are conducted with primary cells, tissues, or small animal models. With limited screening capacity, we need to be selective in the quality of the compound set. In addition to improved physicochemical properties, compounds need to have good solubility, membrane permeability, and display no cytotoxicity at the tested concentrations.
Different compound libraries can be used to suit the aims of phenotypic screening. For repositioning purposes, the collections containing marketed or FDA-approved drug sets, 39 as well as proprietary compound collections of clinical assets and selected candidate compounds with desired exposure and acceptable safety profile would be valuable. If the screens are designed to identify novel drug targets or mechanisms of action, collections of compounds with known-target annotation, 57 or siRNA/shRNA libraries could offer a quick route for target identification. When building annotated compound libraries, compound potency, target selectivity, cellular activity, permeability, and cytotoxicity should all be taken into consideration. It is important to include a few distinctive chemotypes for each target within the library to ensure confidence that the compound’s effect can be attributed to the target modulation rather than off-target effects of specific chemical series. Furthermore, if the screens are aimed at identifying lead molecules for further optimization, a diversified library with desired physiochemical properties would be an ideal choice to start with. Stringent criteria, such as Lipinski’s “rule-of-5” 65 and recent proposals on physicochemical requirements for candidate molecules66 -68 could be considered for the selection of compounds. In Table 4 , we have summarized a list of compound libraries that are either commercially available or accessible from academic centers. In addition, two databases, ZINC 69 and ChemNavigator (http://www.chemnavigator.com), are useful sources to look for commercially available compounds with more chemical diversity.
Nonexhaustive list of available compound libraries.
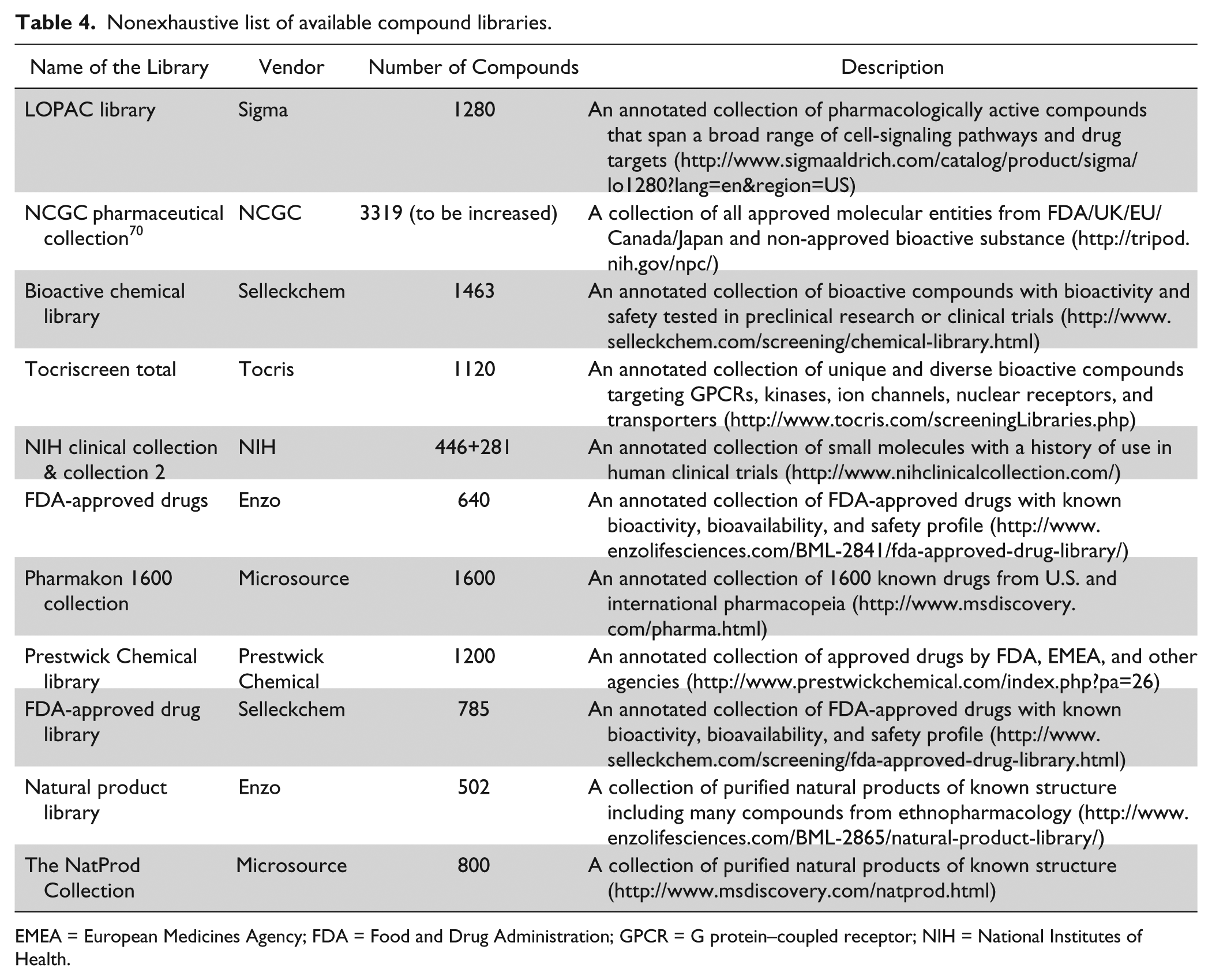
EMEA = European Medicines Agency; FDA = Food and Drug Administration; GPCR = G protein–coupled receptor; NIH = National Institutes of Health.
Considerations on the Models Used for Phenotypic Screening
Choosing the most relevant model for phenotypic screening is critical but challenging. Immortalized cells can be scaled up easily and can also be engineered genetically to build reporter assays. However, for ND drug discovery, these immortalized cell lines may not fully recapitulate the neuronal-specific regulatory systems and reflect similar susceptibility to insults. Primary rodent neurons, which are with similar properties to human neurons, limited by the number of animals one can dissect from, can be a better source of cells for phenotypic screens. 71 Different types of primary neurons could be prepared related to the disease studied, such as cortical or hippocampal neurons for AD 72 and dopaminergic neurons for PD, 73 which allows the study of disease-specific neuronal phenotype. Glial cells and other kinds of cells can also be introduced into primary neuronal culture to model the interactions among different cell types in the nervous system.
Human neurons differentiated from embryonic stem cells (ES), NPC, and iPSC, especially the iPSC derived from diseased patients, offer great potential in phenotypic screening. These cells carry the same genetic makeup as the patients and therefore are ideal cells for compound testing. After the disease-related phenotype is observed in the neurons differentiated from iPSC from patients, 74 phenotypic screening specifically designed to correct the defect thus offers the promise to identify therapeutic agents. As one such example, familial dysautonomia (FD) is caused by a single point mutation in the IKBKAP gene (inhibitor of κ light polypeptide gene enhancer in B-cells, kinase complex-associated protein), which decreases IKBKAP expression via mis-splicing. In FD patient iPSC-derived neural crest cells, reduced IKBKAP mRNA levels were observed, leading to a cellular model suitable for screening of chemicals to increase IKBKAP expression. 64 A qPCR assay was developed to screen a library of 6912 compounds. The screen identified a known α2-adrenergic receptor antagonist, SKF-86466, as an IKBKAP inducer, possibly through regulating cyclic AMP (cAMP) and protein kinase A–dependent cAMP response element-binding protein phosphorylation.
However, this approach requires the phenotypes to be clearly demonstrated in iPSC-differentiated neurons. Taking neurons differentiated from iPSC derived from familial AD patients with APP mutation as an example, reduction of synapses was not observed in these cells, despite some changes of Aβ40 and phosphorylated Tau (pT231) levels. 75 Similarly, dopaminergic neurons from iPSC derived from PD patients, bearing LRRK2 mutations, did not develop any defect compared with the ones from healthy people, although they did show higher sensitivity to stressors. 76 Therefore, to maximize the potential of using iPSC for phenotypic screening, more work needs to be done to ensure a reproducible and translatable in vitro disease phenotype in the test system. 77
Organotypic culture or small animal models provide a way to model the neuronal network connections and the in vivo situation, especially when targeting the noncell autonomous phenotypes. Several successful examples have been provided in the previous sections, such as the C. elegans or Zebrafish screens.45,49,62 One powerful screening approach is to use genetic manipulation, such as GFP labeling, to score the desired phenotype. However, there are several complications with these screening models, such as the species differences of target proteins, the difficulty of achieving assay robustness due to inconsistent compound exposure and culture preparations, and the limited assay throughput and analytical methodologies.
In summary, each model system has its strength and weakness. Choosing the best suitable model for screening requires careful consideration of many factors, including the translatability of the phenotype, the throughput, the choice of compound collection, and the assay format. Furthermore, multiple models for the studied phenotype could be used in the screening either in parallel or sequential to strengthen the confidence in the hit validity.
Assay Methodologies Applied in Phenotypic Screen and Specific Requirement
Technologies have been greatly advanced to enable measurement of cellular phenotypes. In this section, we will discuss the technologies that are the most applicable to study phenotypes associated with neurodegeneration, such as newly developed fluorescent probes or protein tags and automated cell-imaging platforms. These technologies serve as powerful tools to monitor various cellular phenotypes related to disease pathogenesis in a much higher throughput manner.
Various fluorescent probes have been applied to detect the cellular events associated with potential pathological mechanisms of NDs. Mitochondria dysfunction can be monitored by fluorescent probes such as Rhodamine 123, TMRM, TMRE, JC-1, or MitoView 633 78 ; oxidative stress can be detected by fluorescent probes such as dihydroethidium, MitoSOX, H2DCFDA, CM-H2DCFDA, or DAF-FM, which become fluorescent after reacting with superoxide, hydrogen peroxide, or nitric oxide. 79 Cell viability can be simultaneously detected by double staining with Hoechst 33342 (live cells staining) and YoPro-1 (damaged cells staining). It can also be measured via quantification of caspase 3/7 activity using the cell-permeable substrate PhiPhi-lux. 80
Besides probes, GFP/luciferase reporters are useful tools to label specific neurons, although they may not be applicable to primary neurons because of the limited transfection efficiency and the variations between batch-to-batch cultures. One alternative is to label the ES/iPSC cells and use the subsequently differentiated neurons for screening.81,82
Although neuronal survival or neuronal death is commonly used as the endpoint for phenotypic assays, it may not truly reflect neuronal dysfunction in NDs. In the diseased condition, functional changes in synapses and structural changes in neurites occur much earlier than cell death. Monitoring these phenotypes in a dysfunctioning neuron will be valuable for identifying agents for early intervention of neurodegeneration. High-content screening (HCS) based on imaging technology has been more widely applied to monitor neuronal-specific changes, such as neurite length, branching, or synapse formation. For example, the Cellomics ArrayScan VTI platform and the associated BioAapplication algorithm from Thermo Scientific, as described in a HCS assay for Huntington’s disease, 83 offer very comprehensive detection and analysis of neuronal features. High-content analysis also provides the opportunity to quantify the number of synapses using presynaptic marker synapsin staining. In one study, Nguyen et al. 84 found that Aβ induced significant damage to the synapses quantified as the number of synapsin-positive spots. Thus, by detecting various neuronal phenotypic changes simultaneously, HCS is able to verify the most sensitive and robust phenotype and uncover hits with different mechanisms of action. Compared with other technologies, the major advantages of HCS are the high level of multiplexing (simultaneous readout of several parameters) and superior spatial resolution (detecting changes in subcellular level in more defined readouts). However, most current HCS assays are still hampered by the limited throughput, the difficulties in automation 85 and algorithms to decovolute images, which could be improved at least through the application of models with easier readouts such as engineered GFP reporters.52,62
In summary, a disease-relevant, robust, and quantitative phenotypic assay, together with a fit-for-purpose compound library with good physicochemical properties and diversity, are the prerequisites for an optimal and valuable phenotypic screen. In addition, reasonable Z′, good reproducibility, and solid confirmation of the hits should all be achieved to ensure a successful screening campaign, similar to that in target-based HTS. Based on the outcome of phenotypic screening, several follow-up strategies could be applied. These will be discussed in detail in the following section.
Follow-up Strategies for Phenotypic Screens
After a fruitful phenotypic screening campaign, developing the hits to the selection of a compound for clinical testing usually is time-consuming and challenging, similar to that encountered in target-based drug discovery. Different follow-up strategies could be considered based on the outcome of the phenotypic screen.
If the hit compounds from a phenotypic assay are launched drugs or clinical assets, with favourable efficacy in secondary assays, good activity in in vivo disease-relevant animal models, acceptable pharmacokinetic profile, and sufficient therapeutic window, they can be quickly tested in clinical studies. Such attempts to reposition FDA-approved drugs have already been comprehensively reviewed elsewhere 86 and will not be further detailed here.
If the hits have known target annotation, target validation should then be conducted through pharmacological validation using different compounds modulating the same target, or genetic validation by knock down/overexpression in vitro or in vivo. Once the target is confirmed and validated, target-based drug discovery can be initiated. A good example for this is the phenotypic screen on β-cell regeneration, a cell-based model for diabetes. Seven thousand compounds were screened for promoting regeneration of β cells after toxin-induced ablation in a transgenic zebrafish model. 87 Among the five hits identified in the screen, four were involved in adenosine signaling (one adenosine agonist, one adenosine kinase inhibitor, and two PDE3/4 inhibitors). This strongly suggested that the adenosine pathway is involved in β-cell regeneration.
However, many times, the hits from phenotypic screens do not have target annotation. In these cases, target identification is essential to allow a subsequent target-based drug discovery campaign for lead optimization. Progress has been made during the past decades for target deconvolution.88–90 As an example, compound TWS119 was discovered from a phenotypic cell-based screen for neurogenesis using murine ES. 91 Through affinity chromatography and mass spectrometry, the binding protein enriched in TWS119-linked affinity matrix was identified as GSK3β.
Another successful example is the target identification of a quinazoline compound series for spinal muscular atrophy. The compound was initially found via a phenotypic screen for molecules that increased the expression of survival motor neuron protein from a second copy of the survival motor neuron gene (SMN2). 92 Using affinity-based protein array, the target of the compound was identified to be the scavenger mRNA decapping enzyme (DcpS). 93 A lead compound, RG3039, was subsequently developed and tested in a phase 1b trial. 94
Recently, more advanced affinity-based technologies have been developed to allow efficient target deconvolution, such as SILAC (stable isotope labeling by amino acids in cell culture)95,96 and ITRAQ (isobaric tags for relative and absolute quantification).97,98 Both are designed based on the binding affinity between the hit compound and its potential targeting protein, and they are developed to identify enriched proteins in the hit compound–loaded matrix using a mass spectrometry–based proteomics detection method. These approaches have been validated using compounds with known target. A good example is the study on the mechanisms of action of clinical ABL kinase inhibitors. Quantitative profiling using ITRAQ techniques not only confirmed the known targets (ABL, SRC kinase) of the drug imatinib but also identified novel targets (DDR1 and NQO2) of the compound. 98
Another emerging method for target identification is Connectivity Map (C-Map).99,100 C-Map was originally developed as a bioinformatics tool for repositioning of FDA-approved drugs based on disease gene signatures. The general idea of C-Map is to compare the gene expression profile associated with a specific hit from a phenotypic screen to the signature profiles in the C-Map database, so as to identify similar profiles to suggest the potential target. For example, as a follow-up study for the phenotypic screen of neurite outgrowth, 46 MCF7 cells were treated with the hit molecule F05, and a gene expression profile was generated. By comparing to the profiles in the C-Map database, it was discovered that the hit compound had a similar profile to piperazine antipsychotics, which were known to be involved in the calmodulin pathway. 101
In summary, because of the various outcomes of a phenotypic screen, there is no standard procedure that can be routinely applied for hit follow-up. Insight can be obtained from successful cases, in which a compound discovered from phenotypic assay was successfully developed as a marketed drug. As one such example, suberoylanilide hydroxamic acid (SAHA), a launched anticancer agent, was discovered through the process of lead optimization using a phenotypic-based assay for murine erythroleukemia cell differentiation. 102 The target of SAHA was later discovered to be histone deacetylase (HDAC), 103 which has allowed target-based discovery and development of the second generation of HDAC inhibitors. 104 Alhough neurodegeneration has its own uniqueness, some merits or lessons can be learned from these cases and applied to ND drug discovery.
Challenges and Future Perspective
In this review, we have described a path for phenotype-based drug discovery for NDs, including phenotype modeling, compound library selection, and hit follow-up. Each part comes with its own challenges ( Fig. 2 ).
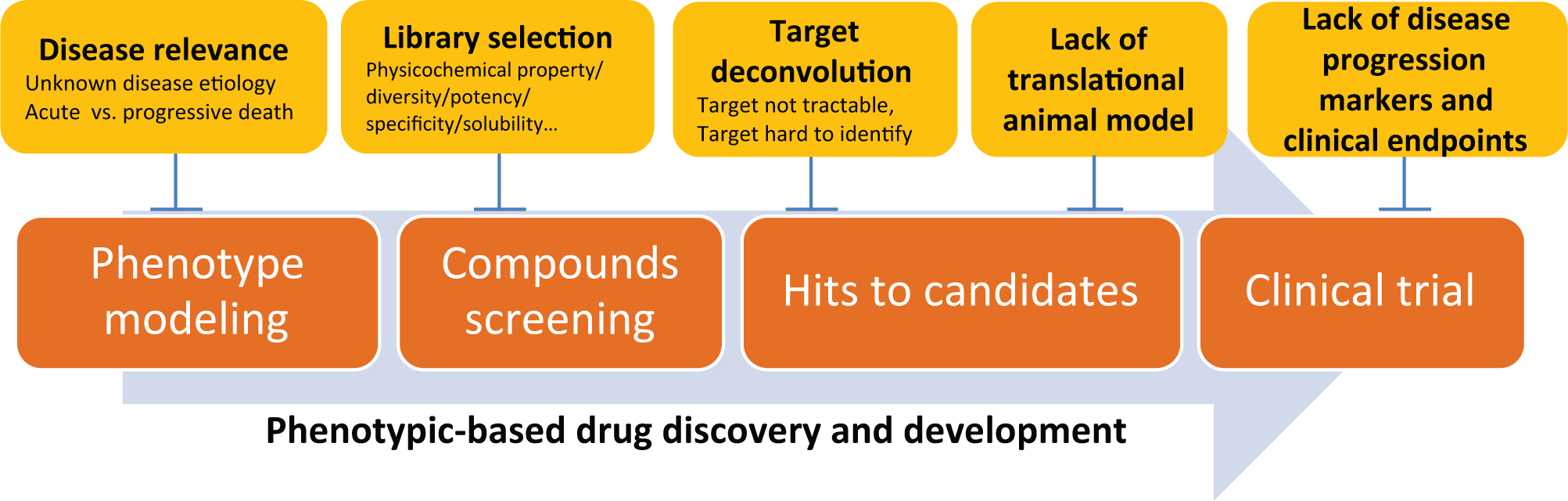
Key challenges (marked yellow) along the path of phenotypic-based drug discovery and development.
The key of previous successes of phenotypic screening for drug discovery is the use of translatable driver disease phenotype. Although it is obvious that among all NDs, neuronal dysfunction or neuronal death is the consistent phenotype to trigger disease progression, it is still unclear what the real cause for neurodegeneration is. In addition, it is reasonable to question whether the acute cellular neuronal death model accurately reflects the chronic progressive nature of ND. To mitigate the risks associated with these gaps, phenotype selection should be guided by insights from human genetics, epidemiology, pathology, and clinical findings. In addition, to reduce the risk associated with disease irrelevance of a specific phenotypic assay, compounds with multiple mechanisms are preferable to ones that modulate only a single phenotype. For example, a compound with both toxin removal and neuroprotection properties may offer better disease-modifying effects and potentially reduce the chance of false-positive.
After a successful phenotypic screening, to further develop the lead compounds, translatable animal models and disease progression markers will be helpful to evaluate their disease-modifying efficacy. Encouragingly, more and more resources and efforts have been devoted to this area to facilitate drug development. As a leading example of these efforts, to enhance translation from preclinical studies to clinical tests, PharmaCog is trying to standardize preclinical models to identify translational endpoints to allow prediction and quick assessment of new drug candidates for AD (http://www.alzheimer-europe.org/Research/PharmaCog). Initiatives, such as the Alzheimer’s disease neuroimaging initiative and Parkinson’s progression markers initiative, have been actively working on the identification of disease progression markers to facilitate clinical testing of potential disease-modifying compounds (http://adni.loni.ucla.edu/; http://www.ppmi-info.org/). Recently, the FDA has drafted a guideline with defined clinical outcome measures for developing drugs for the treatment of the early stage of AD, at which point injury to the brain is considered to be reversible (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm338659.htm). Outcomes from these efforts will pave the way for efficient clinical development and proof-of-concept studies. With clinical proof or disproof, we will be able to revisit the phenotypic assay and adjust accordingly, which will eventually lead to phenotypic screening models closely reflecting the human diseases.
Footnotes
Acknowledgements
We would like to thank Dr David Tattersall for his critical reading and editing of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The review was written in the course of employment at GlaxoSmithKline.