
Abstract
Clinical studies of gene expression are increasingly using the whole blood, peripheral blood mononuclear cells, and leukocyte subsets involved in the innate and adaptive immune responses. However, the small amount of RNA available in the clinical setting is a limitation for commonly used methods such as quantitative polymerase chain reactions (qPCR) and microarrays. Our aim was to design 96 gene assays to simultaneously measure gene expression in the whole blood and seven leukocyte subsets using a new-generation qPCR method—high-throughput nanofluidic reverse transcription qPCR (HT RT-qPCR). The leukocyte subset purity was 94% to 98% for seven subsets and was less for the γδ T-cell receptor subset (80%). The HT RT-qPCR replicate sample measurements were highly reproducible (r = 0.997, p < 2.2 × 10−16), and the ΔΔCt values from HT RT-qPCR correlated significantly with those from qPCR. The control genes were differentially expressed across the eight leukocyte subsets in the control subjects (p = 1.3 × 10−5, analysis of variance). Two analytical methods, absolute and relative, gave concordant results and were significantly correlated (p = 1.9 × 10−9). HT RT-qPCR permits the rapid, reproducible, and quantitative measurement of multiple transcripts using minimal sample amounts. The protocol described yielded leukocyte subsets of high purity and identified two analytic methods for use.
Introduction
Genetics plays a very important role in current research and in clinical medicine. Contrary to the static genome encoded by DNA, messenger RNA (mRNA) transcripts dynamically link DNA with proteins. As profiles of mRNA transcripts are tissue and condition specific, they reflect the often rapid changes occurring with physiological and pathological processes. Over the past decade, microarray technology has been used to study gene expression in a high-throughput way, although the information obtained is qualitative rather than quantitative. It is also still unclear how the results can be applied clinically.1,2 Fluorescence-based quantitative real-time PCR (qPCR) has become the gold standard for quantitatively determining low concentrations of target mRNAs. 3 Although well established and reliable, this method is low throughput, costly, and time-consuming. Furthermore, only a limited number of transcripts can be studied from typical clinical samples.
The next generation of qPCR platforms has addressed several of these limitations associated with qPCR and microarrays. Terms for these new platforms, such as high-throughput reverse transcription qPCR (HT RT-qPCR), as used in this article, and nanofluidic qPCR, are in use. 4 HT RT-qPCR offers high sensitivity and a wide dynamic range, allowing for the detection of single complementary DNA (cDNA) copies. Scaling to nanoliter volumes enables as many as 96 transcripts to be screened simultaneously in as many as 96 samples, performing thousands of reactions in a few hours. This enables high throughput and dramatically reduces the volume of required reagents and clinical samples. In contrast to standards for qPCR experiments described in the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE), 5 guidelines on how to perform next-generation qPCR experiments are still emerging.
The peripheral blood is readily sampled in the clinical setting, and gene expression studies have been increasing rapidly. Initial studies in stroke and in other disease states used either the whole blood or peripheral blood mononuclear cells (PBMCs).6–10 However, the whole blood and PBMCs contain multiple leukocyte subsets that differ in their function, often to the point of having opposing effects, potentially masking many gene expression changes. Specific leukocyte subsets are variably involved in pathogen recognition, immune responses, and tissue repair. As an example, monocytes (CD14+) and granulocytes (CD15+) are involved in nonspecific, innate immune responses, whereas T lymphocytes (CD4+, CD8+) and B lymphocytes (CD20+) are involved in antigen-specific, adaptive immune responses. The γδ T-cell receptor (TCR) T lymphocytes are unique in that they participate in both adaptive and innate immune responses. Studies are increasingly turning to leukocyte subsets to study innate, adaptive, cellular, and humoral immune responses as well as individual cells.8,11–14 As it is likely that these leukocyte subsets substantially differ in their gene expression, it is imperative that mRNA is extracted from carefully selected and homogeneous samples. The control or reference genes used for qPCR also need to be sample specific.
In this study, our aim was to develop and validate a method for measuring the expression of multiple transcripts simultaneously in whole blood and its leukocyte subsets. We (1) assessed and evaluated the next-generation qPCR method, HT RT-qPCR; (2) established feasible methods of leukocyte subset separation; (3) designed and wet tested 96 gene expression assays for qPCR; (4) developed and verified an absolute gene expression analysis method based on cell count; and (5) identified appropriate control genes for relative gene expression analysis in leukocyte subsets.
Material and Methods
Where applicable, the conduct and reporting of the study are in accordance with the MIQE criteria. 5 The Institutional Review Board at the State University of New York (SUNY) Downstate Medical Center approved the study.
Study Participants
The peripheral blood samples of 36 participants were used, including 21 acute stroke patients admitted to the University Hospital of Brooklyn at SUNY Downstate Medical Center and at Long Island College Hospital, within 48 hours from onset, and 15 sex- and race-matched healthy control participants recruited from the local community. Stroke was diagnosed according to the World Health Organization stroke criteria. 15 All patients or their authorized representative gave full and signed informed consent. The details of the participants are shown in Table 1 .
Study Participants.
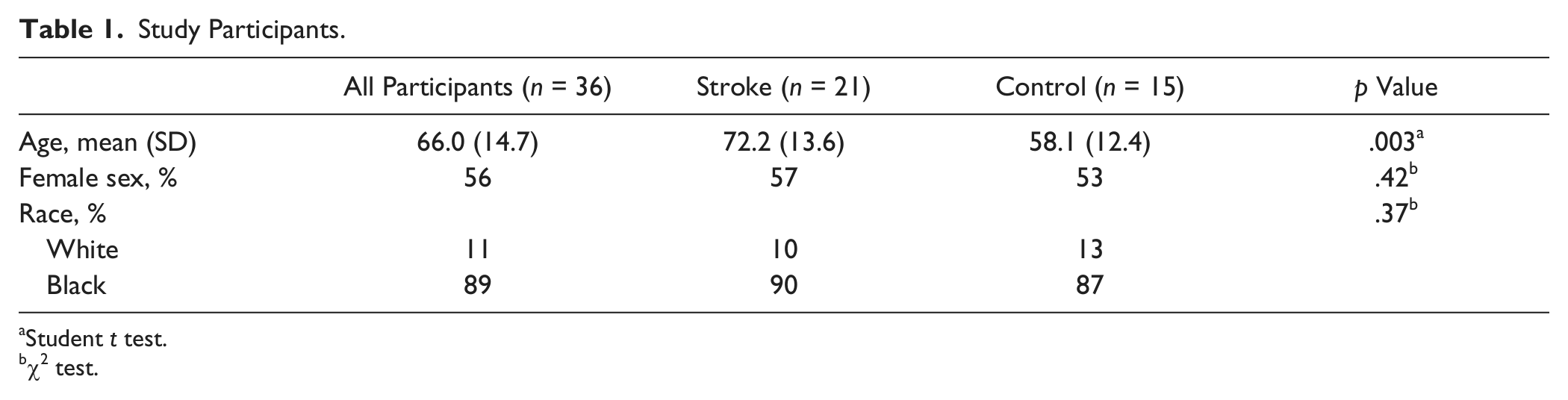
Student t test.
χ2 test.
Leukocyte Separation
Thirty milliliters of whole blood (WB) was drawn into ethylenediaminetetraacetic acid tubes and a complete blood count (CBC) with differential was performed on each participant. Then, 100 µL of WB and up to 2 million cells were collected for RNA extraction from each of PBMCs, granulocytes (CD15+), monocytes (CD14+), T lymphocytes (CD4+), γδTCR+ T lymphocytes, B lymphocytes (CD20+), and T lymphocytes (CD8+). The cellular viability and number were assessed using a hemocytometer and Trypan blue dye staining. Density gradient centrifugation with Histopaque 1077 and 1119 (Sigma-Aldrich, St. Louis, MO) was used to separate PBMCs and granulocytes from the whole blood (
Messenger RNA Isolation
The leukocytes were lysed in Lysing Solution (All-in-One Kit; Norgen Biotek, Thorold, Ontario, Canada) and frozen at −80 °C. Lysis is the first step of the All-in-One Kit isolation protocol. The subsequent protocol steps were performed according to the manufacturer’s protocol after sample thawing on ice. From simultaneously isolated molecules of large RNA (>200 nucleotides), small RNA (<200 nucleotides), DNA, and proteins, only the large RNA fraction containing mRNA was used in this study. The large RNA concentration was measured with the Qubit 1.0 Fluorometer (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. The DNA concentration in the large RNA fraction was measured with the Qubit 1.0 Fluorometer (Invitrogen) to evaluate for potential contamination. The quality of the RNA for blood and for each leukocyte subset was evaluated on 1× 3-(N-morpholino) propanesulfonic acid 1.2% formaldehyde-agarose RNA gel and run under a current of 150 V for 30 min. Antidegradation and anticontamination techniques were applied throughout: these included a sterile environment, RNase pretreatment, and RNase free buffers and solutions. RNase free and RNA low binding tips and tubes were also used.
Reverse Transcription PCR and Primer Design
The High Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA), based on random hexamers, was used according to the manufacturer’s protocol. cDNA was synthesized in reverse transcription PCR (RT-PCR) using the Veriti 96-Well Thermal Cycler (Applied Biosystems, Foster City, CA).
The transcripts were chosen from previously published literature microarray gene expression lists,6–8,16,17 as well as potential control genes18,19 and several negative control genes. The list, transcript names, and sources are in
HT RT-qPCR
HT RT-qPCR was run on the BioMark HD System, using 96 × 96 Fluidigm Dynamic Arrays (Fluidigm, South San Francisco, CA), giving a total of 9216 reactions per plate. Four plates with 96 transcripts were run. The first three contained 94 clinical samples and two duplicate samples of commercial cDNA. The fourth plate contained serial dilutions of commercial cDNA in triplicate (four levels) along with samples from a separate study. The same 96 primer assays were included in the four plates. Specific target amplification (STA) was performed to preamplify 10 µL of the cDNA template of each sample in 14 cycles (10 min at 95 °C followed by 14 cycles of 95 °C for 15 s and 4 min at 60 °C). STA and gene expression measurements were performed according to the manufacturer’s protocol for EvaGreen dye (EvaGreen dye, 20× in water; Biotium, Hayward, CA). The cycling program used consisted of 10 min at 95 °C followed by 40 cycles of 95 °C for 15 s and 1 min at 60 °C. Data were initially processed in Fluidigm Real-Time PCR Analysis software (version 3.0.2) and exported for further statistical analysis to Excel files in “csv” format. The Fluidigm Real-Time PCR Analysis software settings were as follows: a quality threshold of 0.7, a baseline correction of linear derivative, and a cycle threshold (Ct) of auto detectors. On a fourth plate, a triplicate dilution series of the commercial cDNA and NTC was run to evaluate the correlation coefficients (R2) and efficiencies. Primer interactions may occur in multiplex qPCR, and these were evaluated by calculating the efficiency and NTC of each assay. 5 Nonspecific amplification is substantially reduced by STA and melt curve analysis. STA in 14 cycles preamplifies only target genes (96 tested genes in our experiment), thus limiting nonspecific amplification, whereas melt curve analysis automatically excludes all remaining nonspecific PCR products with abnormal melt curves.
qPCR
The qPCR was run on the StepOnePlus Real-Time PCR Systems (Applied Biosystems) using 96-well plates containing a final volume of 20 µL. Optimum reaction conditions for Fast EvaGreen qPCR Master Mix-high Rox were obtained for 10 µL of Master Mix and varying primer concentrations (200, 100, and 50 nM) and 2 µL of cDNA template and topped up with RNase free water (Life Technologies). The primer concentration for each gene is shown in
Data and Statistical Analyses
The “.csv” data and “.xls” data were analyzed using R version 2.15.1. 21 In comparing gene expression between patients and control participants, absolute and relative analytic approaches were used. The absolute analysis was based on the cell count. The transcript Cq value was subtracted from the average of the Cq values of the same transcript for commercial cDNA and then the difference was divided by the cell count. The cell counts were obtained from the laboratory data or from the CBC. The relative analysis used the average of three control genes as the reference. To minimize the plate-to-plate variation for both qPCR and HT RT-qPCR, the expression for each gene was defined as the distance between mean expression (MEx) for all control (CEx) and stroke (StEx) within the cell subset according to the equation (MEx – CEx or MEx – StEx).22,23
For grouped and categorical data t tests, Mann-Whiney U tests and analysis of variance were used to compare groups. For continuous data, Pearson’s product moment correlation coefficients were used. McNemar’s test was used to test concordance, and p-values <.05 were considered statistically significant.
Results
Leukocyte Subset Purity and RNA Quality
The purity of WB, PBMC, CD4, CD14, CD15, CD8, and CD20 ranged between 94% and 98%. The purity of the γδTCR subset was still quite high, but lower at 80% (
Performance of HT RT-qPCR
R2 and Amplification Efficiency
The overall percentage of present calls for the plates was >95%, being 89.8% for plate 1, 91.6% for plate 2, 97.0% for plate 3, and 99.0% for plate 4. Eighty-four percent of transcripts had R2 values >0.95, and 63% had R2 values >0.99. The amplification efficiencies ranged from 60% to 100%. Eight percent of transcripts had amplification efficiencies of 100%, 49% of transcripts had amplification efficiencies more than 80%, and 92% of transcripts had amplification efficiencies more than 70% (
Reproducibility
The reproducibility of HT RT-qPCR measurements was first tested by comparing the Cq values obtained for the replicate commercial cDNA samples on each plate. The Cq values for the cDNA replicate samples were highly correlated with an overall correlation coefficient of 0.987 (p < 2.2 × 10−16), ranging between 0.986 (p < 2.2 × 10−16, plate 1) and 0.997 (p < 2.2 × 10−16, plate 3) ( Fig. 1a ).
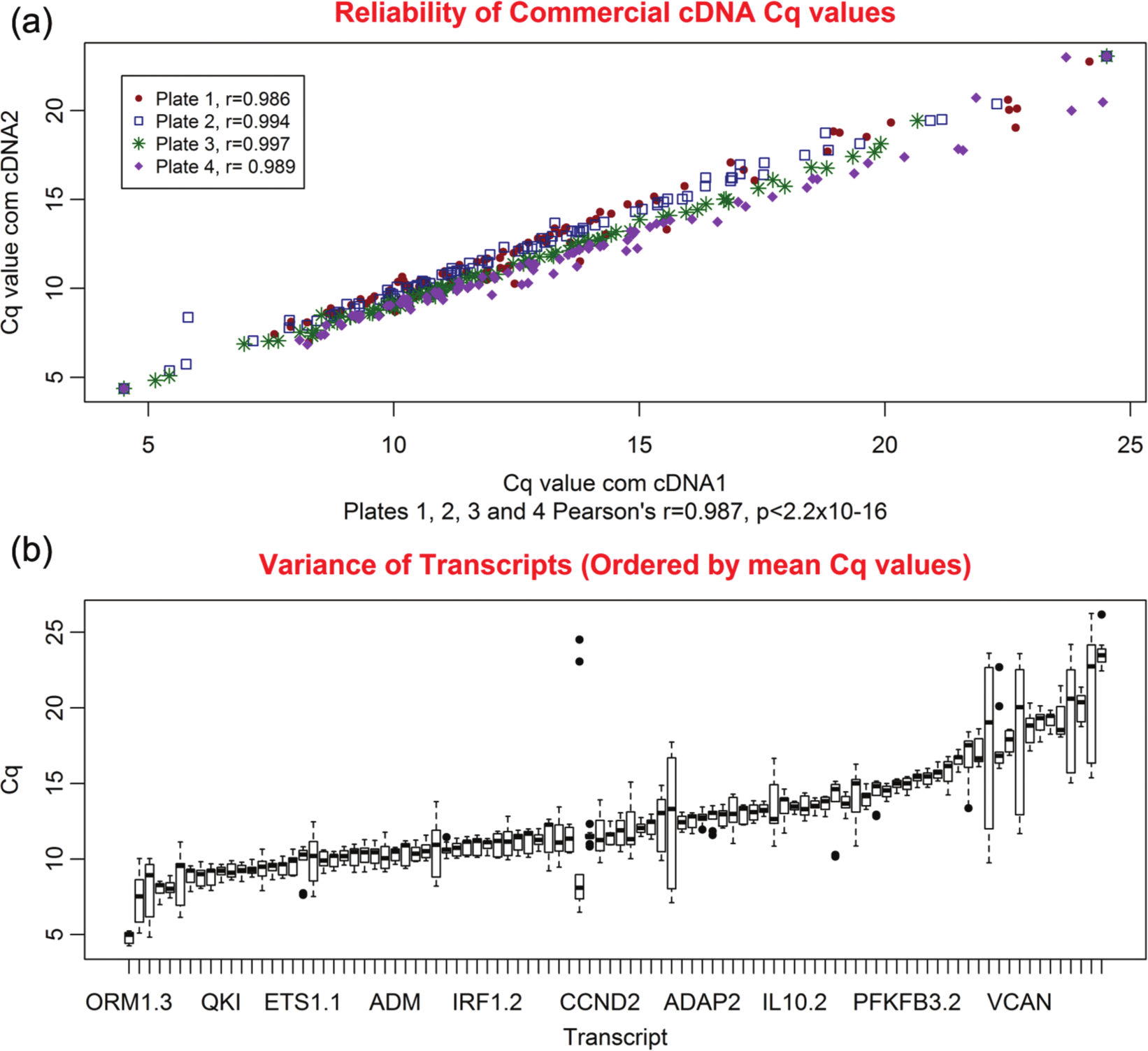
Reproducibility of high-throughput nanofluidic reverse transcription quantitative PCR (HT RT-qPCR) measurements. (
The second measurement of reproducibility was based on the variance of the nine Cq values obtained for each of the 96 transcripts using the commercial cDNA (two replicates on plates 1, 2, and 3 and three replicates on plate 4). The nine measurements, shown in Figure 1b , were expressed as boxplots for each transcript and ordered by median Cq value. In 88.6% of transcripts, the variance was less than the 1.0 Cq value ( Fig. 1b ).
Control Gene Analysis
Ten potential control genes were analyzed, selected from the literature and our preliminary results.16–19 The genes were analyzed with respect to (1) R2, (2) efficiency, (3) variance, and (4) stability by disease state. The efficiency, R2, and variance were based on the commercial cDNA values. Farnesyl-diphosphate farnesyltransferase 1 (FDFT1), fucosyltransferase 4 (α (1,3) fucosyltransferase, myeloid-specific, FUT4), and CD3e molecule (CD3E) were the optimal control genes for all subsets. They had high R2 values, optimal efficiency, and low variance. Expression level of these genes is detectable and varies across studied subsets, with FDFT1 being most stable. Regardless of the acute and inflammatory nature of stroke, expression of these genes was not different between control participants and stroke patients in any of the leukocyte subsets ( Table 2 ). For the analyses involving control genes in last two sections of the Results, the average of these three transcripts was used. GAPDH and B2M differed by disease state, as did 18S rRNA to a lesser degree. B2M and GAPDH also had high variance. In addition, 18S rRNA had a low R2 and B2M had a low efficiency.
Control Gene Characteristics.
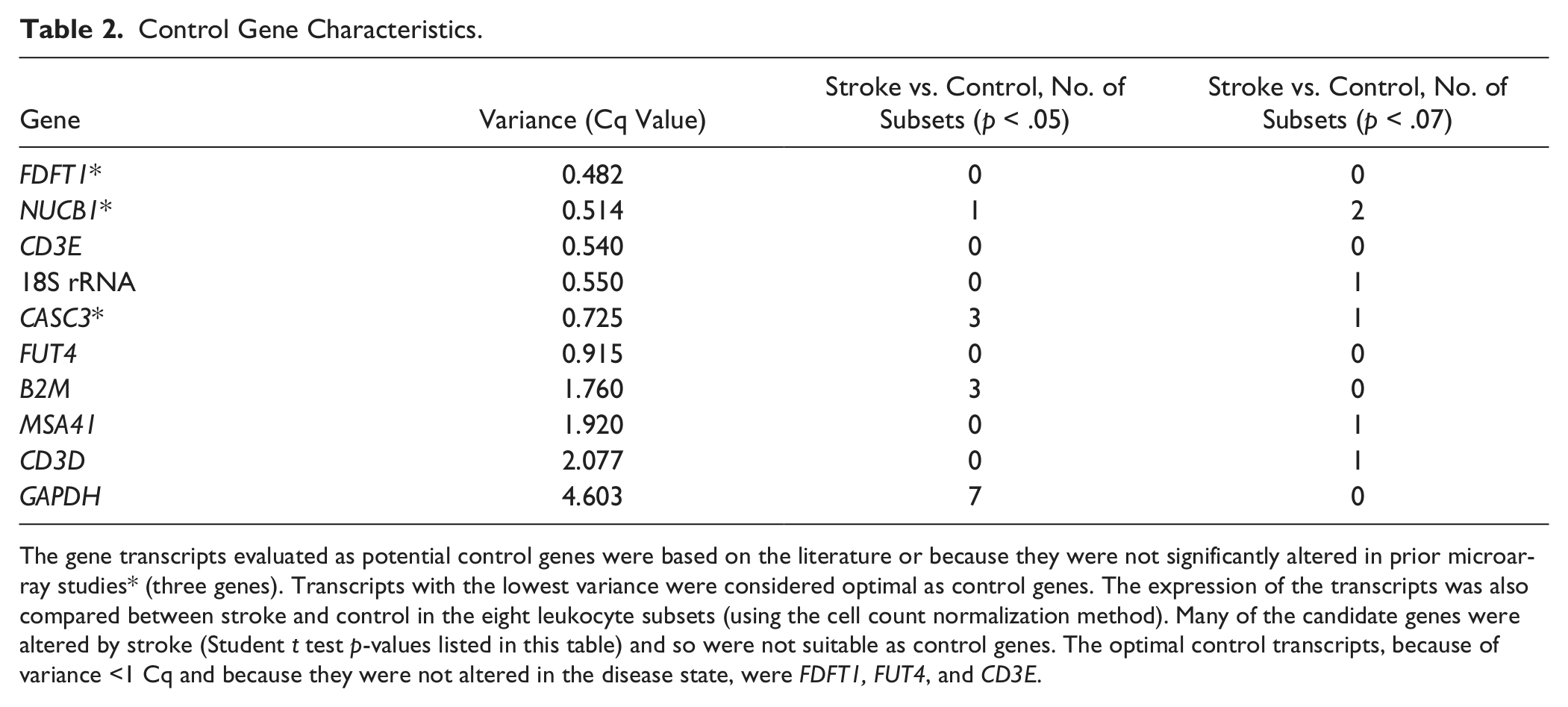
The gene transcripts evaluated as potential control genes were based on the literature or because they were not significantly altered in prior microarray studies* (three genes). Transcripts with the lowest variance were considered optimal as control genes. The expression of the transcripts was also compared between stroke and control in the eight leukocyte subsets (using the cell count normalization method). Many of the candidate genes were altered by stroke (Student t test p-values listed in this table) and so were not suitable as control genes. The optimal control transcripts, because of variance <1 Cq and because they were not altered in the disease state, were FDFT1, FUT4, and CD3E.
Comparison of qPCR and HT RT-qPCR
The levels of expression for seven genes, relative to FDFT1, were compared between HT RT-qPCR and qPCR. The ΔΔCt values from HT RT-qPCR were highly correlated with those from qPCR (p < 10−8). The overall correlation coefficient was 0.78 (p < 2.2 × 10−16). The correlation coefficients for each of the seven transcripts ranged from 0.98 for IL1R2 (p < 2.2 × 10−16) to 0.49 for GAPDH (p = 2.93 × 10−7; Fig. 2 ).
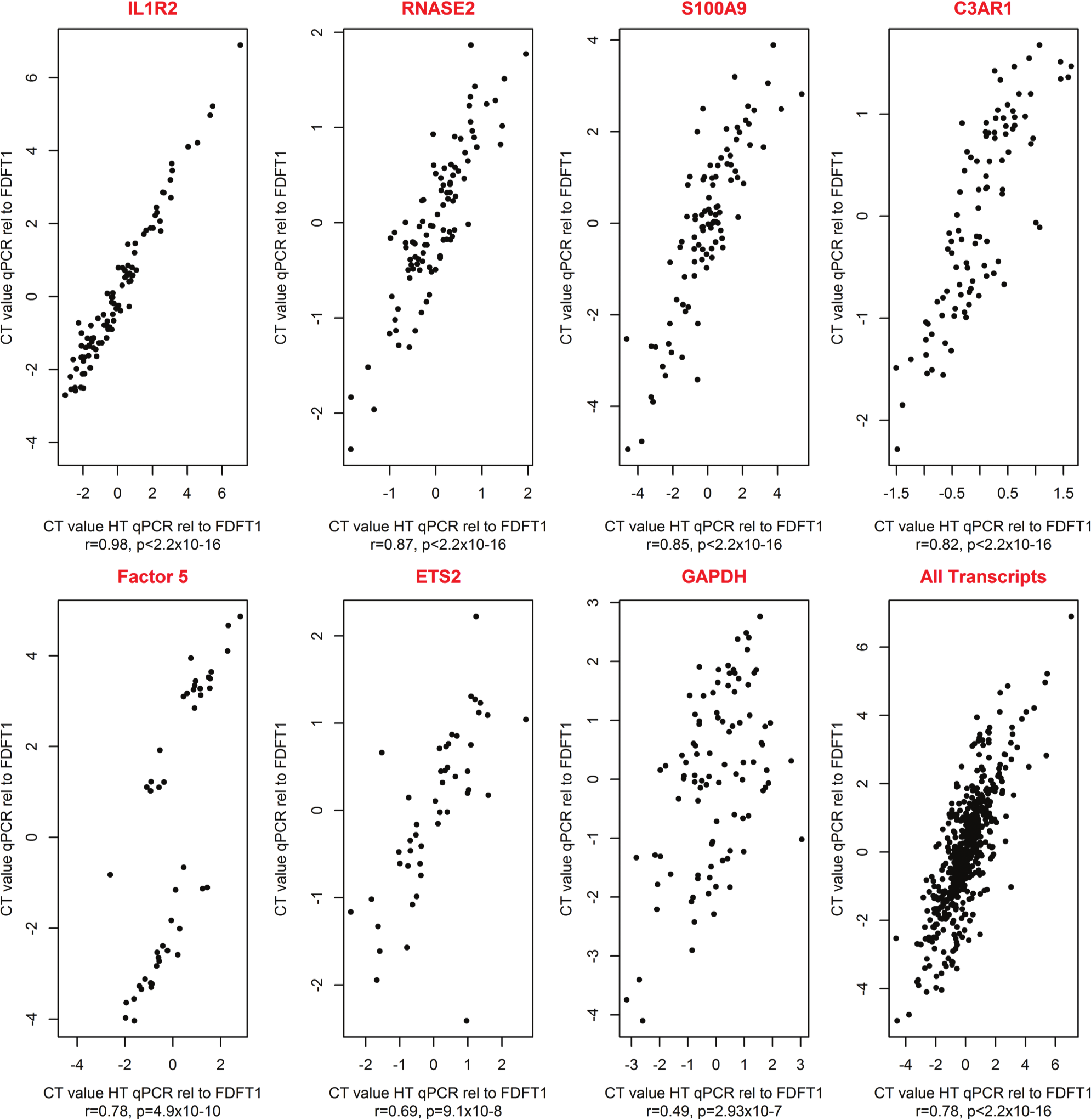
Comparison of high-throughput nanofluidic reverse transcription quantitative PCR (HT RT-qPCR) and qPCR ΔΔCt values. The ΔΔCt values were compared between HT RT-qPCR and qPCR for seven transcripts and measured relative to the control gene FDFT1. The samples for 12 patients and 12 control participants were used and the analyses were run in four leukocyte subsets: peripheral blood mononuclear cells (PBMCs), CD14, CD4, and CD20.
Data Analysis Approaches
Two independent approaches for data analysis were developed and evaluated. The first was absolute, based on the cell count and normalized to commercial cDNA, and the second was relative, measured using the average of the three selected control genes (FDFT1, FUT4, and CD3E). The expression of 16 transcripts was compared between 21 stroke patients and 15 control participants. The 16 transcripts were the 10 genes evaluated as potential reference genes and the 6 stroke-related genes that had been used in the qPCR analyses. The cell count and control gene normalization methods showed similar patterns of upregulation and downregulation. The comparative heatplots are shown in Figure 3 . The percent upregulation and downregulation calls for the two analytic approaches were 88% concordant. McNemar’s test for paired observations—to examine if there was a difference in the upregulation and downregulation calls from the two analysis methods—gave a p-value of .6, indicating no significant difference between the two analyses. The Cq values for the two methods were also correlated (r = 0.50, p = 1.94 × 10−9). By both analytic methods, five of the previously published stroke-related genes (IL1R2, S100A9, F5, ETS2, and C3AR1) were upregulated in the stroke patients relative to the control participants.
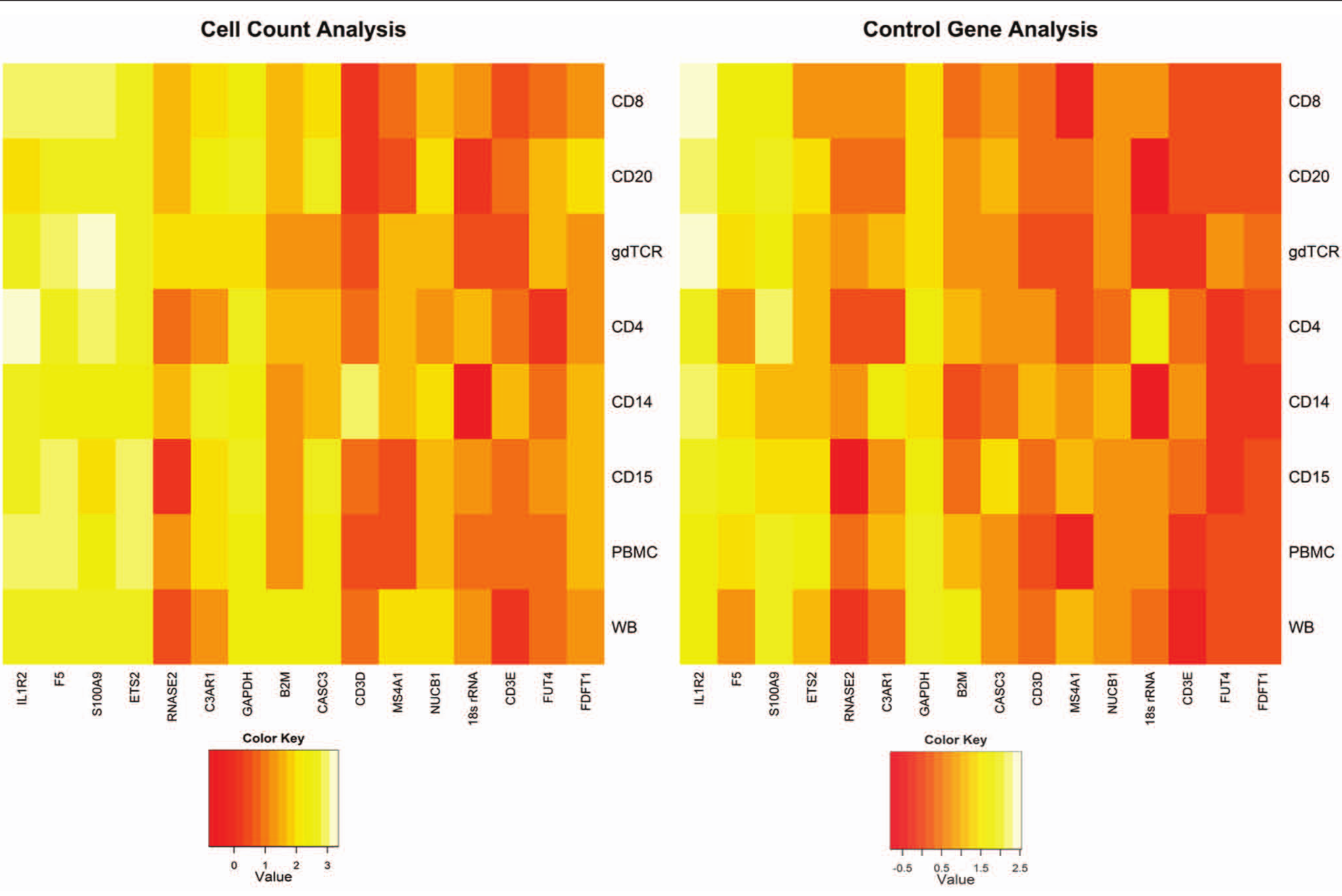
Comparison of cell count–based and control gene analytic methods. Two independent approaches for data analysis were developed and evaluated. The first was absolute, based on the cell count and normalized to the Cq value of the corresponding transcript for the commercial complementary DNA. The second was relative, measured relative to the average of the three selected control genes (FDFT1, FUT4, and CD3E). Using the two methods, the expression of 16 transcripts was compared between 21 stroke patients and 15 control participants. The 16 transcripts were the 10 genes evaluated as potential reference genes and the 6 stroke-related genes that had been used in the quantitative PCR analyses. The 10 control genes are shown on the right and the 6 stroke genes on the left of the figures. The two analyses gave concordant results, and five previously published genes (IL1R2, S100A9, F5, ETS2, and C3AR1) were found to be upregulated in the stroke patients relative to the control participants.
Differential Gene Expression among Leukocyte Subsets
To assess potential differences in gene expression among leukocyte subsets, we measured the expression of the three control genes in the control participants. The expression of the three control genes, singly and on average, differed by leukocyte subset (p = 1.9 × 10−5, analysis of variance). The control gene expression was highest in the whole blood, PBMCs, and CD4 leukocyte subset and lowest in the CD20 subset. The greatest variance in control gene expression was seen in the CD20, CD8, and γδTCR subsets ( Fig. 4 ). The three control genes showed similar differential expression across the leukocyte subsets.
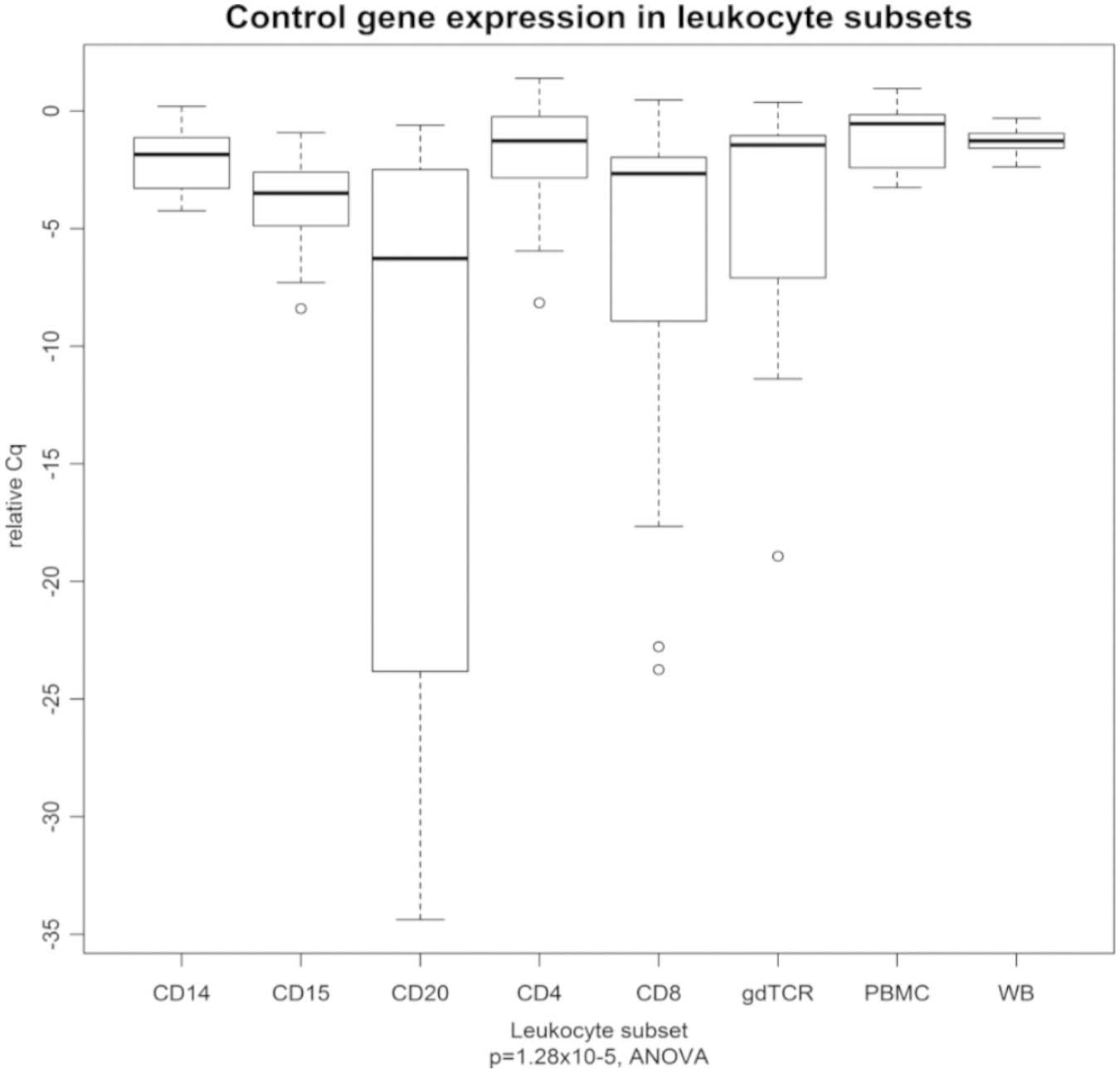
Leukocyte subset differences in gene expression. The expression of the three control genes (averaged) differed by leukocyte subset. The Cq values are presented relative to the commercial complementary DNA, and therefore higher relative Cq values are indicative of higher expression. PBMC, peripheral blood mononuclear cell; WB, whole blood.
Discussion
HT RT-qPCR is a next-generation qPCR method and, despite a limited availability of guidelines, is being reported in a growing number of studies.24–27 In this study, we have assessed and validated HT RT-qPCR measurements of gene expression in the whole blood and its leukocyte subsets. We used the BioMark HD System (Fluidigm) because no other was available at this time. Recently, Devonshire et al. 4 discussed the currently available systems. Using limited amounts of RNA to create cDNA, HT RT-qPCR allowed us to test 96 genes simultaneously and quantitatively in 96 samples, giving a total of 9216 reactions. The amount of cDNA needed for HT RT-qPCR is about 40- to 50-fold less than for regular qPCR. For HT RT-qPCR, 10 µL of cDNA per sample is needed to study 96 genes simultaneously, whereas 6 µL of cDNA is needed to study one gene for qPCR (the variability of this method requires 2 µL of each sample to be run in triplicate). We found HT RT-qPCR to be reliable and valid. We were able to develop an analytic approach to study the absolute levels of gene expression, relative to commercial cDNA and independent of control genes. This absolute analytical method was successfully tested versus a method relative to control genes employing universal control genes across the different leukocyte subsets.
HT RT-qPCR involves the measurement of significantly lower amounts and copy numbers than qPCR and hence has higher sensitivity than qPCR. Because of the nanosize of the reaction chambers and decreased background noise, this technology enables the sensitive and reliable detection of single transcripts. To operate at this low concentration, the additional step of specific target amplification is required. This step is difficult to control in terms of R2 and efficiency. However, in evaluating HT RT-qPCR, we demonstrated extremely high correlations for the replicate samples and established that, unlike qPCR, triplicates are not needed. In terms of validation, we also found close correlation between qPCR and HT RT-qPCR despite some differences in efficiency.
So far, universal control genes for different leukocyte subsets have not been established. HT qPCR permitted 10 control genes to be quantitatively evaluated for all the samples and leukocyte subsets at the same time. With qPCR, this has not been possible due to the limited sample amounts. We confirmed the findings of other investigators18,19 that the expected control genes, such as GAPDH, B2M, and 18S rRNA, did not perform optimally. They showed high standard deviations, and their expression was altered in stroke patients. Three appropriate control genes for use for all eight subsets were identified: FDFT1, FUT4, and CD3E. Although they were expressed at different levels across the leukocyte subsets studied, their expression was not altered by disease state. Their average expression was used in the subsequent analyses. These genes were abundant, had low variance, and were not altered in any of the leukocyte subsets in stroke. The identified control genes may be also suitable for other experiments involving blood subset studies.
The data were analyzed using two independent methods, one relying on the control genes and one relying on the number of cells. Although the methods gave concordant results, some variation in values for the two analytic methods could be due to leukocyte subset differences in the expression of the control genes. The use of the cell count method that controls for input volume gets around this issue. The cell count method has the additional advantages over the standard control gene method of permitting the expression of each gene to be assessed independently, regardless of the efficiencies of other genes. The step of normalization to the expression level of standard commercial cDNA (Biochain) means that batch effects can be removed and results can be compared between plates. The input count is also easily obtained and accurate.
Thirty milliliters of blood permitted the extraction of around 2 million cells per leukocyte subset and provided additional samples for the qPCR validation and permitted rare genes with low expression to be studied (i.e., HPR, MERTK, IL-10, and NAIP). If the HT RT-qPCR had been run without qPCR validation, 15 mL of whole blood would have been enough. Alternatively, if we were to study one abundant subset instead of eight (e.g., the CD3 subset), it is estimated that between 2 and 4 mL per sample would probably be enough to run the HT RT-qPCR, although this has to be run and confirmed for each experiment. Of the two currently available methods for sorting leukocytes—fluorescence-activated cell sorter (FACS) and magnetic beads—we chose to use beads. 28 Using beads permitted us to process more cells with slightly lower purity than FACS. The low numbers of γδTCR cells meant that the purity was affected by minimal numbers of additional cells. The concentration of RNA differed across studied subsets. The concentration was highest in the CD4 T-lymphocyte population and lowest in the CD15 granulocyte population. This may be explained by the fact that the most abundant CBC population (CD15 polymorphonuclear cells, comprising around 60% of the CBC) is mature and differentiated, and its immune response is not believed to be antigen specific. Conversely, the CD4 T-lymphocyte population, the subset with the highest RNA content, is expected to dynamically adjust to varying conditions to develop antigen-specific responses.
HT RT-qPCR has the potential to be used for many lines of clinical and translational research, in stroke and in other disease states. HT RT-qPCR permits further exploration of the results of microarray studies. Blood is a surrogate tissue in a number of disorders, and recent stroke studies have shown that studying different subsets of blood leukocytes opens new avenues in genetic and immunological research.29,30 Differences in gene expression that were found for some of the candidate control genes gives an indication of the promise of the approach to assess cell subset–specific gene expression with microarray technology. Even though the previously published stroke-related gene lists come from studies using the whole blood and PBMCs, strong changes in a number of leukocyte subsets in stroke were found in this study, as shown in the heatplots.
In conclusion, HT RT-qPCR permits rapid, reproducible, and quantitative measurements of multiple transcripts using minimal sample amounts. The protocol described yielded leukocyte subsets of high purity and identified three control genes that may be applied in the study of stroke and other disease states. Furthermore, an analytic approach was developed that eliminates the need for control gene validation and may reliably be applied to all blood or cell line–derived experiments.
Footnotes
Acknowledgements
Dr. Adamski was awarded a scholarship program by Polpharma Scientific Foundation.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr. Baird is supported by National Institutes of Health (NIH) grants R01 EB010087 and R21 MH097639. Dr. Baird is also a co-investigator on R25 NINDS R Train, Neurology Research Education Program and U01 NINDS NeuroNEXT Clinical Trials at SUNY Downstate Medical Center. Dr. Soper is supported by the NIH grant NIBIB R01 EB010087. Dr. Murphy is supported by the NIH grant NIBIB R01 EB010087.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.