
Abstract
Plant-pathogenic bacteria are the causative agents of diseases in important agricultural crops and ornamental plants. The severe economic burden of these diseases requires seeking new approaches for their control, particularly because phytopathogenic bacteria are often resistant to available treatments. The type II secretion (T2S) system is a key virulence factor used by major groups of phytopathogenic bacteria. The T2S machinery transports many hydrolytic enzymes responsible for degradation of the plant cell wall, thus enabling successful colonization and dissemination of the bacteria in the plant host. The genetic inactivation of the T2S system leads to loss of virulence, which strongly suggests that targeting the T2S could enable new treatments against plant-pathogenic bacteria. Accordingly, we have designed and optimized an assay to identify small-molecule inhibitors of the T2S system. This assay uses a double parametric output: measurement of bacterial growth and the enzymatic activity of cellulase, which is secreted via the T2S pathway in our model organism Dickeya dadantii. The assay was evaluated by screening natural extracts, culture filtrates isolated from rhizosphere bacteria, and a collection of pharmaceutically active compounds in LOPAC1280. The calculated Z′ values of 0.63, 0.63, and 0.58, respectively, strongly suggest that the assay is applicable for a high-throughput screening platform.
Introduction
Phytopathogenic bacteria are one of the major threats to our food security. They cause severe plant diseases, such as blight, wilt, soft-rot, and blackleg, in important agricultural crops during their growth, transit, and storage. 1 Currently, treatment of these bacterial infections is based mainly on copper derivatives and antibiotics, including streptomycin and oxytetracycline. These antimicrobials, however, are considered environmental contaminants. In addition, resistance to these compounds has become widespread among phytopathogenic bacteria. 2
A promising approach in antimicrobial therapy involves the identification of new agents that target pathways contributing to bacterial virulence, thus disarming rather than simply killing bacterial cells. One such pathway is the type II secretion (T2S) system. The T2S system is widely conserved among Gram-negative bacteria, including major groups of plant-pathogenic microorganisms from the genera of Erwinia, Ralstonia, Pseudomonas, Xanthomonas, and Xylella. 3 This secretory nano-machine consists of 12 to 16 different proteins spanning the entire bacterial cell envelope. The secretion of exoproteins (secreted proteins, cargo proteins) is a stepwise process that includes their translocation via the cytoplasmic membrane, either by the Sec or the Tat system, and subsequently from the periplasm across the outer membrane by the T2S pathway. 4 The T2S system is responsible for the translocation of a variety of cargo proteins, including cellulases, pectinases, xylanases, lipases/esterases, proteases, endoglucanases, and polygalacturonases. 3 The orchestrated action of these hydrolytic enzymes leads to the degradation of the plant cell walls and enables the bacteria in the successful colonization and dissemination in the plant tissue. Most important, plant-pathogenic bacteria with mutations in the genes encoding the T2S system exhibit severe virulence deficiency in plant models of disease.3,5,6 Accordingly, we propose that the T2S pathway is a molecular target for novel treatments against plant-pathogenic bacteria.
Here, we report the development and application of a quantitative assay amenable for high-throughput screening (HTS) of natural product libraries and small-molecule inhibitors to identify compounds that interfere with the T2S process. The assay uses a double parametric output: measurement of both bacterial growth and the enzymatic activity of cellulase, which is secreted via the T2S pathway (referred to as Out) in our model organism Dickeya dadantii (formerly Erwinia chrysanthemi). The assay conditions have been optimized for a 96-well plate format by culturing D. dadantii in different liquid media followed by assessments of cellulase activity against a fluorescent substrate, resorufin–β-D-cellobioside. 7 The Z′ values calculated from our initial screening procedures of natural products and a defined collection of small-molecule inhibitors exceeded 0.5, strongly suggesting that the assay is robust, sensitive, and applicable for a full-scale HTS platform.8,9
Materials and Methods
Bacterial Strains and Growth Conditions
The bacterial strains used in this study include wild-type D. dadantii A350 (which is a Lac-negative derivative of strain 39377) and its isogenic nonpolar T2S mutant, outD.10,11 Both strains were obtained as a generous gift from Beatrice Py (LCB; CNRS, Marseille, France). Bacterial strains were streaked from −80 °C glycerol stocks onto Luria-Bertani (LB) agar plates (Difco; Becton Dickinson & Company, Franklin Lakes, NJ). Bacteria were grown either in rich LB medium, minimal M9 medium (Difco) supplemented with glycerol (0.5% wt/vol; Sigma Chemicals, St. Louis, MO), 0.1% yeast extract (Difco), and polygalacturonic acid sodium salt (0.5% wt/vol; Sigma Chemicals), thus referred to as PGA medium, or a Pseudomonas minimal salt (PMS) medium containing 0.2% glucose as a carbon source as indicated in the text.5,12 For all growth conditions throughout the experiments and during screening procedures, D. dadantii was maintained at 28 °C.
Optimization of D. dadantii Culture Conditions for the Production of Cellulase in the 96-Well Plate Format
To determine the optimal conditions for bacterial growth and cellulase production in a 96-well microplate format, we evaluated various combinations of media composition (PMS, PGA, or LB), inoculum size (1:50, 1:100, and 1:200 back-dilutions of bacterial cultures into corresponding fresh media), volume of the cultures (50, 100, and 200 µL), presence of DMSO (1%), and either static or agitated (at 70 rpm) incubation. These preliminary experiments (data not shown) established that for the three culture media tested, both robust bacterial growth and cellulase activity were achieved by 1:100 back-dilution of bacterial cultures into fresh respective media (in a final volume of 200 µL per well), followed by static incubation for 24 h. Moreover, both bacterial growth and cellulase activity were unaffected in the presence of 1% DMSO in comparison to the untreated bacterial cultures (data not shown). Thus, these growth conditions were performed for each media (PMS, PGA, and LB) in biological triplicates to calculate statistical parameters determining the suitability of these assays for HTS as described in the paragraph below. For all optimization experiments and subsequent test sample screening, a conserved layout of wells was used for individual 96-well sterile, clear plates with lids (Greiner Bio-One, Monroe, NC). Briefly, the first column (n = 8 wells) contained only culture media (LB, PGA, or PMS) supplemented with DMSO (at 1% final concentration). The wells in the second column (n = 8) functioned as the negative controls for the assay and contained wild-type D. dadantii in the respective media. Columns 3 to 10 (n = 64 wells) contained wild-type D. dadantii and the experimental samples. Columns 11 to 12 (n = 16 wells) contained the outD mutant of D. dadantii and served as the positive controls for the assay. Bacterial growth was assessed after 24 h from inoculation by measuring the turbidity of the cultures (OD600) in the Synergy HT Multi-Mode microplate reader (BioTek, Winooski, VT). Subsequently, 50-µL aliquots from all individual wells were transferred into respective wells on 96-well opaque black plates (Greiner Bio-One) for the fluorescence-based cellulase activity assay as described below.
Fluorescence-Based Cellulase Assay
Cellulase activity was determined by monitoring the hydrolysis of a fluorescent substrate, resorufin–β-D-cellobioside (Marker Gene Technologies, Eugene, OR), in an end-point assay, at excitation and emission wavelengths of 530/25 and 590/35 nm, respectively, 7 using a Synergy HT Multi-Mode microplate reader. The assay mixture in plate columns 2 to 12 consisted of 0.25 mM resorufin–β-D-cellobioside, 50 mM sodium acetate (pH 6.0), and 50 µL of the various bacterial cultures (prepared as described in the previous section) in a total volume of 100 µL per well. 7 Acetate buffer and resorufin–β-D-cellobioside (50 µL total) were also added to the culture media (blanks) in column 1 of the assay plates. Cellulase activity was expressed as relative fluorescence units (RFU). During the assay optimization and screening procedures, the cellulase activity was recorded either every hour for a total of 3 h or after 3 h from the addition of the substrate, respectively. The microplates were protected from light and incubated at 28 °C throughout the experiments.
Preparation of Natural Extracts
Natural extract libraries were generated from field-collected marine and terrestrial Panamanian cyanobacteria, as well as both field collections and laboratory cultures of deep-sea hydrothermal vent-associated macro- and micro-organisms. Cyanobacterial samples were collected by hand from La Amistad, Volcán Barú, and Coiba National Parks in Panama. Ethanol-preserved biomass was repeatedly extracted with dichloromethane-methanol (2:1), concentrated in vacuo, and subjected to normal-phase silica gel chromatography using a stepped solvent gradient (hexanes-ethyl acetate-methanol) to yield nine fractions. When sufficient material was available, these fractions were further separated into five subfractions of reduced complexity by reversed-phase C18 solid-phase extraction (RP18-SPE) using a stepped solvent gradient of MeOH-H2O. Deep-sea hydrothermal vent (DSV) macro-organisms, including tubeworms, palm worms, scale worms, and limpets, were collected from multiple sites along the Juan de Fuca Ridge in the northeast Pacific Ocean. These bulk, ethanol-preserved samples were extracted with dichloromethane-methanol (2:1), concentrated in vacuo, and the resulting organic extracts subjected to fractionation by normal-phase SPE using a stepped solvent gradient (hexanes-EtOAc-MeOH) to yield six fractions. Laboratory cultures of micro-organisms isolated from DSV sediment samples were grown on a 250- to 1000-mL scale in modified seawater medium at 30 °C, with shaking at 200 rpm, until stationary growth phase was reached. Culture material (including cells and culture media) was then extracted three times with equal volumes of EtOAc and combined and concentrated in vacuo. Organic extracts obtained in significant quantities were fractionated into five subfractions by RP18-SPE (MeOH-H2O). All organic extracts and fractions were stored at a concentration of 1 mg/mL in 10% H2O-DMSO. For biological assays, 5 µL of each sample solution was transferred to a 96-well clear microplate (Greiner Bio-One) and stored at 4 °C until sampling.
Preparation of Culture Filtrates from Isolates of Rhizosphere Bacteria
The different culture filtrates used in this study were obtained by culturing bacteria from the Landscape and the Deleterious Rhizosphere Bacterial Isolate Library Collections housed at USDA-ARS (Corvallis, OR). The Landscape Bacterial Isolate Library Collection contains single-colony isolates of unknown taxonomic classification, isolated from both diverse landscapes (agricultural, urban, aquatic) and hosts (roots, leaves, bark, mosses, insects) using semi-selective media for fluorescent Pseudomonas species but without the addition of antibiotics, except for cycloheximide. 13 The Deleterious Rhizosphere Bacteria Isolate Collection consists of single-colony isolates of primarily Pseudomonas species derived from the rhizosphere of grasses and cereals grown in the Willamette Valley, Oregon. These isolates were obtained using a semi-selective medium for fluorescent Pseudomonas species. 13
To prepare the culture supernatants, bacteria were grown overnight (28 °C, 200 rpm) in 900 µL of LB medium in 2.0-mL 96-deep-well polypropylene plates (Axygen Scientific, Union City, CA). Subsequently, bacterial cells were separated from the culture supernatants by centrifugation (3500 rpm, 15 min at 4 °C). Supernatants were filtered using a 0.20-µm syringe filter device (Acrodisc; Pall Corporation, Ann Arbor, MI) and placed into corresponding wells of sterile 96-well deep-well plates. The collection containing 320 culture filtrates was stored at 4 °C until sampling.
Compound Library
The LOPAC1280 compound library, consisting of 1280 pharmacologically active compounds, was purchased from Sigma Chemicals.
Assay Validation by Screening Natural Extracts, Culture Filtrates, and the LOPAC1280 Library
The sterile, clear 96-well plates and black opaque plates were barcoded prior to all assays. The samples and controls for the assay were arranged in each 96-well plate in the order described above. The natural products, culture filtrates, and synthetic molecules were manually delivered to individual wells of columns 3 to 10 in each 96-well plate for final concentrations per well of 25 µg/mL, 10% (v/v), and 10 µM, respectively. Subsequently, a MultiFlow BioTek plate dispenser was used to deliver the media and bacterial cultures of D. dadantii wild-type or outD isogenic mutant, each diluted 1:100 into fresh LB (supplemented with 1% DMSO for negative and positive controls), to make up a final volume of 200 µL per well. The plates were loaded onto the BioTek plate stacker, and the initial OD600 of each well was measured. The microplates were incubated statically at 28 °C, and after 24 h, both bacterial growth and cellulase activity were examined following the procedures described above (for the optimization of culture conditions and the cellulase assay).
Validation of the Sensitivity and Throughput of the Assay
The performance of the assay was assessed by determining statistical parameters: signal-to-background (S:B) and signal-to-noise (S:N) ratios, signal window (SW), and the Z′ factor using equations described previously.8,9 In addition, the coefficient of variation (CV) was calculated to determine the intraplate and interplate variability on two control populations: the negative and positive controls (wild-type and outD mutant D. dadantii, respectively) cultured in LB supplemented with 1% DMSO. All experimental data were analyzed using Prism 6.0 (GraphPad Software, San Diego, CA) and Microsoft Excel (Microsoft Corporation, Redmond, WA).
Results and Discussion
Design of the Assay for Screening of the T2S Inhibitors
We have chosen the phytopathogenic bacterium D. dadantii as our model organism because the structural organization and function of the Out system, as well as its secreted cargo proteins, have been extensively studied.3,11,14 Moreover, D. dadantii is a causative agent of soft-rot disease in all major groups of dicotyledons and has served as a model in studies elucidating bacterial virulence factors in plant pathogenicity. 5
We reasoned that the delivery of the T2S-exoproteins could be blocked by an inhibitory molecule in multiple ways, including (1) plugging the secretion pore, (2) affecting steps in the assembly of the secretion machinery, (3) disturbing the recognition of the cargo protein by the T2S complex, (4) disrupting interactions between the individual constituents of the T2S complex, (5) interfering with either the Sec or the Tat transport systems located within the cytoplasmic membrane, or (6) inhibiting synthesis of the T2S system ( Fig. 1 ). Therefore, to design an assay for HTS that would allow the identification of an inhibitory molecule acting in any of these possible scenarios, we decided to employ a readout assay that verifies the ultimate effect of the T2S, the presence of a cargo protein in the extracellular environment. The T2S is responsible for translocation of a large repertoire of exoproteins, but cellulase was the T2S-dependent protein of choice because its hydrolytic activity can be quantitatively and reliably assessed using a recently developed substrate, resorufin–β-D-cellobioside. 7 A long-wavelength highly fluorescent product, resorufin, is generated upon cleavage of the substrate by active cellulase, and thus the cellulolytic activity could be readily monitored as the amount of released fluorescence (RFU). Cellulase is secreted by the T2S system in all major groups of phytopathogenic bacteria, 3 and thus this assay could subsequently be used to assess the specificity of an identified inhibitory molecule toward T2S in a variety of bacterial species. Moreover, proteomic analyses demonstrated that this hydrolytic enzyme is one of the five most abundant proteins present in culture supernatants isolated from wild-type D. dadantii grown under standard laboratory conditions. 14
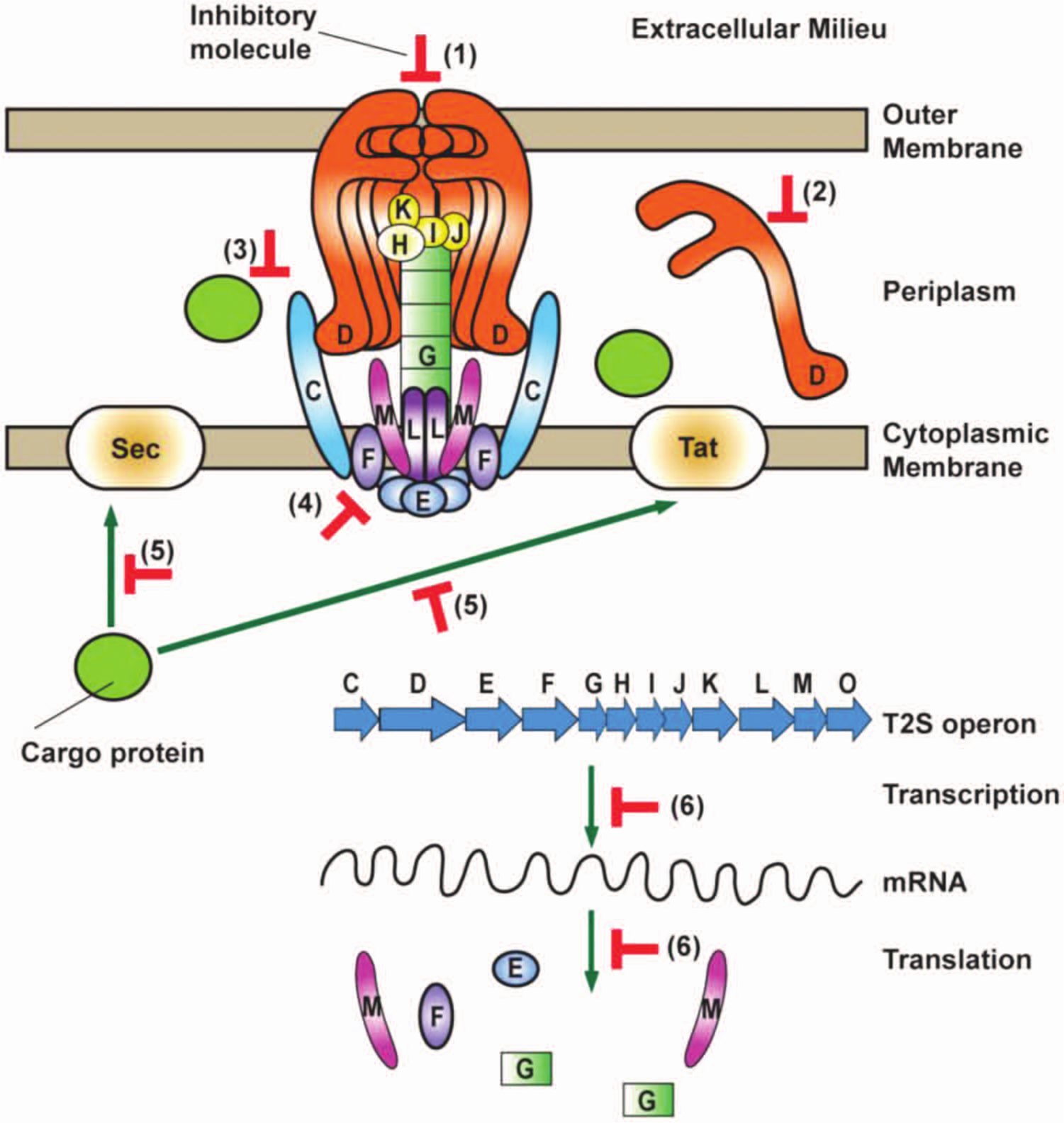
Inhibitory molecules may block the T2S system in multiple ways. The T2S machinery consists of 12 to 16 proteins that form a multiprotein complex spanning the entire bacterial cell envelope (designated here from C to O). The translocation of a cargo protein is a two-step process that involves transport via the cytoplasmic membrane provided by the Sec or the Tat system and subsequent recognition and secretion of the exoprotein by the T2S system across the outer membrane. A small-molecule inhibitor may block the secretion via many ways, including (1) plugging the secretion pore (D), (2) interfering with the steps of the assembly of the secretion machinery, (3) affecting the recognition of the cargo proteins by the T2S, (4) disrupting interactions between the individual constituents of the T2S complex, (5) disabling the function of either the Sec or the Tat transport systems located within the cytoplasmic membrane, or (6) inhibiting synthesis of the T2S system.
Assay Optimization and Validation
To determine robust culture conditions that allow optimal D. dadantii growth and cellulase production in the microplate format, we tested a number of conditions, including PMS, PGA, and LB media containing different carbon sources, varying initial inoculum size of D. dadantii, and either moderately aerated or static incubation (as described in Materials and Methods). These preliminary experiments showed that for the three culture media tested, the optimal conditions were achieved by 1:100 back-dilution of bacterial cultures, maintaining a minimum volume of 100 µL of the culture per well, and a static incubation of the microplates (data not shown). Furthermore, we aimed to determine the suitability of the optimized conditions for HTS. The wild-type and the isogenic outD knockout strain of D. dadantii were cultured in PMS, PGA, or LB with shaking for 24 h at 28 °C. Subsequently, bacteria were subcultured into appropriate fresh media at a 1:100 ratio and delivered into individual 96-well plates as explained in Materials and Methods. Briefly, all wells in column 1 (n = 8) were inoculated with the media (sterility and background control), the wild-type D. dadantii was added to the wells in columns 2 to 10 (n = 72), and wells in columns 11 to 12 (n = 16) contained the T2S mutant (a positive control for the assay). The initial turbidity was measured at OD600, and plates were incubated at 28 °C for 24 h to allow the bacteria to multiply. Afterward, bacterial growth was examined, and 50 µL of the well contents was transferred into corresponding wells on a black opaque 96-well plate. The cellulolytic activity was monitored in an end-point assay every hour after the addition of resorufin–β-D-cellobioside ( Fig. 2 ). Concurrent with previous observations, the hydrolytic activity of cellulase was associated with the wild-type D. dadantii, while in contrast, fluorescence detected in the outD knockout strain was only slightly increased above the background levels ( Fig. 2A – C ). 5 Although all bacterial cultures reached similar OD600, dramatic differences were observed for the cellulase activity measured in different media (data not shown and Fig. 2 , respectively). The lowest cellulase activity was observed in the wild-type D. dadantii cultured in PMS and reached 272.2 ± 21.2 RFU (mean ± SEM in 3 h after the addition of the substrate; see Fig. 2A ), whereas culturing the bacteria in either PGA or LB resulted in 12- and 3.4-fold increases in the cellulolytic activity, respectively, in comparison to PMS medium ( Fig. 2B , C , respectively). The differences in the cellulolytic activity were likely due to the amount of cellulase produced and secreted by the bacteria in response to various nutrients present in the media. The highest cellulase activity was observed in the PGA medium that contained polygalacturonic acid salt, a known inducer of cellulase production. 5 Moreover, there was a linear increase in the cellulase activity in all tested growth conditions for up to 2 and 5 h from the addition of the substrate in PMS, PGA, and LB media, respectively ( Fig. 2A – C , data not shown).
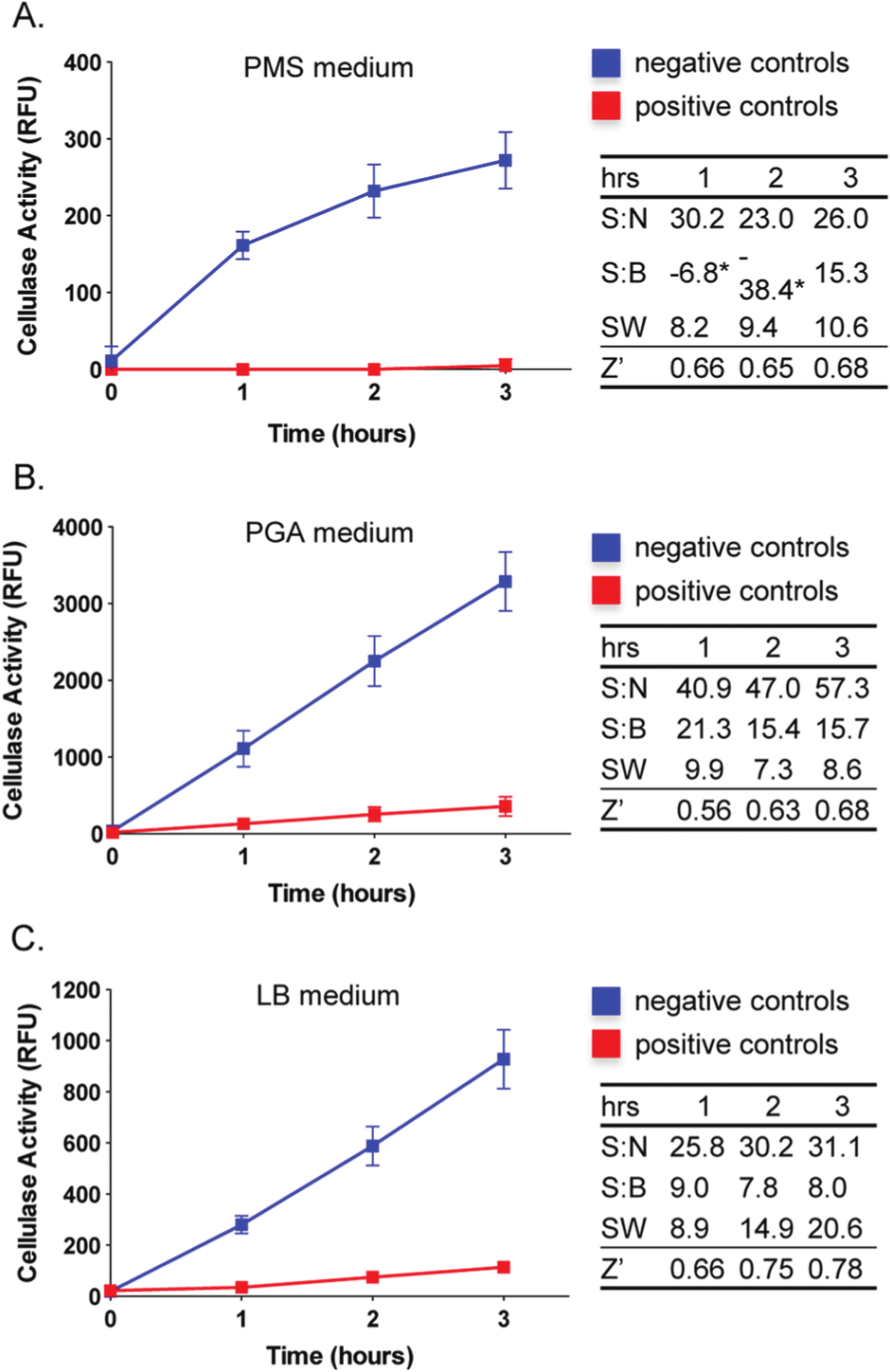
Optimization of the T2S-dependent cellulase secretion assay for a high-throughput screening platform in a 96-well plate format. Dickeya dadantii was cultured in 96-well plates in different liquid media: Pseudomonas minimal salt (PMS) (
To evaluate which of the culture conditions was the most suitable for HTS, we determined values of statistical parameters commonly used in the assessment of assay performance and sensitivity, including S:B and S:N ratios, SW, and the Z′ factor.8,9 These analyses, performed at three different time points of the experiments and in biological triplicates, demonstrated that the S:N ratios were excellent at all time points across tested growth conditions ( Fig. 2 , right panel). The S:B ratios achieved the highest values for the cellulase activity measured after the bacteria were cultured in PGA medium and reached the values of 21.3, 15.4, and 15.7 at 1, 2, and 3 h from the addition of the substrate, respectively ( Fig. 2B ). However, the cellulase assays carried out in LB medium showed the most suitable characteristics for HTS, including the highest values of signal window of 8.9, 14.9, and 20.6 and excellent Z′ factor values of 0.66, 0.75, and 0.78 at the respective first, second, and third experimental time points ( Fig. 2C ). In particular, the highest values were observed at the 3-h time-point, and thus all subsequent experiments were conducted with these optimized settings.
Suitability of the Assay for Screening of Diverse Collections of Compounds
The identification of small-molecule inhibitors displaying the desired biological activity requires that the screening procedures are performed on a large scale with diversified collections of libraries containing both synthetic small- molecule inhibitors and natural products. Therefore, to further evaluate the full applicability of the developed T2S-inhibition assay for a high-throughput format, we aimed to test a diverse set of natural products (organic extracts and bacterial culture filtrates) and pharmacologically active pure compounds. All screening procedures were performed using the same plate layout as described in Materials and Methods.
Organic extracts used in our experiments were isolated from field-collected marine and terrestrial Panamanian cyanobacteria and laboratory cultures of deep-sea hydrothermal vent-associated organisms. The results of this screening assay are shown in Figure 3A , B . None of the tested organic extracts exhibited inhibitory effects on bacterial growth and cellulase activity. The turbidity of the cultures reached OD600 of 0.3 ± 0.003 (mean ± SEM, Fig. 3A ), whereas the cellulase activity was 707.6 ± 10 RFU (mean ± SEM) for the negative controls and the experimental samples ( Fig. 3B ). The assay performance parameters S:N, S:B, SW, and Z′ factors of 31.9, 6.6, 11.0, and 0.63, respectively, demonstrated an excellent suitability for screening natural product extract libraries.8,9 It is also worth noting that the assay tolerated higher concentration of DMSO (2.4%), and the cellulase activity decreased by only 24% in comparison to the assays performed in the presence of 1% DMSO.
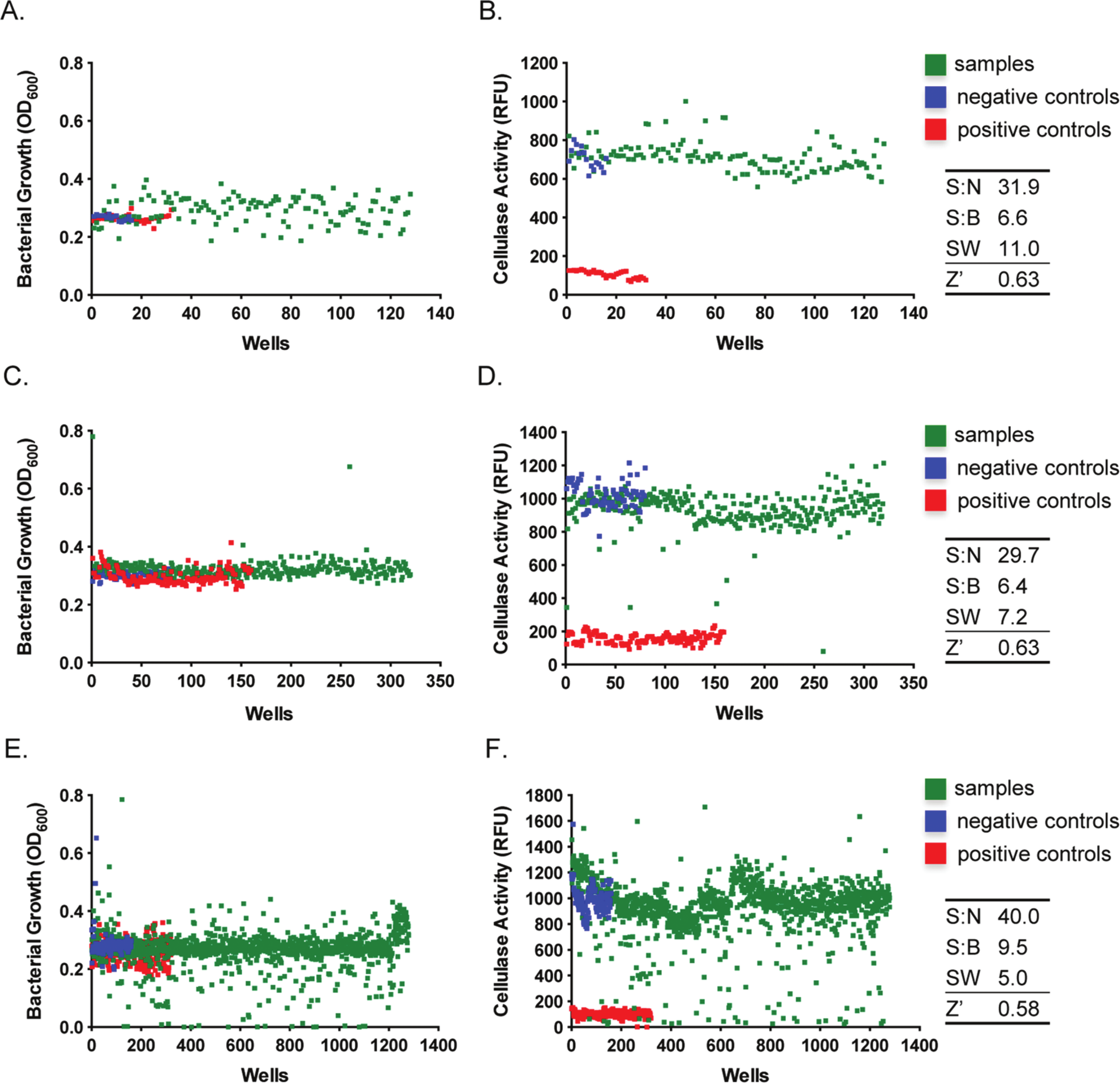
The T2S-dependent cellulose secretion assay is suitable for screening of crude natural products, culture filtrates, and synthetic molecules. (
To further test our assay system, we examined culture filtrates prepared from bacterial isolates obtained from the Landscape and the Deleterious Rhizosphere Bacterial Isolate Library Collections (as described in Materials and Methods). Rhizosphere bacteria, particularly Pseudomonas species, are well known for the secretion of a variety of secondary metabolites with a broad spectrum of bioactivities, including antimicrobials, quorum-sensing agents, and siderophores. 15 These culture supernatants could be used as inexpensive biocontrol agents against phytopathogenic bacteria if they contain a compound targeting the T2S system. We conducted a screen of 320 culture filtrates at the final concentration of 10% (v/v) in the assay solutions. The overall assay performance was very good with the S:N, S:B, SW, and Z′ factor values of 29.7, 6.4, 7.2, and 0.63, respectively ( Fig. 3C , D ). None of the tested culture filtrates had a harmful effect on D. dadantii growth, and in fact a few supernatants seemed to stimulate bacterial proliferation ( Fig. 3C ). Nevertheless, five culture filtrates exhibited a 50% and greater inhibitory effect on cellulase activity ( Fig. 3D ). Studies are under way to verify the inhibitory effects of these culture filtrates in secondary screening assays.
Finally, we determined the suitability of our assay for screening libraries of defined small-molecule inhibitors by utilization of Sigma’s Library of Pharmacologically Active Compounds, LOPAC1280. A total of 1280 compounds, each at a final concentration of 10 µM, were subjected to the screen ( Fig. 3E , F ). Consistent with previous screening procedures, the performance of the T2S-inhibition assay was excellent with a Z′ factor of 0.58, S:N of 40.0, S:B of 9.5, and SW of 5 ( Fig. 3F ). In addition, we examined the intra- and interplate variability of the assay because of a large number of compounds screened during this experiment. The variability of detected cellulase activity was assessed in both negative (wild-type D. dadantii) and positive (outD knockout strain) controls. Within a single plate, the calculated average of cellulase activity for negative controls was 1220 RFU with a standard deviation of 143.9 (CV, 11.8%), whereas a slightly higher intraplate variability was observed for the positive controls with an RFU of 130.1 ± 22.49 (mean ± standard deviation; CV, 17.3%). The calculated interplate variation was very similar for both controls, and the RFU values reached 985 ± 101.0 (CV, 10.3%) and 102.5 ± 21.7 (CV, 21.2%) for the wild-type and outD strain of D. dadantii, respectively. Together, these statistical analyses demonstrated the good quality of the assay.
Furthermore, our experiment confirmed the bactericidal activities against D. dadantii of 14 known antibiotics included in the LOPAC1280 library. Twenty additional compounds also significantly inhibited bacterial growth ( Fig. 3E ). A total of 14 compounds inhibited cellulase activity by 50% or greater and did not have a deleterious effect on bacterial proliferation ( Fig. 3F ). These compounds could be potential inhibitors of the T2S system but may also simply affect the activity or production of cellulase itself. Therefore, to eliminate false-positive hits, the assessment of the activity of purified cellulase in the presence of these compounds and verification of activities of other T2S-dependent exoproteins (e.g., xylanases, proteases, lipases) should be performed. Reduced or no activity of these additional secreted proteins could indicate that the lead compounds block the T2S machinery. As we discussed earlier, the chemical ligand could affect just about every step within the T2S pathway, and thus the T2S appears as an attractive molecular drug target ( Fig. 1 ). Nevertheless, the detailed mechanism of inhibition would need to be determined. This could be achieved by multiple approaches. For instance, quantitative real-time PCR or a reporter gene fusion to the chromosomal region containing the T2S promoter could be used to verify whether these compounds block the transcription of the genes encoding the T2S operon. Interference with the expression of the T2S genes at the posttranscriptional levels could be assessed by immunoblotting analyses with sera raised against T2S constituents, while the appropriate localization of the individual T2S components might be examined by fractionation of cellular contents and subsequent immunoblotting experiments or by fluorescent microscopy. 16 Furthermore, subtractive proteomics of periplasmic fractions and supernatants isolated from wild-type, wild-type treated with the lead compound, and T2S-isogenic mutant strain would shed light on whether the compound interferes with the T2S, Sec, or Tat systems. Finally, one could use affinity chromatography and surface plasmon resonance technology to determine whether the inhibitory molecule affects the protein-protein interactions within the T2S complex or recognition of the cargo proteins. 17
In summary, we report herein the development, optimization, and validation of a quantitative assay that could be used in an HTS platform designed to identify inhibitors of the T2S complex. The T2S system is a common virulence mechanism used by many Gram-negative bacteria to deliver toxins and hydrolytic enzymes that contribute to the disease in humans, animals, and plants. 4 Thus, the identified inhibitory molecules might serve as starting points for pharmaceutical and agricultural programs to develop novel compounds against drug-resistant bacteria, as well as molecular probes to gain insights into the mechanisms underlying the T2S process.
Footnotes
Acknowledgements
We are very grateful to Beatrice Py (LCB, CNRS, France) for generously sharing the D. dadantii strains used in this study.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the Agricultural Research Foundation (Oregon State University, Corvallis, OR) FA364N and start-up funds to Aleksandra E. Sikora. Preparation of natural products extracts was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases (5R21AI085540-02) and an Oregon Sea Grant under award number NA10OAR4170059 (project number R/BT-48) from the National Oceanic and Atmospheric Administr-ation’s National Sea Grant College Program, U.S. Department of Commerce, and by appropriations made by the Oregon state legislature. The statements, findings, conclusions, and recommendations are those of the authors and do not necessarily reflect the views of these funders.