
Abstract
Transporter proteins are known to play a critical role in affecting the overall absorption, distribution, metabolism, and excretion characteristics of drug candidates. In addition to efflux transporters (P-gp, BCRP, MRP2, etc.) that limit absorption, there has been a renewed interest in influx transporters at the renal (OATs, OCTs) and hepatic (OATPs, BSEP, NTCP, etc.) organ level that can cause significant clinical drug-drug interactions (DDIs). Several of these transporters are also critical for hepatobiliary disposition of bilirubin and bile acid/salts, and their inhibition is directly implicated in hepatic toxicities. Regulatory agencies took action to address transporter-mediated DDI with the goal of ensuring drug safety in the clinic and on the market. To meet regulatory requirements, advanced bioassay technology and automation solutions were implemented for high-throughput transporter screening to provide structure-activity relationship within lead optimization. To enhance capacity, several functional assay formats were miniaturized to 384-well throughput including novel fluorescence-based uptake and efflux inhibition assays using high-content image analysis as well as cell-based radioactive uptake and vesicle-based efflux inhibition assays. This high-throughput capability enabled a paradigm shift from studying transporter-related issues in the development space to identifying and dialing out these concerns early on in discovery for enhanced mechanism-based efficacy while circumventing DDIs and transporter toxicities.
Introduction
Transporters are transmembrane proteins that act as pumps and gatekeepers for cellular entry and exit of drugs. They have a global effect on absorption, distribution, metabolism, and excretion (ADME) and can control drug exposure levels in efficacy and safety organs.1,2 Genetic polymorphisms in drug transporters such as OATP1B1 can increase the variability of pharmacokinetics between individuals and are also implicated in several adverse toxicological effects such as hyperbilirubinemia and cholestasis. 3 Transporters are also a major mechanism for causing drug-drug interaction (DDI) and as a result have been the focus of regulatory agencies and reported with labeling language to control adverse drug reactions. 4 Given these considerations, transporters have the potential to have a significant effect on drug design and discovery. Therefore, understanding transporter activities from discovery to clinical development will provide an invaluable impact on addressing adverse drug reactions.
Over the past 5 years, there has been progress to address transporter-mediated drug-drug interactions by regulatory agencies. The Food and Drug Administration (FDA) and European Medicines Agency (EMA) as well as the International Transporter Consortium issued recommendations for transporter studies to guide drug development and labeling. The FDA and EMA recommend that new drugs be routinely assessed for DDIs mediated by transporters.5,6 Therefore, our goal was to establish high-throughput functional inhibition assays for human transporter targets to enable structure-activity assessment and provide support to project teams facing issues related to transporter DDI, hyperbilirubinemia, and other transporter-related toxicology issues.
Over four decades, there has been an evolution of mammalian models for the study of transporter activity. More than 50 years ago, using perfused kidneys, isolated tubules, and liver slices models, researchers discovered transport mechanisms for elimination of organic anions and cations. 7 In the mid-1970s, surface labeling studies with colchicine-resistant Chinese hamster ovary cells revealed the P-glycoprotein (P-gp) ATPase. 8 In the early 1980s, methods were developed to isolate vesicles from renal tubules, which identified distinct transport systems in the apical and basolateral membranes. 7 In 1987, with the onset of molecular biology techniques, expression cloning in Xenopus oocytes was the defining event that marked the renaissance of the transporter field.7,9 Using this technique, individual transporters involved in absorption, distribution, and excretion were cloned throughout the 1990s, and subsequent in vitro techniques using recombinant cell lines enabled detailed transporter studies. 7 These in vitro techniques coupled with the mouse P-gp crystal structure in 2009 significantly enhanced our understanding of the structure-activity relationship (SAR) of mammalian transporters. 10 The evolution of transporter models from ex vivo to in vitro provides opportunities to track SAR in early drug discovery. However, a capacity gap for generating transporter SAR data existed in industry.
Transporters can be categorized into two distinct superfamilies: (1) the solute carrier class or the influx transporters that mediate drug uptake by movement across electrochemical gradients and (2) the ATP-binding cassette (ABC) superfamily or efflux transporters that mediate drug efflux by ATP hydrolysis. 11 Reagent generation and technology selection for assay development depend on the transporter class. There are several main types of screening technologies to assess transporters.12,13 Ligand binding filtration or scintillation proximity assays can evaluate compound binding to transporters in membrane-based assay systems. However, these assays do not access functional transporter activity and are costly due to radiolabeled probes. 13 ATPase assays are available to analyze ABC efflux transporters but are challenged by the nonlinear relationship of ATPase activity and transport rate. 4 Vesicular transport assays may have background interference and a cost factor. For cell-based assays, substrate uptake and efflux assays historically have been performed using radiolabeled probes versus use of fluorescent probes due to lack of suitable fluorescent substrates. In addition, polarized cell monolayer assays historically have lacked specificity and have associated high costs, and sandwich cultured hepatocytes are very challenging assays to establish. 4 The majority of published literature on higher-throughput transporter assays reflect 96-well transporter assay formats.14–20
Here we report the establishment of three streamlined functional assay formats to increase capacity for transporter inhibition assays. Because of the complexity and current gaps for transporter SAR, the guiding principles for technology selection were the ability to address flexibility, information content, and physiological relevance/connectivity in addition to cost, throughput, sensitivity, and efficiency. Novel fluorescent transporter endpoint and high-content imaging assays were developed in whole cells to analyze organic anion transporters OATP1B1 and OATP1B3 uptake and P-gp efflux in 384-well format. For nonfluorescent substrates, we adapted a radioactive filtration assay for influx transporters in whole cells expressing the sodium/bile acid cotransporter also known as the Na+-taurocholate cotransporting polypeptide (NTCP). We also established 384-well miniaturized radioactive transporter filtration assays using inverted membrane vesicles for efflux transporters such as bile salt export pump (BSEP) and multidrug resistance–associated protein 2 (MRP2). These assay formats have been completely automated on laboratory robots. The innovation of transporter technology solutions has led to a high-throughput bioassay infrastructure amenable for understanding transporter SAR to affect both the discovery and development space.
Methods and Materials
Chemicals
[3H]Taurocholic acid and [3H]estradiol 17-β-D-glucuronide were purchased from PerkinElmer (Waltham, MA). Fluorescein-methotrexate and calcein AM were from Invitrogen (Carlsbad, CA). All reference compounds were from Sigma (St. Louis, MO), including bromsulphthalein (BSP), cyclosporin, rifampicin, cimetidine, saquinavir, piroxicam, valinomycin, taurocholic acid, glyburide, reserpine, vinblastine, and furosemide. All other chemicals were from commercial sources of reagent grade.
Cells
OATP1B1, OATP1B3, and NTCP HEK293 cells were established and maintained as described previously. 21 Briefly, recombinant HEK293 cells expressing either human OATP1B1, OATP1B3, or NTCP transporters were cultured in Dulbecco’s modified minimum essential medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and hygromycin (100 µg/mL). Cultures were maintained in a humidified atmosphere containing 5% CO2 at 37 °C and were passaged at a 1:5 ratio every 3 to 4 days. Madin-Darby canine kidney (MDCK)–MDR1 cells were obtained from the Netherlands Cancer Institute (Amsterdam, the Netherlands) and were cultured as described above.
BSEP and MRP2 Vesicle Preparation
Cloning and Insect Cell Expression of Human MRP2 and BSEP
The open reading frames of human MRP2 and BSEP were cloned into a modified pFastBac1 vector (Invitrogen) by PCR and sequence verified. Baculovirus was generated for each construct using the Bac-to-Bac baculovirus expression system (Invitrogen) according to the manufacturer’s protocol. Briefly, recombinant bacmid was isolated from transformed DH10Bac Escherichia coli competent cells (Invitrogen) and used to transfect Spodoptera frugiperda (Sf9) insect cells (Invitrogen). Baculovirus was harvested 72 h after transfection, and a virus stock was prepared by infecting fresh Sf9 cells at a virus/cell ratio of 1/1000 for 66 h.
For membrane vesicle preparations, Sf9 cells from Expression Systems (Davis, CA) routinely grown in ESF921 insect medium (Expression Systems) were infected with the virus stock in shaker flasks at a virus/cell ratio of 1/100 for 66 h, yielding ~80% infection efficiency. The cells were harvested by centrifugation, washed once with lysis buffer (10 mM Tris-HCl, pH 7.5, 0.25 mM MgCl2, 250 mM sucrose, 3 mM KCl, and EDTA-free protease tablet) and stored at −70 °C.
Membrane Vesicle Preparation of Human MRP2 and BSEP
Frozen insect cell pellets expressing either human MRP2 or BSEP were thawed and resuspended in lysis buffer. Cells were then lysed by nitrogen cavitation, and EDTA was added to the lysed cell solution for a final concentration of 1mM. Membrane fraction was collected after a low-speed centrifugation and layered over a 35% (w/w) sucrose solution in 10 mM Tris-HCl, pH 7.5, and 1 mM EDTA. The interface was collected after centrifugation at 40,000 rpm for 2 h in a Ti45 rotor in XL-90 ultracentrifuge (Beckman, Brea, CA). The membrane fraction was collected and washed twice in formulation buffer by centrifugation at 40,000 rpm for 1 h. The membrane pellet was resuspended in formulation buffer and passed 20 times through a 27-gauge needle for vesicle formation. The final concentration of the membrane vesicle was determined by Bradford assay using BSA as standards. To limit batch-to-batch variation, each batch of membrane preparation was assessed for transporter activity through membrane titration experiments. The membrane quantity to achieve optimal assay signal-to-noise ratio and consistent reference compound inhibition values was determined.
OATP1B1/OATP1B3–Mediated Fluorescein-Methotrexate Uptake Inhibition Assays
OATP1B1 (6 × 104 cells/well) and OATP1B3 (1.2 × 105 cells/well) cells were seeded in PureCoat Amine black/clear-bottom 384-well plates (BD Biosciences, San Jose, CA) in culture media (DMEM supplemented with 10% FBS) and incubated in a humidified atmosphere containing 5% CO2 at 37 °C for 24 h. The cells were then washed twice with 80 µL of warm assay buffer (HBSS with 10 mM HEPES) using the ELx405 Select Deep Well Microplate Washer (Biotek, Winooski, VT). After wash, 20 µL of assay buffer was added to each well by Multidrop Combi Reagent Dispenser (Thermo Scientific, Waltham, MA), and then 200 nL of test compound (11 point, threefold serial dilution) was added to each well by the Labcyte Echo Liquid Handling Platform (Labcyte, Sunnyvale, CA). The positive control for background was 50 µM BSP and for total signal was 1% DMSO vehicle. Ten microliters of 3 µM fluorescein-methotrexate (Invitrogen) in assay buffer was then added to each well with the Multidrop Combi Reagent Dispenser. The reaction was incubated at room temperature for 40 min, followed by four washes with 80 µL of ice-cold assay buffer using the ELx405 Select Deep Well Microplate Washer. Eighty microliters of assay buffer was left in each well after the final wash, and the plates were read on the PerkinElmer EnVision Multilabel Reader. High-content imaging assays were performed the same way, except 5 µg/mL of Hoechst 33342 (Invitrogen) was added together with fluorescein-methotrexate. Images were acquired by the Cellomics ArrayScan VTI high-content screening plate reader (Thermo Scientific). Data analysis was performed using the CellHealthProfling BioApplication on the ArrayScan.
P-gp–Mediated Calcein AM Efflux Inhibition High-Content Imaging Assay
MDCK-MDR1 cells were seeded at 1.5 × 104 cells/well in culture media (DMEM with 10% FBS) on PureCoat Amine black/clear-bottom 384-well plates and incubated for 24 h in a humidified atmosphere containing 5% CO2 at 37 °C. The cells were then washed twice with 80 µL of warm assay buffer (HBSS with 10 mM HEPES). After washes, 20μL of assay buffer was added to each well by Multidrop Combi Reagent Dispenser and 200 nL of test compound was then added to each well by the Labcyte Echo Liquid Handling Platform. Twenty microliters of a 3 µM stock of calcein AM in assay buffer was then added to each well with the Multidrop Combi Reagent Dispenser. The reaction was incubated in a humidified atmosphere containing 5% CO2 at 37 °C for 40 min. The cells were then washed four times with 80 µL of ice-cold assay buffer by the ELx405 Select Deep Well Microplate Washer. The plates were then read on the PerkinElmer EnVision Multilabel Reader. High-content imaging assays were done essentially the same way, except 5 µg/mL of Hoechst 33342 was added together with calcein AM. Images were acquired by Cellomics ArrayScan VTI HCS Reader. Data analysis was performed using the CellHealthProfling BioApplication on the ArrayScan.
NTCP-Mediated [3H]Taurocholate Whole-Cell Uptake Inhibition Assay
HEK cells stably expressing NTCP were seeded in white scintillation 384-well plates (Nunc, Cat. No. 142762) that were precoated with poly-D-lysine (Sigma, Cat. No. P6407) in culture media (DMEM supplemented with 10% FBS) at 7.5 × 104 cells/well and were incubated in a humidified atmosphere containing 5% CO2 at 37 °C for 24 h prior to assay. The cells were then washed twice with 80 µL of warm assay buffer (HBSS with 10 mM HEPES) by the ELx405 Select Deep Well Microplate Washer. Twenty microliters of assay buffer was added to each well by Multidrop Combi Reagent Dispenser after the second wash, and 200 nL of test compound was added to each well by the Labcyte Echo Liquid Handling Platform. [3H]Taurocholate was supplemented with cold taurocholate in assay buffer to make 2× substrate solution, which had a radioactivity of 4 µCi/mL and a total taurocholate molar concentration of 2 µM. Twenty microliters of 2× substrate solution was then added to the plate with the Multidrop Combi Reagent Dispenser. The reaction was incubated at room temperature for 40 min, followed by washing four times with 80 µL of ice-cold assay buffer by the ELx405 Select Deep Well Microplate Washer. Five microliters of 0.1 N NaOH containing 0.1% SDS (w/v) was added to each well, and cells were lysed by incubation at room temperature for 60 min. Seventy-five microliters of Ultima Gold scintillation cocktail (PerkinElmer) was added to each well, and the scintillation mixture was then incubated at room temperature overnight to stabilize the signal. Radioactivity was measured on the PerkinElmer TriLux Microplate Scintillation and Luminescence Counter.
BSEP-Mediated [3H]Taurocholate Vesicle-Based Efflux Inhibition Assay
BSEP vesicles were diluted in assay buffer (10 mM Hepes-Tris, 100 mM KNO3, 10 mM Mg[NO3]2, 50 mM sucrose). The dilution factor was determined by titrating each batch of vesicles for optimal assay performance. Eight microliters of diluted BSEP vesicles was added into each well of a 384-well clear round-bottom plate (REMP, Oberdiessbach, Switzerland) with the Multidrop Combi Reagent Dispenser, and 200 nL of test compound was dosed to each well by the Labcyte Echo Liquid Handling Platform. [3H]Taurocholate was supplemented with cold taurocholate in assay buffer to make 5× substrate solution, which had a radioactivity of 10 µCi/mL and a total taurocholate molar concentration of 5 µM. Four microliters of 5× substrate solution and 8 µL of 10 mM ATP in assay buffer were then added to each well by the Multidrop Combi Reagent Dispenser. The reaction was incubated at room temperature for 40 min. Eighty microliters of cold wash buffer (10 mM Hepes-Tris, 100 mM KNO3, 50 mM sucrose) was then added to stop the reaction. The mixture was transferred by the VPrep Liquid Handling Platform (Velocity11) to a MultiScreen HTS-FB Plate (Millipore, Billerica, MA) that was prewetted by 0.1% PEI in wash buffer. The filter plate was washed four times with 100 µL of cold wash buffer by a modified Embla Microplate Washer (Molecular Devices, Sunnyvale, CA). The plate was then dried at 60 °C for 30 min. Radioactivity was measured on the TriLux Microplate Scintillation and Luminescence Counter (PerkinElmer) after the addition of 20 µL MicroScint 20 scintillation fluid (Perkin Elmer).
MRP2-Mediated [3H]Estradiol 17-β-D-glucuronide Vesicle-Based Efflux Inhibition Assay
MRP2 vesicles were diluted in assay buffer (50 mM MOPS-Tris, 70 mM KCl, 7.5 mM MgCl2). The dilution factor was determined by titrating each batch of vesicles for optimal assay performance. Seven microliters of diluted MRP2 vesicles was added into each well of a 384-well clear round-bottom plate (REMP) with the Multidrop Combi Reagent Dispenser, and 200 nL of test compound was dosed to each well by the ECHO Liquid Handling Platform. [3H]Estradiol 17-β-D-glucuronide was supplemented with cold estradiol 17-β-D-glucuronide in assay buffer to make a 5× substrate solution, which had a radioactivity of 10 µCi/mL and total estradiol 17-β-D-glucuronide molar concentration of 5 µM. Four microliters of 5× substrate solution, 8 µL of 10 mM ATP, and 1 µL of 200 mM glutathione in assay buffer were then added to each well by Multidrop Combi Reagent Dispenser. The reaction was incubated at room temperature for 40 min. Eighty microliters of cold wash buffer (40 mM MOPS-Tris and 70 mM KCl) was then added to stop the reaction. The mixture was transferred by the Velocity11 VPrep Liquid Handling Platform (Agilent, Santa Clara, CA) to a MultiScreen HTS-FB Plate (Millipore, Cat. No. MZFBN0W50) that was prewetted by 0.1% PEI and 50 µM of cold estradiol 17-β-D-glucuronide in wash buffer. The filter plate was washed four times with 100 µL of cold wash buffer using a modified Embla Microplate Washer. The plate was dried at 60 °C for 30 min. Radioactivity was measured on the TriLux Microplate Scintillation and Luminescence Counter after the addition of 20 µL MicroScint 20 scintillation fluid (PerkinElmer, Cat. No. 6013627).
Data Analysis
Results were plotted as percentage inhibition of the average positive control signal. The IC50 was defined as the concentration of test compound corresponding to 50% inhibition derived from the 11-point fitted curve as determined using a four-parameter logistic regression model.
The Z′ value as a criterion for data quality was calculated according to Zhang et al. 22 with the following equation: Z′=1 – (3*SDhigh control + 3*SDlow control)/(|Meanhigh control – Meanlow control|), where SD = standard deviation. A Z′ value of 1 represents an ideal assay, whereas a Z′ value between 0.5 and 1 indicates a good assay quality. The coefficient of variation (%CV), defined as the percentage ratio of standard deviation to signal magnitude, was calculated as 100*SDcontrol counts/Meancontrol counts. 22
The coefficient of determination (R2) was used for determining the degree of linear correlation of variables in regression analysis and was calculated using defined equations in Microsoft Excel.
The Student t test was used to calculate p values using defined equations in Microsoft Excel.
Results and Discussion
High-Throughput, Whole-Cell Fluorescent Substrate Uptake and Efflux Inhibition Assays
Inhibition of OATP1B1/OATP1B3-Mediated Uptake of Fluorescein-Methotrexate
A recombinant HEK293 cell line expressing human OATP1B1 or OATP1B3 transporters was used as a model system for establishing a 384-well, high-throughput fluorescence uptake inhibition assay. 21 HEK-293 cells grow fast, are easy to handle for cell culture, and lack or express endogenous transporter proteins at very low levels and thus minimize nonspecific activity due to other transporter interference. 23 Once transporter DNAs are stably transfected in HEK-293 cells, the recombinant cell lines maintain transporter activity over passages, offering consistent transport studies. For these reasons, HEK-293 cells are the most widely used cells for transporter transfection.
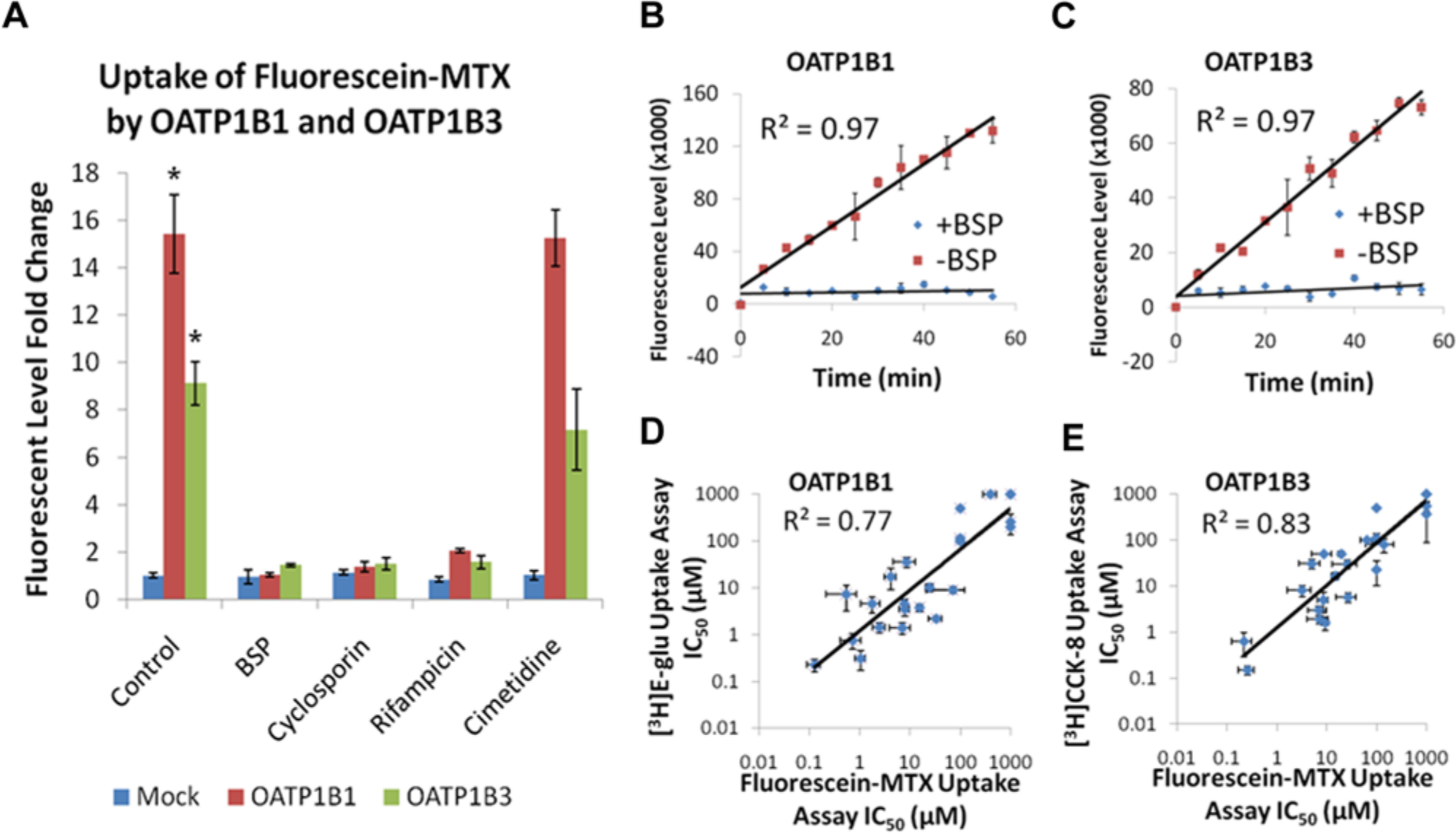
The first step in OATP1B1 and OATP1B3 assay development was to identify a fluorescent substrate for the transporters. Screening a matrix of fluorescent-labeled probes, the substrate fluorescein-methotrexate demonstrated active uptake and significant assay window in both OATP1B1 and OATP1B3 cell lines (p = 0.003;
Fig. 1A
). Mock cells stably transfected with empty vector control were a measure of specificity and showed negligible nonspecific activity (
Fig. 1A
). This uptake was completely inhibited by known OATP inhibitors such as bromosulfophthalein (BSP; 50 µM), cyclosporin (100 µM), and rifampacin (100 µM), whereas the negative control cimetidine (100 µM) displayed no inhibitory activity (
Fig. 1A
). Hepatic uptake mediated by OATPs has been shown previously to be linear for only 2 min at 37 °C, which is not amenable for high-throughput methodology.
21
Therefore, it was critical to investigate uptake kinetics at room temperature to establish if incubation time could be increased using these conditions. Characterization of fluorescein-methotrexate absorption kinetics at room temperature revealed that the fluorescence level remains in the linear range up to 60 min for both OATP1B1 and OATP1B3 transporters (
Shown are 384-well OATP1B1 and OATP1B3 fluorescent-substrate uptake inhibition assays. (
The Z′ factor serves as an indicator of assay quality and takes into account the variability in sample data as well as the dynamic range between the high (total counts) and low (background counts) data populations. 22 For cell-based assays, a Z′ value of 0.5 is an acceptable measure of favorable assay quality. The Z′ values for the OATP1B1 and OATP1B3 assays were 0.8 and 0.7 across 22 independent experiments, respectively (data not shown). The %CV, defined as the percentage ratio of standard deviation to signal magnitude, is used to quantify data variability and the precision available in the assay to identify hits with confidence. 22 The lower the %CV, the higher the assay precision. The OATP1B1 and OATP1B3 assays displayed %CV values equal to 5 across 22 independent experiments.
Following initial assay validation, a set of compounds composed of known OATP inhibitors from the literature and in-house chemistries were assessed in the fluorescein-methotrexate inhibition assay. These data were subsequently compared with data generated by gold-standard radioactive uptake inhibition assays in 24-well format using [3H]estradiol 17-β-D-glucuronide and [3H]CCK-8 as substrates.
21
Inhibition data generated for this compound set using the fluorescein-methotrexate uptake assays agreed well with those determined using [3H]estradiol 17-b-D-glucuronide and [3H]-CCK-8 with R2 correlation coefficient values of 0.77 and 0.83, respectively (
High-content screening uses fluorescence microscopy and automated image analysis algorithms to extract biologically meaningful measurements from cellular images. High-content screening approaches have been used to study cell signaling, cell physiology, in vitro toxicology, and organism physiology using such applications as protein expression, phosphorylation, cell cycle, apoptosis, nuclear translocation, receptor internalization, and transcription. 25 Although this platform has been primarily used to study target classes such as G-protein–coupled receptors, kinases, and enzymes, the use of this technology to investigate drug transporter function has not been reported.
Identification of the OATP1B1 and OATP1B3 fluorescent probe allowed novel application of high-content image analysis for transporter studies. In this experiment, co-incubation of OATP1B1 expressing HEK293 cells with fluorescein-methotrexate resulted in uptake of the green fluorescent compound into the cells, which was measured using the FITC channel of the Cellomics ArrayScan VTI high-content imager ( Fig. 2 ). An OATP1B1 inhibitor such as BSP prevented the uptake of fluorescein-methotrexate by the transporter in a concentration-dependent manner with complete inhibition of the transporter at a concentration of 5 µM ( Fig. 2 ). The OATP1B1 high-content assay displays a consistent signal-to-noise ratio of 47 and average Z′ and %CV values of 0.8 and 6%, respectively. High-content imaging analysis of fluorescent probe uptake presents us with unique opportunities to study cell populations and subcellular localization and conduct real-time kinetic analysis to determine rate of transport. Theoretically, one can label specific substrates in different colors to enable multiplexing of several transporters in the same assay well, which can further drive down assay costs. This multiplexing capability presents a major advantage over the radioactive uptake assays.
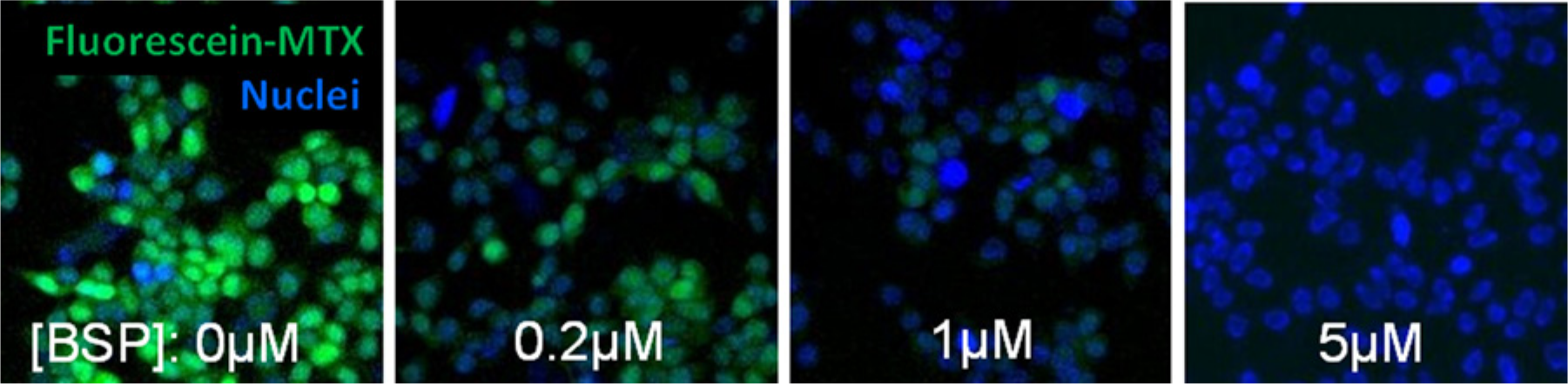
Novel 384-well functional high-content imaging assay for transporter uptake inhibition. OATP1B1/HEK cells were incubated with 1.5 µM fluorescein-methotrexate (green) for 30 min at room temperature in the absence or presence of increasing concentrations of bromsulphthalein. High-content images were acquired by Cellomics ArrayScan VTI. Nuclei are stained with DAPI (blue).
Inhibition of P-gp–Mediated Calcein AM Efflux Using High-Content Image Analysis
The high-content imaging assay format was also employed for whole-cell efflux studies. In this model, canine kidney cells engineered to overexpress P-gp were used. Calcein acetoxymethyl ester (AM) is a P-gp substrate and nonfluorescent compound that traverses the cell membrane and is converted to a fluorescent form (calcein free acid) by cellular esterases.
26
In cells expressing P-gp, calcein-AM is extruded by the multidrug transporter before its intracellular conversion. When calcein-AM efflux is blocked by a P-gp inhibitor such as cyclosporin A, fluorescent calcein rapidly accumulates with increasing concentrations of inhibitor in a concentration-dependent manner (
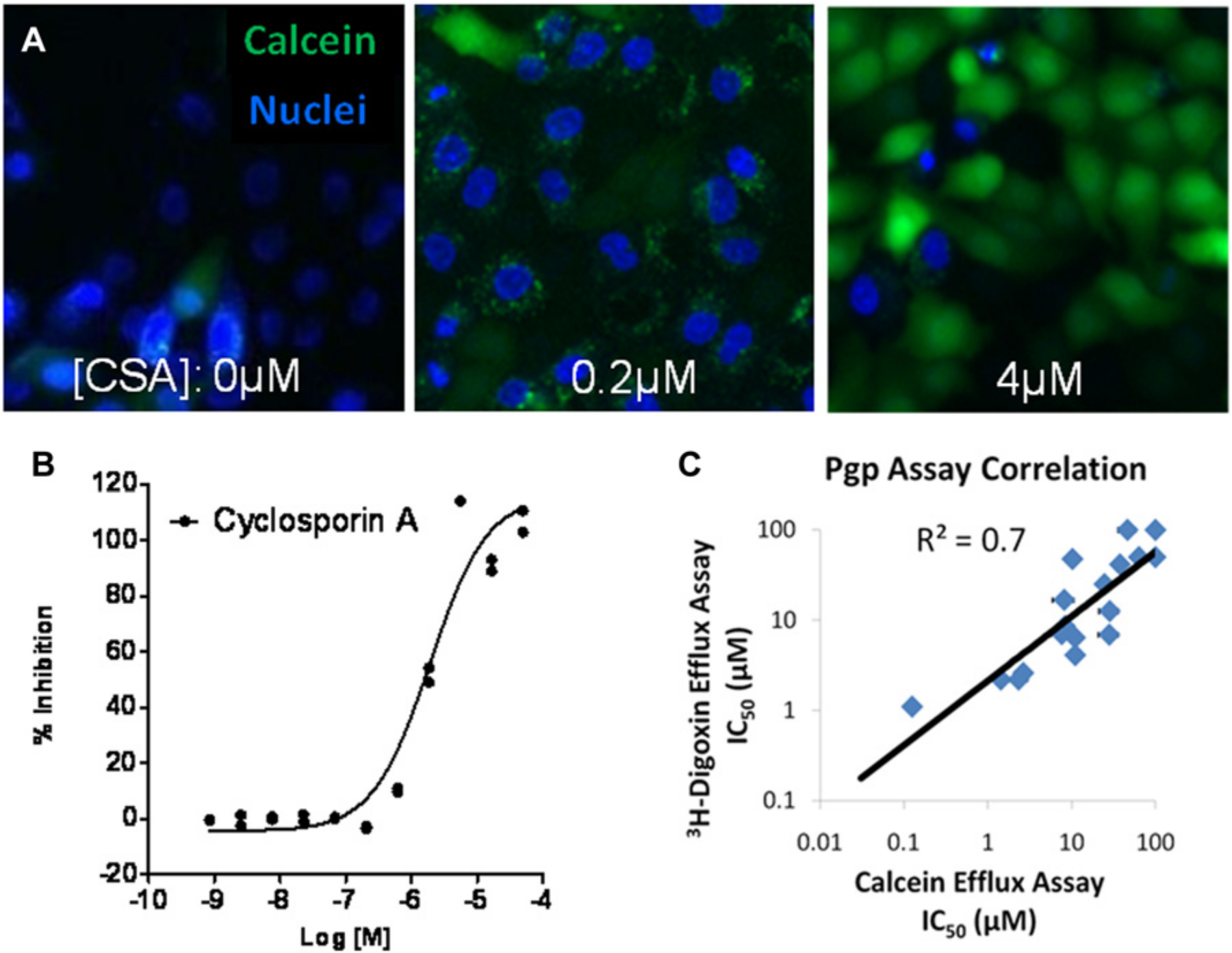
Rapid, 384-well high-content imaging assays for whole-cell efflux transporter studies. (
High-Throughput, Whole-Cell Radioactive Substrate Uptake Inhibition Assay
Inhibition of NTCP-Mediated Uptake of [3H]Taurocholate
For transporters such as NTCP, suitable fluorescent probes for whole-cell inhibition assays were unavailable. Therefore, the key objective was to increase capacity by miniaturizing the assay from a 24-well to a 384-well format. 21 Use of a 384-well poly-D-lysine–coated tissue culture plate optimized for scintillation counting was critical to miniaturizing the assay to ensure cell adhesion upon multiple wash steps and to preclude absorption admission when measuring radioactivity. In the miniaturized format, the [3H]taurocholate substrate maintained linearity up to 70 min ( Fig. 4A ) at room temperature, and the intra-assay variability was small, allowing the production of high-quality dose-response data ( Fig. 4B ). Therefore, the NTCP inhibition assay was performed by incubating the cells with [3H]taurocholate for 40 min, when uptake is still within a linear range. IC50 values of reference compounds were within the twofold acceptable range comparing the 384-well format versus historical 24-well assay format ( Fig. 4C ). An assessment of the robustness of the 384-well assay in a production setting was represented by the Z′ value (a criterion for data quality) of 0.8 obtained between three consecutive experiments and a consistent signal-to-noise ratio of 11-fold (data not shown).
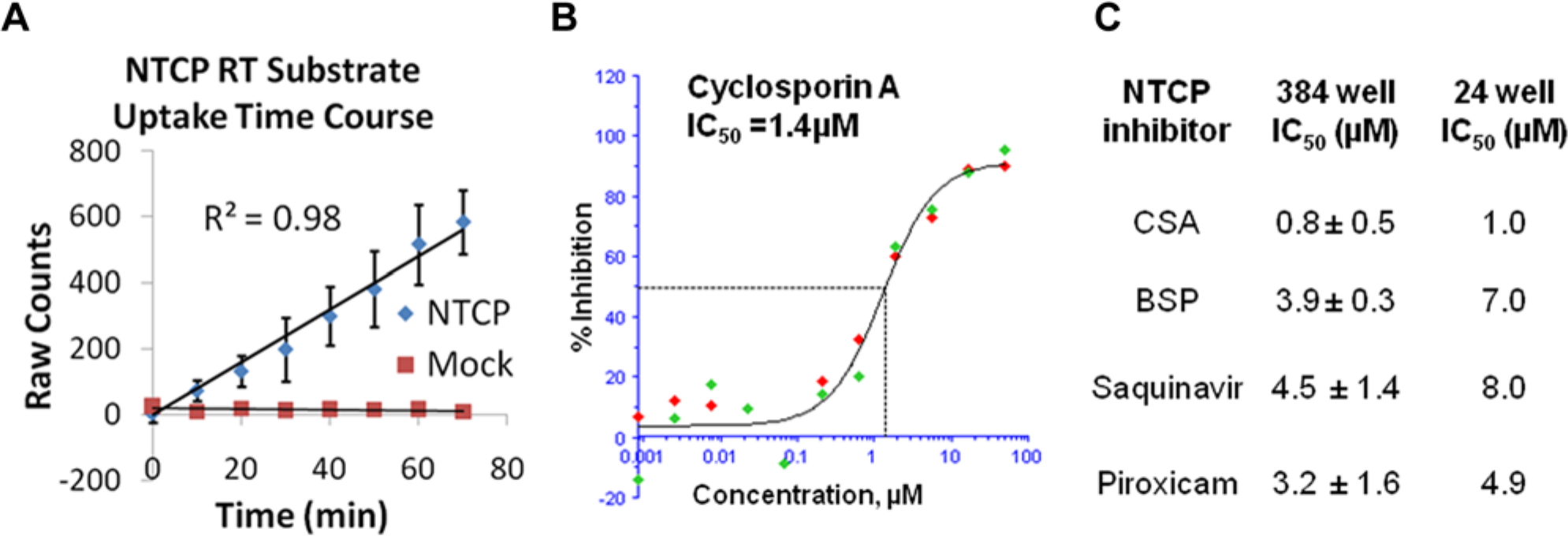
A 384-well Na+-taurocholate cotransporting polypeptide (NTCP) radioactive substrate uptake assay. (
High-Throughput, Vesicle-Based Efflux Transporter Inhibition Assays
BSEP-Mediated [3H]Taurocholate and MRP2-Mediated [3H]Estradiol 17-β-D-Glucuronide Vesicular Efflux Inhibition Assays
BSEP and MRP2 are efflux transporters requiring inverted membrane vesicles and ATP for measuring activity. BSEP and MRP2 membrane vesicles were prepared from baculovirus-infected Sf9 cells expressing the transporters in relatively high levels to obtain sufficient signal. The ATP requirement was illustrated in vesicle titrations measuring [3H]taurocholate (1 µM) uptake in BSEP vesicles (
Fig. 5A
) and [3H]estradiol 17-β-D-glucuronide (1 µM) uptake in MRP2 vesicles (
Fig. 5D
), which show high signal-to-noise ratios in the presence of ATP versus ADP. Optimal protein concentrations for the highest signal-to-noise ratios were determined by vesicle titration (
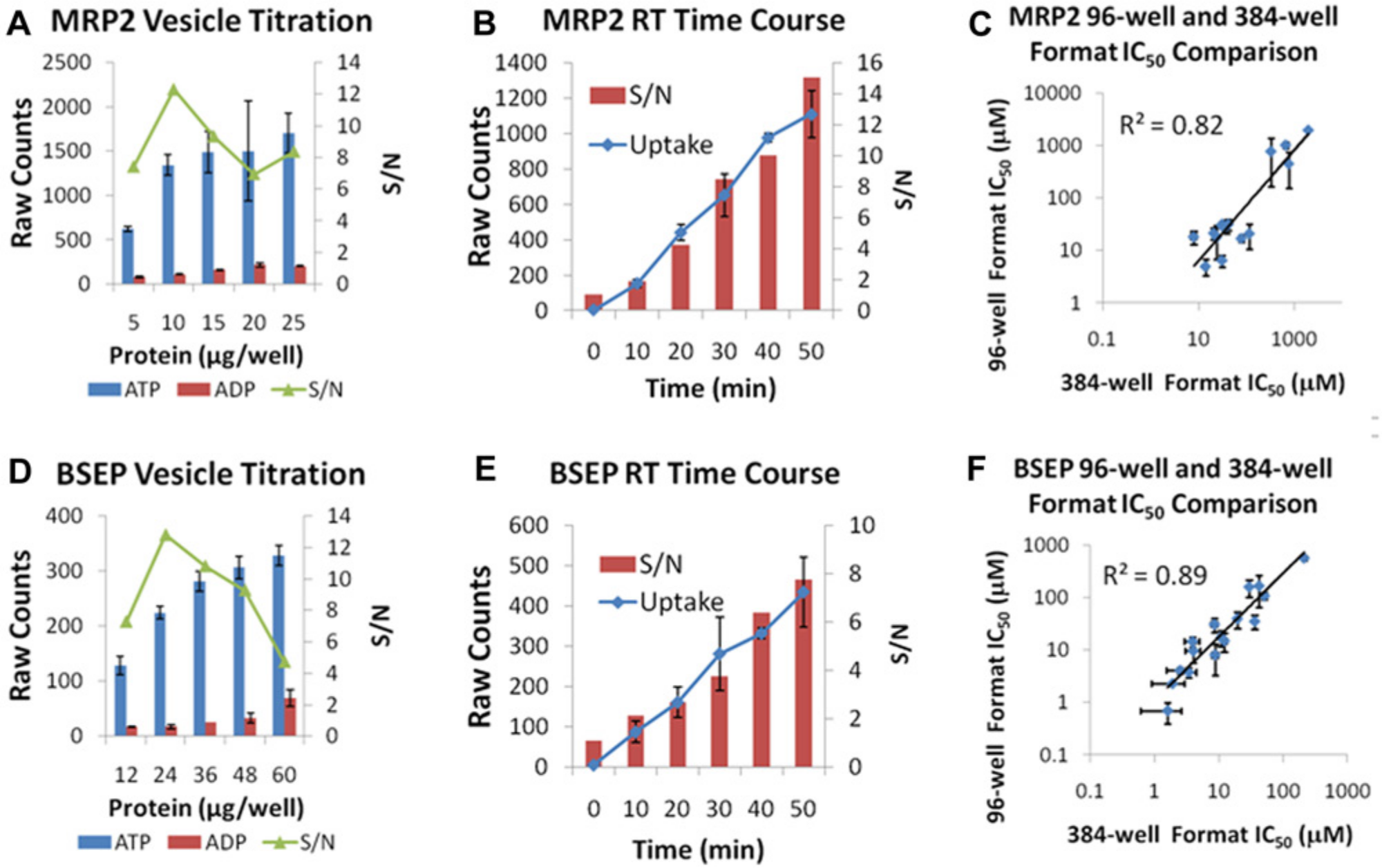
Vesicle uptake assays: direct determination of transport inhibition of efflux transporters. (
Automation of Transporter Inhibition Assays
The transporter assay process was developed by leveraging an existing automation platform with 384-well filtration capability to fully automate the transporter inhibition assays in the three assay formats.28,29 Ability to conduct assays at room temperature was a key enabler for assay automation. For example, observing BSEP assay reproducibility over three weekly runs, the automated assay demonstrated excellent reproducibility of reference compounds with solid assay statistics from run to run as measured by signal-to-background and Z′ ( Fig. 6A ). With the precision of the robot, data are highly correlated (R2 = 0.999) between experiments, as observed by screening compounds in experiments conducted on different days ( Fig. 6B ). With these automation solutions in place, we can further expand the transporter assay suite to develop internal capability well beyond basic regulatory requirements.
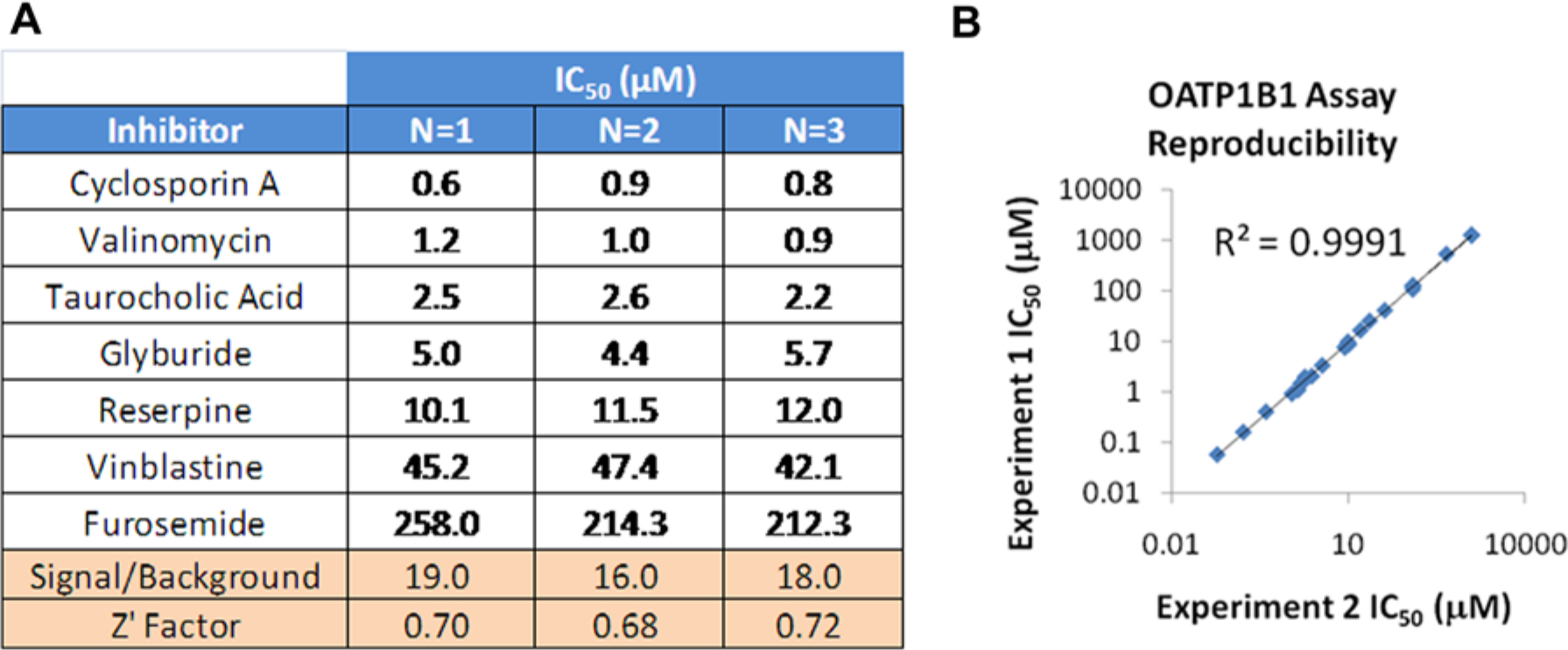
Leveraging automation systems to support transporter panel screening. (
In summary, a capacity gap solution has been implemented for tracking transporter SAR related to drug-drug interactions. A high-throughput panel of transporter inhibition assays was launched to monitor transporter activity for all programs across the portfolio. Substrates were identified to enable fluorescent 384-well transporter inhibition assays in a novel high-content imaging format. Miniaturized and robust radioactive whole-cell uptake and vesicle-based efflux inhibition assays in 384-well format were validated for production mode. Automation systems were leveraged to support fully automated transporter panels. This high-throughput capability has enabled a robust process to study transporter-related DDI issues in the early discovery space, which allows for dialing out these concerns in drug candidates rather than just highlighting potential issues in the development space.
Establishment of a 384-well high-throughput transporter assay suite has allowed us to expand the landscape of safety profiling to monitor transporter-related drug safety issues with high capacity at reduced cost. These high-throughput transporter assay formats can easily be used for assessing transporters as drug targets also. Because tissue distribution or organ-specific entry of a drug is facilitated or hindered by transporters, transporter SAR can provide additional information for decisions on advancing the most efficacious compounds with the best pharmacokinetic properties and with better insights governing oral bioavailability. 30 By leveraging new technology, it is now possible to conduct mechanistic studies to dissect mode of action for understanding drug safety and efficacy.
The future vision for transporter capabilities includes the development of high-throughput assays for all clinically relevant transporters (safety and drug-drug interactions) well beyond regulatory requirements. The establishment of high-throughput LC/MS capability to enable substrate assays for mechanistic studies with nonradiolabeled compounds will also be critical in defining transporter events. Because there is limited literature addressing transporter species variation, the creation of cross-species transporter cell lines will be critical to predict in vivo toxicity study outcome and address exposure differences between species. Execution of transporter strategies should be a tiered approach for early assessment of transporter interaction potential to improve the quality of new chemical entities and generation of appropriate transporter data to develop the right clinical plan for development compounds.
Understanding transporter activities from discovery to clinical development will provide invaluable impact to address adverse drug reactions and define risks associated with DDIs. By enabling transporter SAR, chemistry teams will gain valuable information for the design of compounds with enhanced mechanism-based efficacy while circumventing DDIs and other transporter toxicities.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.