
Abstract
An R345W mutation in fibulin-3 causes its inefficient secretion, increased intracellular steady-state levels, and the macular dystrophy, Malattia Leventinese (ML), a disease similar to age-related macular degeneration. It is unknown whether R345W causes ML through increased intracellular levels, by the secretion of a potentially aggregation-prone protein, or both. To identify small molecules that alter the secretion of fibulin-3, we developed ARPE19 retinal cell lines that inducibly express wild-type (WT) or R345W fibulin-3 fused to an enhanced Gaussia luciferase (eGLuc2). Screening of the Library of Pharmacologically Active Compounds demonstrated that these cell lines and the GLuc assay are suitable for high-throughput chemical screening. Two estrogen-related compounds enhanced fibulin-3 secretion, whereas a diverse series of small molecules reduced fibulin-3 secretion. A counterscreen identified compounds that did not substantially alter the secretion of unfused eGLuc2, demonstrating at least partial selectivity for fibulin-3. A secondary assay using untagged fibulin-3 confirmed that the top three inhibitory compounds reduced R345W fibulin-3 secretion. Interestingly, in untagged fibulin-3 studies, one compound, phorbol 12-myristate 13-acetate, reduced R345W fibulin-3 secretion while minimally enhancing WT fibulin-3 secretion, the desired activity and selectivity we sought for ML. The identified compounds could serve as tools for probing the etiology of fibulin-3–related diseases.
Keywords
Introduction
Malattia Leventinese (ML) is a rare, autosomal dominant macular dystrophy that is caused by a R345W mutation in fibulin-3, a disulfide-rich, secreted glycoprotein normally found in the extracellular matrix. ML is characterized by many of the same pathological phenotypes as age-related macular degeneration (AMD), the most prominent of which is the formation of lipid- and protein-rich extracellular deposits (drusen) underneath the cells of the retinal pigmented epithelium (RPE). Drusen consist of lipids, such as esterified cholesterol and phosphatidylcholine, and proteins such as vitronectin, apolipoprotein E, and complement components. 1 Although not a major component of drusen, fibulin-3 accumulates within RPE cells and between the RPE and drusen in AMD and ML patients, 2 linking aggregated fibulin-3 with drusen formation and subsequent retinal degeneration. Previously, we3,4 and others 2 have demonstrated that R345W fibulin-3 is misfolding prone, is inefficiently secreted, and exhibits higher intracellular levels. However, it is still not clear how R345W fibulin-3 causes ML. Some hypothesize that it is the chronic increase in its intracellular levels (potentially causing prolonged endoplasmic reticulum [ER] stress) that is responsible, whereas others believe that secretion of a misfolding- and aggregation-prone fibulin-3 could contribute to extracellular drusen formation and proteotoxicity by an unknown mechanism.
Evidence from mouse transgenic studies indicates that targeted replacement of the mouse wild-type (WT) fibulin-3 gene with human R345W fibulin-3 results in AMD-like symptoms, including basal laminar deposits, RPE atrophy, and complement activation.5,6 In contrast, ablation of the fibulin-3 gene in mice resulted in no eye-related phenotype, indicating that removal of fibulin-3 (whether WT or mutant) does not trigger ML or AMD-like symptoms. 7 These results provide strong evidence that R345W fibulin-3 causes ML through a gain–of–toxic function mechanism (possibly through prolonged ER stress and/or extracellular aggregation-linked proteotoxicity), and they suggest that selectively preventing R345W fibulin-3 secretion while targeting it for degradation without perturbing WT fibulin-3 is likely to be an ideal therapeutic strategy for treating ML.
In prior studies, we have demonstrated that the R345W fibulin-3 secretion defect can be partially remedied by a reduced cell growth temperature, primarily acting through translational attenuation.3,4 Furthermore, the pool of R345W fibulin-3 whose secretion is enhanced as a consequence of translational attenuation appears to have a “WT-like” fold based on its disulfide bonding pattern and migration in nonreducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels.3,4 Although translational attenuation was informative from a mechanistic point of view because it suggested that modulation of the proteostasis network could alter the balance between R345W fibulin-3 folding and secretion versus degradation, translational attenuation itself is not a potential therapeutic strategy for ML. Thus, we sought to identify two categories of small-molecule secretion modulators as potential therapeutic lead compounds for fibulin-3–associated diseases: (1) compounds that enhance the secretion of R345W fibulin-3 exhibiting a “WT-like” fold and (2) small molecules that inhibit R345W secretion while hopefully promoting its intracellular degradation (preferably without affecting WT secretion). Small molecules that increase intracellular degradation of mutant fibulin-3 at the expense of folding and secretion could not only serve as lead compounds for the treatment of ML, but compounds that reduce fibulin-3 secretion in general (without necessarily being selective for WT vs. R345W) may also be useful for the treatment of certain gliomas, whose growth and invasion are dependent on fibulin-3.8,9
To identify fibulin-3 secretion modulators, we developed an assay to measure fibulin-3 secretion in a high-throughput manner. Conventionally, trafficking of proteins (specifically fibulin-3) through the secretory pathway has been measured by Western blotting or by pulse-chase radioactive labeling followed by immunoprecipitation and Western blotting.2,10 However, recently, the small, naturally secreted luciferase, Gaussia luciferase (GLuc), has been used to quickly, sensitively, and inexpensively measure gene expression 11 and ER-Golgi dynamics,12,13 and it has also been used to follow the secretion of specific proteins of interest, including fibulin-3, by creating GLuc fusions.3,4,14 GLuc is substantially more sensitive (>20 000 fold) than conventional secreted reporter proteins (secreted embryonic alkaline phosphatase 12 ) and is inherently more practical for following secreted proteins than other reporters, which typically fold in the cytosol (green fluorescent protein, firefly luciferase, Renilla luciferase, etc.). Thus, we chose to follow WT and R345W fibulin-3 secretion by fusing these proteins to GLuc, enabling quantification of the fusions’ GLuc luminescence inside and outside of the cell.
It is well known that different cell lines have distinct abilities to handle misfolding-prone proteins, 15 which is likely due to differences in their subcellular proteostasis network capacities. Therefore, we generated a set of novel, physiologically relevant RPE-based cell lines (Tet-On ARPE19) for identifying small-molecule modulators of fibulin-3 secretion. These cell lines enable regulated expression of the WT or R345W fibulin-3 gene fused to an enhanced GLuc (eGLuc2), with characteristics suitable for high-throughput screening (HTS). Control of these genes with tetracycline/doxycycline also circumvents potential cytotoxicity and changes to stress-responsive signaling resulting from constitutive expression. Using these cell lines, we reproducibly screened a small chemical library in a high-throughput fashion with extremely low coefficients of variation and high Z′ scores. We were successful in identifying both enhancers and inhibitors of fibulin-3 secretion, some of which appear to be selective for affecting R345W secretion over WT fibulin-3 secretion in secondary assays. These are the first compounds that have been demonstrated to alter fibulin-3 secretion. If they possess suitable pharmacological properties, they may be useful for probing the molecular mechanism underlying ML and for scrutinizing the hypothesis that reducing fibulin-3 secretion could ameliorate ML and fibulin-3–dependent gliomas.
Materials and Methods
Cloning
Humanized Gaussia luciferase (GLuc) was mutated (M43I and M110I) to yield a luciferase enzyme with glow-like properties (eGLuc2) and thus more suitable for HTS.16,17 Genes encoding for eGLuc2, fibulin-3 fused to a C-terminal eGLuc2, or untagged fibulin-3 or fibulin-5 were generated in the pENTR1A Dual Selection entry plasmid (Life Technologies, Carlsbad, CA). eGLuc2 and fibulin-3–eGLuc2 genes were shuttled into the pLenti4/TO/V5-DEST vector (Life Technologies) while untagged fibulin-3 and WT fibulin-5 genes were shuttled into the pAd/CMV/V5-DEST vector by LR Clonase II recombination (Life Technologies). All constructs were sequenced to confirm their identity.
Cell culture
Human embryonic kidney (HEK-293T) cells were grown in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum (FBS; Omega Scientific, Tarzana, CA), glutamine, and 1% penicillin/streptomycin (PSQ; Cellgro). Human retinal pigmented epithelium cells (ARPE19; American Type Culture Collection [ATCC] CRL-2302; ATCC, Manassas, VA) were cultured in high-glucose DMEM/F12 50:50 (Cellgro) supplemented with 15 mM HEPES and containing 10% FBS and PSQ. All cells were cultured under typical conditions (37 °C, 5% CO2). Transfection of HEK-293T cells was performed as described previously. 3
Virus production
Vesicular stomatitis virus (VSVG) pseudotyped lentiviral particles were produced by cotransfecting LentiX cells (Clontech, Mountain View, CA) with the structural plasmids necessary for virus production: Rev, RRE, and VSVG along with the pLenti4/TO lentivirus constructs. LentiX cells were transfected for 24 h, and media containing lentivirus particles were collected 48 h and 72 h posttransfection. Viral particles were concentrated by centrifugation (~40 000 g for 2 h at room temperature [RT]) prior to storage at −80 °C (until use). Adenovirus was generated according to the instructions from the ViraPower Adenoviral Gateway Expression Kit (Life Technologies).
Generation of Tet-On ARPE19-inducible, eGLuc2-based fibulin-3 and control cell lines
To generate a physiologically relevant tetracycline/doxycycline-inducible cell line, ARPE19 cells were initially infected with lentivirus encoding for the tet repressor (TR; originating from the pLenti6/TR vector) and a stable, clonal cell line (Tet-On ARPE19) with high levels of the TR was generated after selection in selective antibiotic (blasticidin, 10 µg/mL). The Tet-On ARPE19 cell line was then used to generate the subsequent cell lines that inducibly express WT fibulin-3–eGLuc2, R345W fibulin-3–eGLuc2, or unfused eGLuc2. These heterogeneous stable cell lines were generated by infection with the corresponding eGLuc2-pLenti4/TO lentivirus followed by selection with zeocin (200 µg/mL).
Non-HTS GLuc assay to monitor fibulin-3 secretion/intracellular accumulation
GLuc/eGLuc2 secreted from transiently transfected HEK-293T cells was measured as described previously.
3
For follow-up studies in ARPE19 cells, cells were plated at a density of 20 000 cells/well of a clear-bottomed, black 96-well plate (Costar 3603; Corning, Corning, NY) overnight followed by treatment with small molecules and doxycycline for 24 h. For dose-dependent studies, secreted fibulin-3–eGLuc2 was monitored by taking 50 µL aliquots of conditioned media and reacting them with 50 nL GLuc substrate (per reaction) diluted in 10 µL GLuc buffer (per reaction; New England Biolabs [NEB], Ipswich, MA) in a flat-bottomed black assay plate (Costar 3915; Corning). Measurement of intracellular eGLuc2 was achieved by ≥4× freeze/thaw in 50 µL Dulbecco’s phosphate-buffered saline (DPBS) using a dry ice/ethanol bath. Intracellular levels were measured directly in the plate, employing the same substrate/buffer ratio as described above. Immediately after mixing, luminescence was measured in a Tecan Safire II (Tecan, Upsala, Sweden) using a 100-ms integration time. Importantly, freezing and thawing the soluble eGLuc2 enzyme two, three, and four times (the latter being the average number of freeze/thaw cycles that we used) does not substantially reduce GLuc activity, lowering its luminescence by 4.6%, 2.4%, and 7.6% relative to untreated GLuc, respectively (
Primary HTS assay
WT and R345W fibulin-3–eGLuc2 fusion Tet-On ARPE19 cells were plated in flat-bottomed white 384-well plates (Costar 3570; Corning) at a density of 3500 to 5000 cells/well using a Multidrop liquid dispenser (Thermo Scientific, Waltham, MA) in a total media volume of 20 µL (containing 1 µg/mL doxycycline to induce expression of the gene of interest). Shortly after seeding, the cells were treated with compounds (10 µM final concentration in DMSO [0.5%, final]) from the Library of Pharmacologically Active Compounds (LOPAC; Sigma Aldrich, St. Louis, MO) via pintooling (100 nL pins). Cells were treated with compounds for 24 h, after which fibulin-3–eGLuc2 secretion was measured by a modified BioLux GLuc assay (NEB). Briefly, a GLuc assay reagent master mix was generated containing the equivalent of 10 nL GLuc substrate per well diluted in 2.2 µL of buffer/media per well (1 µL GLuc buffer and 1.2 µL full media). To measure secreted fibulin-3–eGLuc2, 2.2 µL/well of GLuc assay reagent was directly administered into the 384-well plate by a BioRAPTR flying reagent dispenser (Beckman Coulter, Fullerton, CA). Whole-well eGLuc2 luminescence was measured immediately after GLuc assay reagent addition with an EnVision Multilabel Reader (Perkin Elmer, Wellesley, MA) using a 100-ms integration time. Note: while coelenterazine is a cell-permeable substrate, and thus a whole-well read should report on total eGLuc2 (secreted and intracellular), we find that GLuc assay reagent administration in this fashion largely reports on secreted eGLuc2. In untreated wells (no DMSO or small molecule), 97% of the whole-well signal on average arises from secreted eGLuc2 (
GLuc assay performance was calculated by determining the Z factor (Z′) using the following equation 18 :
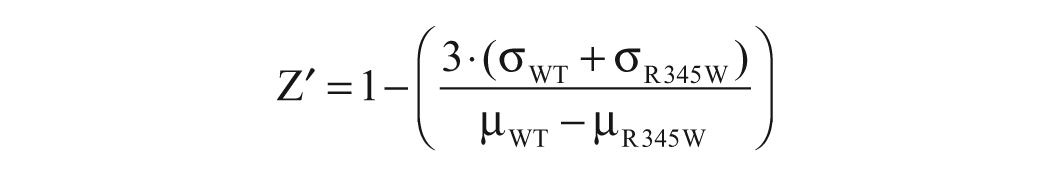
where µWT and σWT are the mean and standard deviation (SD), respectively, of vehicle-treated cells expressing WT fibulin-3–eGLuc2 (i.e., the high control, which was the highest signal we hypothesized R345W secretion could be raised), and µR345W and σR345W are the mean and standard deviation of vehicle-treated cells expressing R345W fibulin-3–eGLuc2 (i.e., the low control). The Z′ value was obtained for each 384-well plate of the LOPAC library. The coefficient of variation (CV) of each eGLuc2 cell line was calculated by dividing the SD by the average signal in DMSO-treated wells and expressing it as a percentage of the average. The LOPAC library was screened in triplicate on separate days. Each screen assayed the LOPAC compounds individually (10 µM), and hits were identified if the compound increased or decreased the WT or R345W fibulin-3–eGLuc2 fusion signal by greater than 3 SD of the mean.
Counterscreening and dose-response assays
Hit compounds were excluded from further analysis if they caused significant changes to unfused eGLuc2 secretion (±3 SD). In addition, compounds that reduced fibulin-3–eGLuc2 secretion were evaluated for toxicity and excluded if deemed detrimental to cell viability by a resazurin assay (defined by a signal 3 SD below the vehicle-treated wells). For follow-up dose-response studies, select compounds identified as hits were purchased as fresh powder: phorbol 12-myristate 13-acetate (PMA; Fisher Scientific, Waltham, MA, cat. number: FL-01-0605), ARP 101 (Sigma, cat. number: A8356), and XCT790 (Sigma, cat. number: X4753).
Secondary assay: Western blotting
For secondary assay experiments following the secretion of untagged fibulin-3, ARPE19 cells were seeded at a density of ~100 000 cells/cm2 in 12- or 24-well plates for 48 h. Cells were then infected with adenovirus encoding untagged fibulin-3 or fibulin-5, as a control, for an additional 48 h. Media was replaced with fresh media containing 2% FBS (instead of 10% FBS) supplemented with hit compounds for an additional 24 h. Then, 40 µL (typically 1/20th of the total volume) of conditioned media was subjected to Western blotting. Conditioned media aliquots were boiled in reducing Laemmeli buffer, separated on a Tris-Gly SDS-PAGE gel, and transferred to a nitrocellulose membrane. Membranes were probed with one or more of the following primary antibodies: rabbit anti–fibulin-3 (FBLN3, 1:500; ProSci, Poway, CA), rabbit anti-DNAJB10 (ERdj3, 1:1000; Proteintech, Chicago, IL), and mouse anti–fibulin-5 (FBLN5, 1:1000; Proteintech). Blots were imaged on a LI-COR Odyssey infrared imager (LI-COR, Lincoln, NE) using either a goat anti-mouse 680-nm or goat anti-rabbit 800-nm secondary antibody (LI-COR).
Quantitative PCR (qPCR) analysis to evaluate stress-responsive signaling activation
After treatment with compounds, the mRNA of specific stress-responsive genes from ARPE19 cells was analyzed by qPCR. Twenty-four hours after treatment with small molecules, ARPE19 cells were harvested by trypsinization and washed once in DPBS. RNA was extracted from the cells using the RNeasy kit (Qiagen, Germantown, MD) according to the manufacturer’s protocol. Then, 500 ng RNA was reverse transcribed using the Quantitect Reverse Transcription kit (Qiagen). The resulting cDNA was diluted 4-fold with DNase/RNase-free water before being used for amplification.
qPCR primers were selected using Primer3 software (http://frodo.wi.mit.edu/) and were optimized for amplification of 130- to 180-bp amplicons with annealing temperatures centered on 62.5 °C. cDNA was amplified using Sybr Green Fast Master Mix (Roche, Piscataway, NJ). cDNA amplification was quantified using an Applied Biosystems 7900 qPCR machine (Life Technologies). The resultant data were analyzed using the RQ manager (Life Technologies) followed by analysis in Data Assist (Life Technologies).
Results
Reporter cell line generation and validation
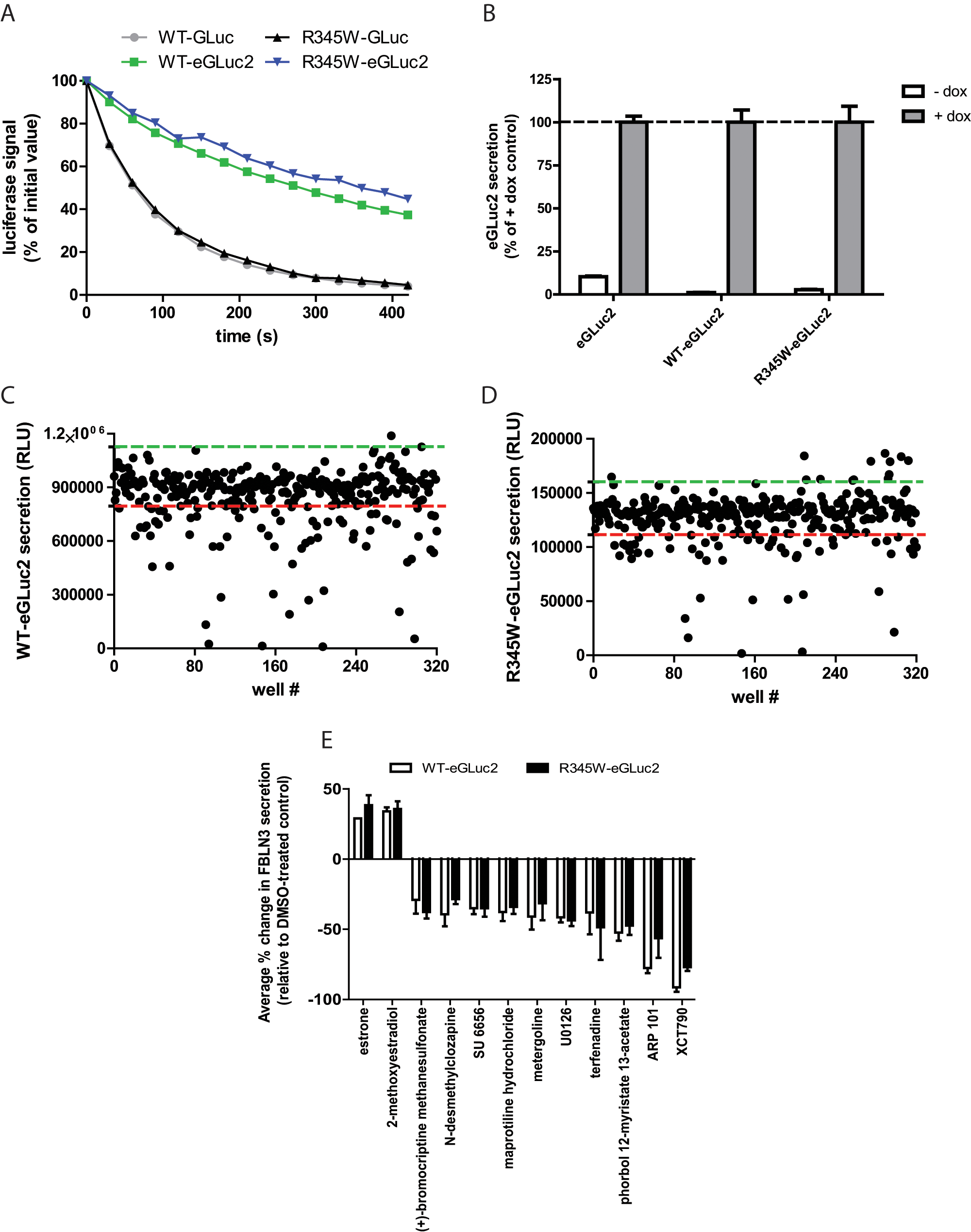
GLuc is a relatively newly characterized luciferase enzyme that was originally isolated from the marine conepod, Gaussia princeps. 19 Because of its brightness and sensitivity, GLuc has been used as a reporter of a variety of biological processes, 20 including following the fate of fibulin proteins.3,4 However, GLuc catalyzes its luminescent reaction with flash-like kinetics, resulting in a relatively unstable and quickly decaying signal, which is undesirable for HTS. 17 To slow the decay of the GLuc luminescent signal, we mutated GLuc at two oxidation-prone methionines (M43I and M110I16,17) to generate eGLuc2. eGLuc2 was fused to the C-terminus of WT and R345W fibulin-3 to follow their secretion. Mutating GLuc to eGLuc2 reduced the specific activity of GLuc by approximately half. 16 However, the luciferase signal decay of eGLuc2 was substantially reduced compared to GLuc ( Fig. 1A ).
(
The fibulin-3–eGLuc2 fusions were introduced into a Tet-On ARPE19 cell line by lentivirus infection and stable, heterogeneous pools were generated by selective antibiotic resistance. In the absence of doxycycline (dox), the expression of the eGLuc2 genes was tightly repressed ( Fig. 1B ); the amount of “leaky” expression in the absence of dox ranged from 1.1% (WT fibulin-3–eGLuc2) to 10% (eGLuc2) of the respective luminescence signal with dox treatment ( Fig. 1B ) .
LOPAC screening
As an initial proof of principle that our cell-based fibulin-3–eGLuc2 reporter assay could be used to identify regulators of fibulin-3 secretion, we chose to screen the LOPAC library from Sigma Aldrich. This library contains 1280 compounds that affect a variety of cellular processes covering many of the major drug target classes, including G-proteins, ion channels, and kinases. We miniaturized the GLuc assay into 384-well plates and obtained a robust, reproducible secretion assay with average CVs of <9% across all cell lines ( Table 1 ). The Z′ values, which indicate the quality of the assay (reflecting the assay window and precision), were all exceptional—consistently above 0.70 ( Table 2 ). The variability of the data, as well as the assay window between WT and R345W fibulin-3– eGLuc2 secretion, is demonstrated in Figure 1C , D .
Average Coefficient of Variation (CV) for Each Cell Line, Across All Runs.

WT, wild-type.
Average Z′ Value per Plate, All Runs.
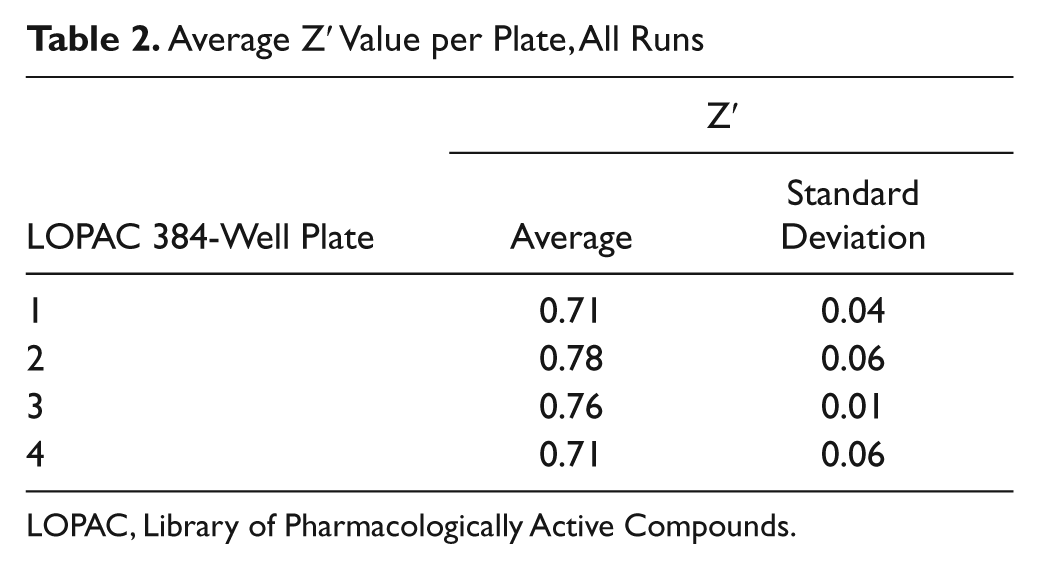
LOPAC, Library of Pharmacologically Active Compounds.
After screening the LOPAC library against WT or R345W fibulin-3–eGLuc2–expressing cells in three independent experiments (and against eGLuc2 in two independent counterscreening experiments), we identified a series of hit compounds (>3 or <3 SD of the vehicle-treated control) that influenced fibulin-3–eGLuc2 secretion without significantly affecting eGLuc2 secretion or causing significant toxicity (>3 SD below vehicle-treated samples as measured by the resazurin assay). To reduce the potential for false positives, in follow-up screening, we selected compounds that reproduced in three of three experiments in either WT or R345W fibulin-3–eGLuc2–expressing cells. Using these criteria, we identified 20 hits, resulting in a hit rate of 1.56%. For simplicity, we have presented the two compounds that enhanced WT and R345W fibulin-3–eGLuc2 secretion and the top 10 compounds that reduced WT and R345W fibulin-3–eGLuc2 secretion (
Fig. 1E
). Interestingly, the two compounds that were found to significantly enhance WT and R345W fibulin-3–eGLuc2 secretion were both estrogen-related compounds (estrone and 2-methoxyestradiol;
Fig. 1E
and
Confirmatory dose-dependent assessment of select hit compounds
The top three compounds that reduced fibulin-3 secretion—PMA, ARP 101, and XCT790—were purchased as fresh powders, and their ability to alter fibulin-3–eGLuc2 secretion (and intracellular accumulation) was confirmed/evaluated in dose-dependent studies ( Fig. 2A – F ).
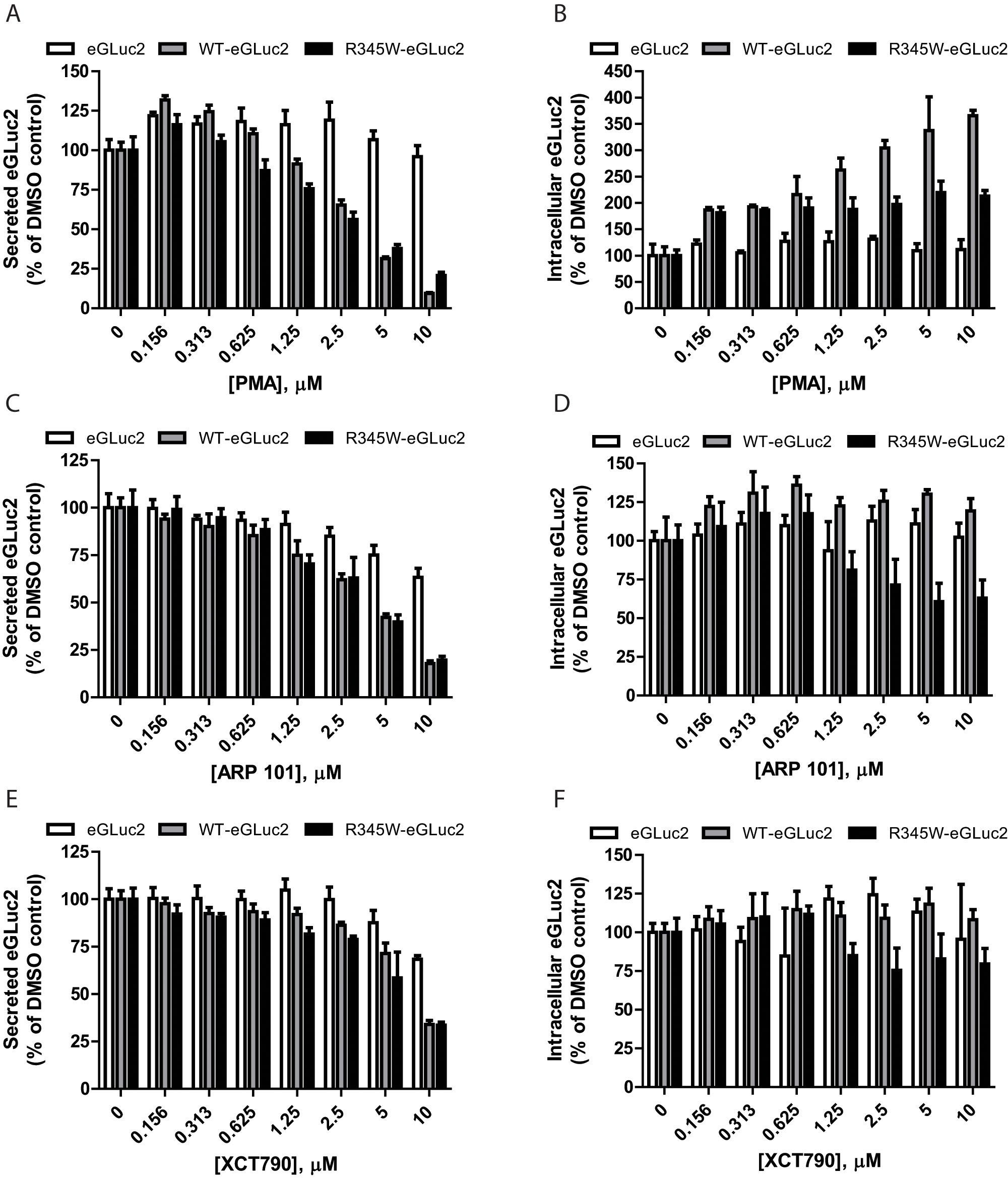
Dose-dependence confirmation of select fibulin-3–reducing compounds. ARPE19 cells expressing eGLuc2, wild-type (WT) fibulin-3–eGLuc2 (WT-eGLuc2), or R345W fibulin-3–eGLuc2 (R345W-eGLuc2) were treated with the indicated compounds for 24 h, after which the secreted (
According to the dose-dependent follow-up GLuc assay, PMA reduced WT and R345W fibulin-3–eGLuc2 secretion to the greatest extent among the three hit compounds at 10 µM to 9.5% ± 0.5% and 20% ± 2% of the DMSO control, respectively ( Fig. 2A ). PMA also demonstrated the highest amount of specificity, barely affecting eGLuc2 secretion (96% ± 7% of the DMSO control at 10 µM; Fig. 2A ). PMA increased the intracellular steady-state levels of WT and R345W fibulin-3–eGLuc2, up to 365% ± 10% and 213% ± 11% of DMSO-treated controls, respectively ( Fig. 2B ).
ARP 101 demonstrated partial selectivity for reducing WT and R345W fibulin-3–eGluc secretion down to 18% ± 1% and 20% ± 2% (at 10 µM) of the DMSO control, respectively ( Fig. 2C ), while having less of an effect on eGLuc2 secretion (63% ± 5% of the secretion of the DMSO control; Fig. 2C ). The absolute effects of ARP 101 on fibulin-3 secretion were similar to those of PMA; however, the effect of ARP 101 on the fate of intracellular fibulin-3 was distinct. ARP 101 did not cause a substantial increase in any of the steady-state intracellular levels of the variants tested ( Fig. 2D ). In fact, ARP 101 substantially reduced the intracellular levels of R345W fibulin-3 (63% ± 12% of the control) when compared with either WT (119% ± 8% of the control) or eGLuc2 (102% ± 9% of the control; Fig. 2D ), a desirable characteristic.
XCT790 (10 µM) reduced both WT and R345W–fibulin-3–eGLuc2 secretion to 34% ± 2% of the DMSO control ( Fig. 2E ) and reduced eGLuc2 secretion to 68% ± 2% of the DMSO control. In contrast to both PMA and ARP 101, XCT790 had little impact on intracellular steady-state levels ( Fig. 2F ).
A secondary immunoblot assay to verify luciferase-based results
To confirm that the effects of the hit compounds on the secretion of WT and R345W fibulin-3–eGLuc2 fusion proteins were not a consequence of the fusion protein employed for the primary screen, we performed immunoblotting on secreted WT and R345W fibulin-3 lacking the eGLuc2 tag in ARPE19 cells ( Fig. 3A ). The specificity of the compounds was evaluated by monitoring the secretion of two additional proteins, fibulin-5 and ERdj3, after hit compound treatment ( Fig. 3A ). Fibulin-5, another member of the fibulin protein family, shares >50% sequence homology with fibulin-3, as well as a near-identical domain organization. 21 ERdj3 is an ER heat-shock protein 40 co-chaperone that partitions between the ER and extracellular space, and its expression can be altered during particular conditions of ER stress. 22
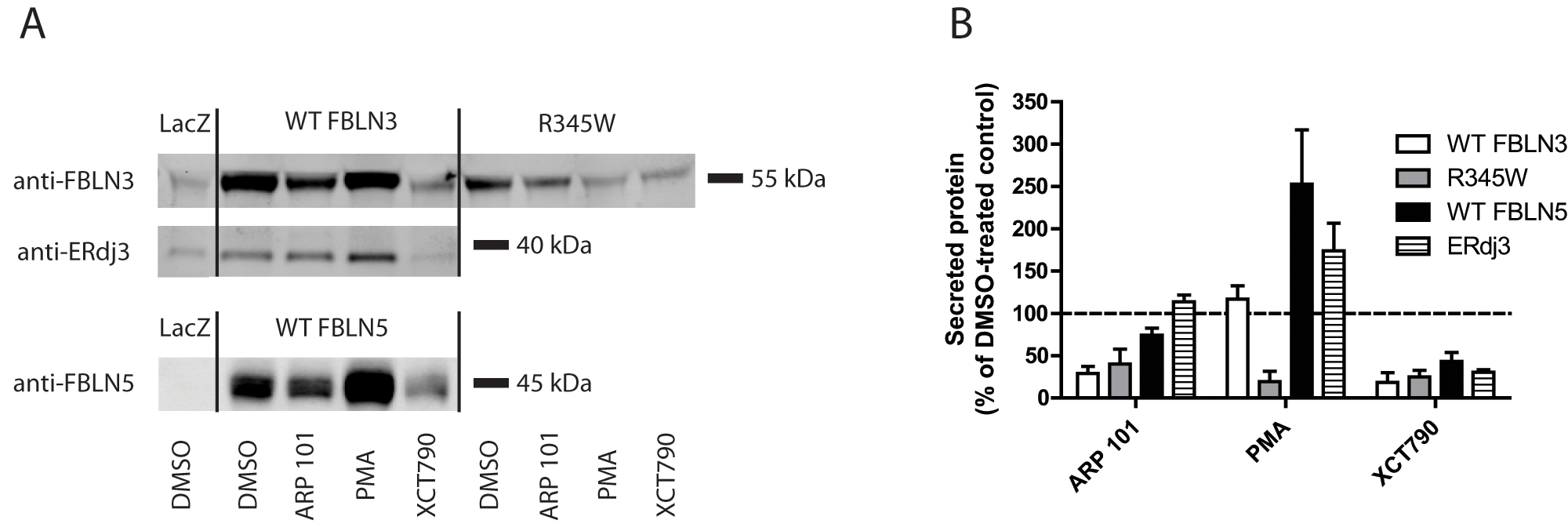
Hit compounds alter the secretion of untagged fibulin-3. (
Treatment of ARPE19 cells with ARP 101 (10 µM) for 24 h reduced WT and R345W fibulin-3 secretion to 29% ± 8% and 40% ± 18% of the DMSO control, respectively ( Fig. 3A , B ). ARP 101 had little or no effect on fibulin-5 or ERdj3 secretion (74% ± 8% and 114% ± 8% of control levels, respectively; Fig. 3A , B ). Treatment with the ERRα inverse agonist XCT790 resulted in substantial reduction in the secretion of each protein assessed, on average by ~71% (29% of DMSO control levels; Fig. 3A , B ). These observations were consistent with the eGLuc2-based studies ( Fig. 1E and Fig. 2C – F ).
In contrast to the consistent results observed with ARP 101 and XCT790 treatment with respect to fibulin-3–eGLuc2 versus untagged fibulin-3 secretion, PMA actually slightly enhanced untagged WT fibulin-3 secretion (117% ± 16% of DMSO control levels;
Fig. 3A
,
B
) but still maintained a profound inhibitory effect on untagged R345W secretion (19% ± 12% of DMSO control levels;
Fig. 3A
,
B
). This selectivity for reducing the R345W mutant was also bolstered by the observation that PMA did not decrease ERdj3 or fibulin-5 secretion but, instead, increased their secretion (174% ± 32% and 253% ± 64% of the DMSO control, respectively;
Fig. 3A
,
B
). The inhibitory effect of the hit compounds on R345W secretion was not simply due to changes in endogenous or overexpressed fibulin-3 mRNA levels (
The effect of hit compounds on stress-responsive signaling in ARPE19 cells
We hypothesized that the identified compounds may exert their effects on fibulin-3 secretion through activation of one or more of the cell’s stress-responsive signaling pathways. For example, we have previously demonstrated that selective activation of one of the arms of the unfolded protein response (UPR) in the ER can alter fibulin-3 secretion. 4 Furthermore, Roybal and colleagues 10 demonstrated that the R345W mutant likely activates the inositol requiring enzyme 1 (IRE1) and the activating transcription factor 6 (ATF6) arm of the UPR more so than WT fibulin-3, and that this activation may play a role in the pathogenesis of ML. Therefore, we used qPCR to assess changes in transcript levels of genes associated with activation of stress responses after treatment with hit compounds. As surrogate reporters of the activation of stress-responsive signaling, we looked for increases in the transcript levels of 78-kDa glucose-regulated protein (GRP78), increased by activation of the ATF6 arm of the UPR; ERdj4, increased by activation of the IRE1 arm of the UPR; asparagine synthase (ASNS), increased by activation of the PKR-like ER kinase (PERK) arm of the UPR; and heme oxygenase 1 (HMOX1), increased by an antioxidant response.
Each hit compound altered the transcript levels of the selected stress-responsive pathways distinctly. ARP 101 treatment did not change the transcript levels of GRP78 ( Fig. 4A ) or ERdj4 ( Fig. 4B ) when compared with controls, indicating a lack of activation of ATF6 and IRE1 arms of the UPR, respectively. However, ARP 101 did increase ASNS levels to ~7.2-fold above the control ( Fig. 4C ), suggesting activation of PERK or one or more of the three other known eukaryotic initiation factor 2 α (eIF2α) kinases. ARP 101 also caused significant induction of HMOX1 (~9.5 fold), most likely indicating induction of a cellular antioxidant response.
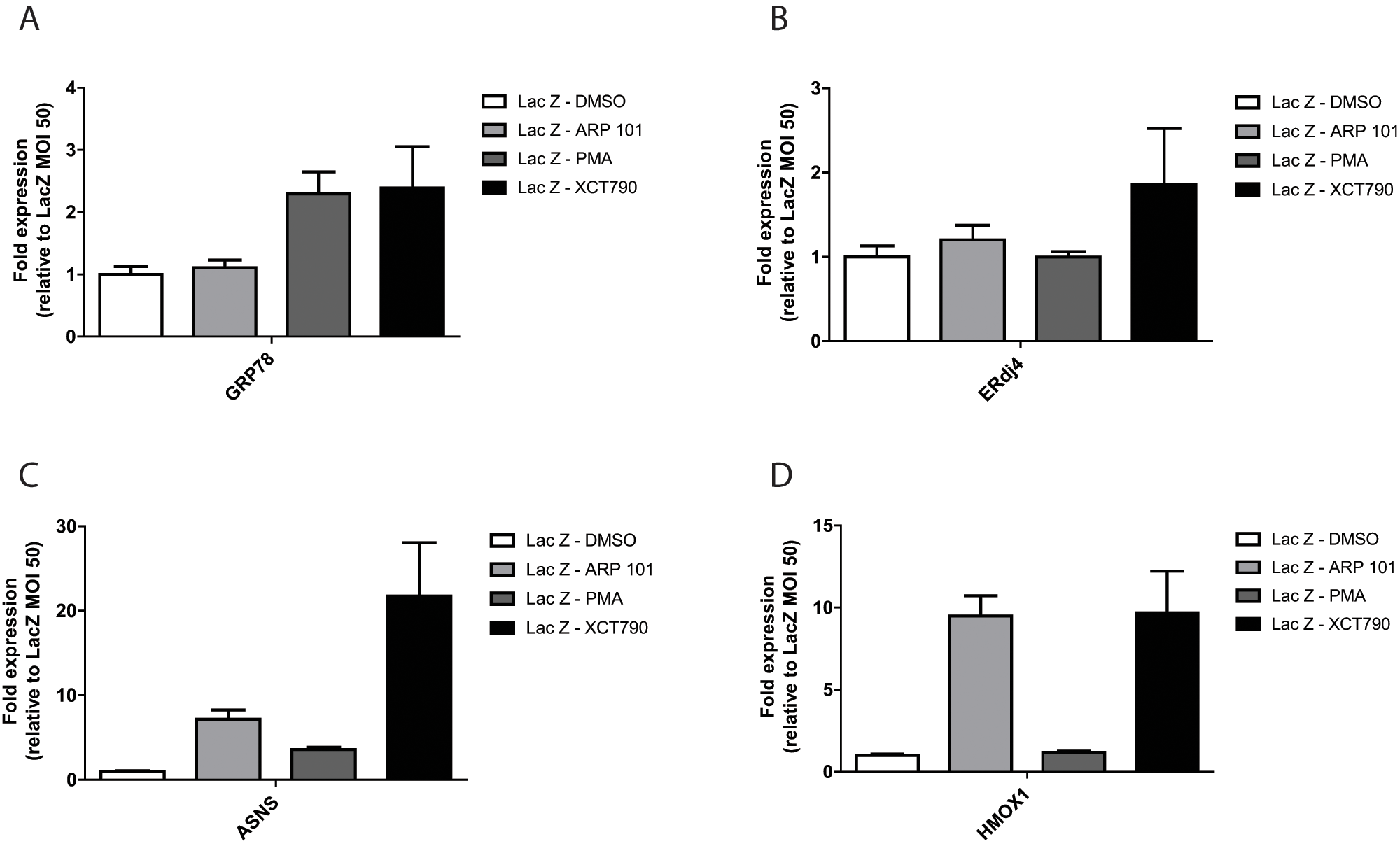
The influence of hit compounds on stress-responsive signaling transcripts. ARPE19 cells were infected with adenovirus encoding for LacZ for 48 h followed by treatment with the designated compound (10 µM, 24 h). RNA was extracted from cell pellets and gene expression was analyzed by quantitative PCR (see
In contrast, PMA induced changes only in GRP78 transcript levels (~2.3 fold; Fig. 2A ) and minimal changes to ASNS (~3.6 fold; Fig. 4C ) when compared with the other compounds. These results suggest that within the context of the UPR, PMA may selectively activate the ATF6 arm with little or no impact on other UPR arms, and that selectivity may be key in preferentially regulating WT versus R345W fibulin-3 secretion.
XCT790 induced an increase in each of the stress-responsive transcripts and did so to the largest extent compared with the other hit compounds (
Fig. 4A
–
D
). XCT790 increased GRP78 (~2.4 fold;
Fig. 4A
), ERdj4 (~1.9 fold;
Fig. 4B
), ASNS (~21.8 fold;
Fig. 4C
), and HMOX1 (~9.7 fold;
Fig. 4D
) transcript levels, likely indicating global UPR activation (similar to treatment with the classic ER stressors, tunicamycin and thapsigargin) in addition to triggering activation of the oxidative stress response. These results are in accordance with our secretion data (
Fig. 3A
,
B
) demonstrating that XCT790 nonselectively reduces the secretion of fibulin-3, ERdj3, and fibulin-5. These data suggest that XCT790 treatment (at 10 µM for 24 h) likely causes pleiotropic effects on a variety of pathways, ultimately culminating in cellular stress. XCT790 has a highly activated Michael acceptor structure (
Discussion
In the research described herein, we have developed and miniaturized a dox-regulated fibulin-3–eGLuc2–based assay to quantify fibulin-3 secretion. We have demonstrated that this cell-based assay can be used in a high-throughput manner to identify novel compounds that regulate the fate of WT and/or R345W fibulin-3 rather selectively by altering folding and/or secretion and/or degradation. We expect that this assay can also be used to identify genetic modifiers of fibulin-3 secretion in a manner similar to that used in our chemical screen. Furthermore, we anticipate that fusion of this enhanced GLuc to other secreted proteins of interest could facilitate the identification of new small molecules or genetic modifiers that regulate the cellular folding and secretion versus degradation quality control decision of other proteins of interest. However, care should be exercised when fusing eGLuc2 to small or non–disulfide-bonded secreted proteins as eGLuc2 could dominate the folding kinetics/thermodynamics of the protein of interest. In addition, fusion to eGLuc2 may potentially change the interacting proteostasis network components, thus likely influencing the ultimate fate of the protein of interest (e.g., folding and secretion vs. degradation).
The compounds we identified in our screen that reduce fibulin-3 secretion appear to have differential selectivity (affecting fibulin-3 vs. fibulin-5 or ERdj3 secretion; Fig. 3A , B ) and specificity (WT vs. R345W fibulin-3 secretion; Fig. 3A , B ). They also affect stress-responsive signaling differentially ( Fig. 4A – D ), which could occur by a variety of mechanisms, yet all reduce fibulin-3 (WT and/or R345W; Fig. 3A , B ) secretion. These findings suggest that multiple proteostasis network components and pathways govern the fate of fibulin-3, and, therefore, it is likely that a variety of compounds will influence fibulin-3 secretion/degradation.
Consistent with this hypothesis, the three top fibulin-3–reducing compounds originate from different drug classes and have significantly different proposed mechanisms of action. XCT790, an ERRα inverse agonist, was the most nonspecific in all follow-up studies, likely due to global activation of stress-responsive signaling pathways resulting from proteome modification-linked misfolding/aggregation. The triggering of these pathways by XCT790 could cause translational attenuation in addition to retaining proteins within the ER, likely causing a reduction in cell proliferation or robustness. 23 Nonetheless, it is likely that regulation of the estrogen receptor or estrogen-related metabolites truly affects fibulin-3 secretion, in accordance with past studies demonstrating that fibulin-1 and fibulin-3 can be regulated transcriptionally and posttranscriptionally by estradiol and 17β-estradiol.24,25 These observations, combined with our estrogen-related hits (estrone, 2-methoxyestradiol; Fig. 1E ), provide validation that our assay can identify previously discovered fibulin- regulating compound families.
In addition to XCT790, we identified ARP 101, a small molecule that has been characterized previously as an MMP-2 inhibitor, 26 as a confirmed inhibitor of fibulin-3 secretion that has higher specificity for affecting fibulin-3 than either fibulin-5 or ERdj3 ( Fig. 3A , B ). These findings link the manipulation of an extracellular matrix (ECM) protease with alterations in the secretion of another ECM protein (fibulin-3). Previous groups have found that expression of fibulin-3 downregulated MMP-2 protein levels. 27 Given our observations, it is intriguing to speculate that the converse could be true: that MMP-2 (or inhibition of MMP-2) could in turn alter fibulin-3 secretion or protein production. Alterations of fibulin-3 secretion due to MMP-2 inhibition could influence, in part, functions that were originally thought to be governed solely by MMP-2 inhibition itself, such as cancer metastasis and angiogenesis (reviewed in Page-McCaw et al. 28 ), both of which can be regulated by fibulin-3.8–10
Arguably, the most interesting of the hit compounds we discovered was PMA. In untagged fibulin-3 secretion studies, PMA caused a decrease in R345W but not WT fibulin-3 secretion ( Fig. 3A , B ). Furthermore, it does not decrease fibulin-5 or ERdj3 secretion, demonstrating selectivity and specificity ( Fig. 3A , B ). At low concentrations for short durations, PMA is typically considered a protein kinase C (PKC) activator (reviewed in Liu and Heckman 29 ). However, it is important to note that the actual mechanism of action by which a drug works under screening/follow-up studies may differ significantly from its annotated mechanism. For example, at high concentrations or during prolonged treatments, PMA causes depletion of PKC and thus acts effectively as a PKC inhibitor, not activator. 30 Thus, it is likely that the PMA treatment concentration (10 µM) and duration (24 h) caused significant PKC inhibition, which may be key in regulating the secretion of fibulin-3. In support of this notion, we identified additional PKC inhibitors (tamoxifen and 1-(5-isoquinolinylsulfonyl)-3-methylpiperazine dihydrochloride) as reducers of fibulin-3 secretion. Although these compounds reduced WT and/or R345W in three of three experiments, they were not among the top 10 fibulin-3–reducing compounds.
An alternative mechanism by which PMA may cause the selective decrease in R345W fibulin-3 secretion is through increasing the transcription/translation of fibulin-3 (
Seven of the 12 hits we have presented in this report have been or are currently in clinical trials for maladies unrelated to ML (http://clinicaltrials.gov). These hit compounds, which may not have the optimal characteristics to serve as potential therapeutics for ML, could nonetheless shed light on the mechanism of action by which mutant fibulin-3 causes ML. Transitioning these hits into mouse models of ML5,6 could serve as an important proof of principle for determining the therapeutic strategy that could benefit ML patients (i.e., enhancement of “WT-like” R345W fibulin-3 secretion or selectively reducing R345W fibulin-3 secretion and targeting it for degradation).
In conclusion, we have demonstrated that fusion of eGLuc2 to the C-terminus of fibulin-3 serves as a sensitive means to identify compounds that alter the cellular secretion of fibulin-3. We identified a set of interesting compounds that have been validated by using untagged forms of fibulin-3 in a physiologically relevant cell line (ARPE19). By using this cell-based assay to screen the LOPAC library, we have identified a compound (ARP 101) that appears to be relatively selective for reducing fibulin-3 secretion (with no preference for WT or R345W) while selectively lowering mutant fibulin-3 intracellular levels, as well as a compound (PMA) that selectively reduces R345W secretion while having only slight effects on WT fibulin-3 secretion. We anticipate that these compounds (or derivations thereof) will be useful for understanding and potentially treating fibulin-3–related diseases, including ML, and the fibulin-3–dependent gliomas.8,9
Footnotes
Acknowledgements
The authors thank R. Luke Wiseman and Joel Buxbaum for useful discussions. They also thank Lusine Abgaryan for her technical assistance with the HTS machinery.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Lita Annenberg Hazen Foundation, the Skaggs Institute for Chemical Biology, Grant UL1 RR025774 from the Scripps Translational Science Institute (J.W.K.), Grant AG018917 from the National Institutes of Health (J.W.K.), and Grant U54 MH0845121 from the National Institutes of Health (H.R.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.