
Abstract
Fragment screening, an emerging approach for hit finding in drug discovery, has recently been proven effective by its first approved drug, vemurafenib, for cancer treatment. Techniques such as nuclear magnetic resonance, surface plasmon resonance, and isothemal titration calorimetry, with their own pros and cons, have been employed for screening fragment libraries. As an alternative approach, screening based on high-performance liquid chromatography separation has been developed. In this work, we present weak affinity LC/MS as a method to screen fragments under high-throughput conditions. Affinity-based capillary columns with immobilized thrombin were used to screen a collection of 590 compounds from a fragment library. The collection was divided into 11 mixtures (each containing 35 to 65 fragments) and screened by MS detection. The primary screening was performed in <4 h (corresponding to >3500 fragments per day). Thirty hits were defined, which subsequently entered a secondary screening using an active site-blocked thrombin column for confirmation of specificity. One hit showed selective binding to thrombin with an estimated dissociation constant (KD) in the 0.1 mM range. This study shows that affinity LC/MS is characterized by high throughput, ease of operation, and low consumption of target and fragments, and therefore it promises to be a valuable method for fragment screening.
Introduction
Introduction in the 1990s of the concept of fragment screening opened up for a new era in drug discovery. 1 Fragment-based drug design is based on finding small organic fragment molecules that bind weakly to the target in the mM to µM range. By merging, linking or growing fragments, high-affinity drug leads can be generated. A number of published reviews emphasize the importance of this area.2–6 Recently, the first drug based on fragment screening was approved under the brand name of Zelboraf (vemurafenib). This drug is used to treat late-stage metastatic melanoma by blocking B-RAF kinase activity.7,8 Also, transient binding drugs can take advantage of fragment-screening approaches. 9
Fragments (molecular weight [MW] of typically 150–300 Da) generally bind with low-affinity setting requirements on procedures to be used for screening. Issues need to be addressed, including throughput and the consumption of protein target and fragments, as a high concentration of fragments (up to 1 mM) is used in most techniques to detect fragment-protein binding. Another consideration is the specificity of the method and the ability to detect the fragment hits to minimize false-positives. Several methods are currently available, with their inherent pros and cons, which can be used in fragment-screening procedures. These include X-ray crystallography, 10 nuclear magnetic resonance (NMR), 11 surface plasmon resonance (SPR), 12 isothermal titration calorimetry (ITC), 13 affinity selection–mass spectrometry (AS-MS), 14 and functional biochemical assays. In addition, virtual screening procedures have been used with varying success to select potential fragments to be experimentally tested. 15 Comparison of methods, such as NMR versus SPR, has been performed recently. 16
Since the introduction of weak affinity chromatography (WAC)17,18 in which weakly binding monoclonal antibodies were used to separate similar carbohydrate antigens, this technique has been applied to the separation and characterization of complex mixtures. WAC has been evaluated as a tool in drug discovery for high-throughput analysis of drugs binding to albumin,19,20 and a recent study demonstrated the screening of a small collection of fragments on trypsin and thrombin. 21
This work represents the first study of high-throughput fragment screening of a commercial library using WAC technology in which 590 fragments were screened toward the serine protease thrombin. Thrombin is a well-established target for anticoagulation 22 in the pharmaceutical industry and was selected to be used as a model protein. The fragment screening was performed on a commercial high-performance liquid chromatography (HPLC)/mass spectrometry (LC/MS) platform in which thrombin was immobilized in sub-milligram amounts in capillary columns. The technique did not require high concentrations of fragments to detect transient binding, and therefore the consumption of fragments was low (in the nanogram range). Throughput was high, and the 590-compound library was analyzed in less than 4 h. Based on this study, we are convinced that fragment screening by affinity LC/MS will be a valuable and complementary tool in early drug discovery.
Materials and Methods
Chemicals
All chemicals were of analytical grade or higher and purchased from Sigma Aldrich (St. Louis, MO), if not otherwise stated. Human α-thrombin (2400 IU/mg) was a gift from Octapharma (Stockholm, Sweden). D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone (PPACK) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Fragment Library
A library of 3200 fragments (PO6300-FBL-3200) was purchased from TimTec LLC (Newark, DE). The MW of the fragments in the library ranged from 75 to 289 Da, with an average of 216 Da; clogP, as provided by the supplier, ranged from −4.24 to 2.52. The number of heavy atoms was between 5 and 21.
The original library was scaled down to a collection of 590 fragments for the experimental screening. This was done by splitting the 3200 fragments into three groups based on their expected charge in electrospray ionization. Fragments with carboxyl or hydroxyl functionalities were assumed to be negatively charged and constituted one group of 483 compounds, whereas fragments containing nitrogen were placed in another group assumed to be positively charged and including 1952 compounds. Fragments that contained both types of the above-mentioned functionalities were placed in a separate group of 765 compounds. Fragment subgroups of 35 to 64 members were then arbitrarily composed within each group by selecting fragments that covered the MW range of the library using the random selection option in Maestro (v.9.1; Schrödinger Suite 2010, Schrödinger LLC, New York, NY). Compounds with identical MW were placed in separate subgroups to distinguish them in the MS analysis. This splitting resulted in three subgroups with an expected negative charge (107 compounds in total), six subgroups with an expected positive charge (384 compounds), and two subgroups that could be both positive and negative (99 compounds). The resulting selection of 590 fragments had an MW range from 75 to 288, an average MW of 212, clogP from −3.11 to 2.52, and number of heavy atoms from 5 to 21.
The TimTec fragments were delivered dissolved in DMSO with a concentration of 1.5 mM. Subgroup mixtures were prepared by mixing equal volumes of the selected fragments without further dilution. The six positive mixtures contained 65 fragments each, including 3- aminobenzamidine (3-ABA) or 4-aminobenzamidine (4-ABA), which served as reference ions for the MS detection. The concentration of each fragment in these mixtures was 0.023 mM. The three negative mixtures contained 35, 36, and 36 compounds, resulting in a concentration of 0.043 and 0.042 mM of each fragment. Because of poor ionization of benzamidines in the negative mode, these reference binders were not included in negative mixtures. The two mixtures that were analyzed in both positive and negative modes contained 50 and 51 compounds at a concentration of 0.030 and 0.029 mM of each fragment, including either 3-ABA or 4-ABA as reference ions.
Thrombin Affinity Capillary Columns
Porous and spherical silica particles (Kromasil, Eka Chemicals, Bohus, Sweden), in average 5 µm in diameter, 300 Å in pore size, and 100 m2/g in surface area, were used as chromatographic support. Diol functionalities were introduced onto the silica surface by derivatization according to previously published methods. 23 The resulting diol silica particles were packed into capillary columns (35 × 0.5 mm) by Agilent Technologies (Waldbronn, Germany).
Thrombin was then immobilized in situ onto the diol silica support particle by reductive amination as described previously. 24 The process was performed on an Agilent 1200 HPLC system with thermostated column and sample tray (Agilent Technologies). The diol silica was first oxidized into aldehyde silica with 10 × 20 µL injections of periodic acid (100 mg/mL in water) at 22 °C. The flow was stopped for 10 min after each injection. The procedure proceeded with a coupling step in which the aldehyde silica was reacted with a solution containing thrombin (8.7 mg/mL) and sodium cyanoborohydride (10 mg/mL) in sodium phosphate buffer (100 mM, pH 7.0) by 10 × 10 µL injections using a reaction time of 60 min after each injection. The temperature was kept at 12 °C. The preparation of the column material was finalized by deactivation of remaining aldehyde groups with ethanolamine (6.7 mg/mL), using sodium cyanoborohydride as a reducing agent and the same procedure as the above coupling step for thrombin. Two thrombin columns were prepared: column 1 and column 2. Column 1 was used in primary screening and column 2 in secondary screening.
Primary Fragment Screening
The Agilent capillary 1200 HPLC system was used as the analytical platform together with thrombin column 1. The system was controlled, and data were acquired by LC/MS ChemStation software (Rev. B.04.01 SP1; Agilent Technologies). Injection volume was 0.4 µL, and flow rate was 14 µL/min. The column temperature was set at 22 °C. The mobile phase for this screening step was 20 mM ammonium acetate, pH 7.0 (AmAc). The HPLC instrument was equipped with both an ultraviolet (UV) diode array detector and a single quadrupole mass detector (Agilent Technologies) that was run in the selected ion monitoring (SIM) acquisition mode. The mass spectrometer was an Agilent 6130 series quadrupole LC/MS that can detect a mass (m/z) range 2 to 3000; the mass accuracy is ±0.13 u within the calibrated mass range in scan mode. Ionization was carried out with an atmospheric electrospray ionization interface that employed a nebulizer pressure of 10 psig and a capillary voltage of 3000 V in the positive phase and 2200 V in negative phase. The ionization phase was set according to the expected charge of fragments in each subgroup, either negative or positive, or both. Drying gas (N2) had a temperature of 250 °C and a flow of 7 L/min. Fragmentor voltage was 90 V, and the quadrupole temperature was 100 °C. Analysis time for each injection was 20 min. Target masses for SIM detection were selected to match each fragment mixture component, and up to 65 signals were collected in a single run.
The 11 different fragment mixtures were analyzed on column 1 in one primary screening campaign. Blank samples of pure DMSO were run at identical conditions as the fragment mixtures to check for ions originating from the sample solvent. About 10% of the fragments were not detected by MS or UV when analyzed in mixtures. These compounds were reanalyzed individually on column 1. Samples were prepared by diluting the original solution (1.5 mM in DMSO) with water to achieve the same concentration as they had in the mixtures. The analytical conditions were identical to the analysis of the mixtures except for the selection of target ions that were adjusted for each sample to cover only the target ions of interest in both positive and negative phases. The analytical cycle in this case was extended to 40 min.
Fragments with the highest retention from the primary screening were considered as tentative hits, and the 30 most retained were selected for the secondary screening. These tentative hits were reanalyzed as single solutions on column 1 to compare the retention time between analyses in mixture and in single solution. The preparation and analytical conditions of these samples were the same as above.
Secondary Fragment Screening
The selected 30 tentative hits from the primary screening were subjected to a secondary screening on thrombin column 2. The fragments were injected individually at the same concentration as they had been in mixtures. Samples were prepared by dilution of the original solutions with water. The analytical conditions were identical to primary screening except that the MS ionization was performed on both positive and negative phases and only ions of the fragment of interest were acquired. In the secondary screening, analyses were also performed in 10 mM sodium phosphate, 150 mM NaCl, pH 7.4 (phosphate-buffered saline [PBS]), and monitored by UV detector at 254 ± 8 nm using a reference wavelength of 360 ± 50 nm. The PBS was used to achieve an environment resembling physiological conditions, although the compound affinity ranking in AmAc and PBS correlated well. 21
The thrombin in column 2 was then inactivated with the known thrombin inhibitor PPACK, which reacts with thrombin in its active site and forms covalent bonds to Ser195 and His57, residues that contribute to the thrombin catalytic triad. 25 The inhibition procedure was performed on the Agilent capillary 1200 HPLC system. An injection of 8 µL of PPACK (5 mg/mL in PBS) was introduced to thrombin column 2 at a flow rate of 10 µL/min. The mobile phase flow was stopped for 6 min after each PPACK injection, and thereafter the retention time of 4-ABA was checked. The procedure was repeated four times until the retention time of 4-ABA did not decrease further. The 30 tentative hits were run individually on column 2 before and after inactivation of thrombin by PPACK in both AmAc and PBS.
Data Analysis
The retention time (tR) of a fragment was measured by the peak apex of corresponding extracted ion chromatograms. The adjusted retention time (t′R) was the primary ranking parameter, and t′R = tR – to, where to is the void time as measured by DMSO. Negative values of t′R were reported as zero.
The specific retention time (tspec) of each fragment was calculated from its t′R on column 2 before inactivation subtracted with t′R after inactivation. The true hits were defined as fragments that had tspec >30% of its t′R. The dissociation constant (KD) was calculated by the equation 23
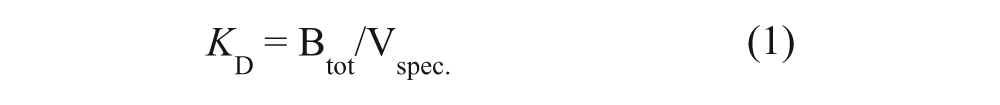
Btot is the total amount in moles of active target estimated by frontal chromatography with 3-ABA, and Vspec is the specific retention volume; Vspec = tspec × flow rate. It is important to point out that equation 1 is an approximation because it is valid only under linear adsorption isotherm ([L] << KD; [L] is the fragment concentration).
Frontal Affinity Chromatography
Frontal affinity chromatography 26 was carried out on column 1 and 2 with PBS as a mobile phase at a flow rate of 14 µL/min, using the HPLC 1200 platform equipped with a photodiode array detector (Agilent Technologies). UV detection was performed at 254 ± 8 nm using a reference wavelength of 360 ± 50 nm. Data acquisition was performed by ChemStation software (Rev. B.04.01 SP1; Agilent Technologies). The compound 3-ABA was injected on the thermostated column (22 °C) with a volume of 80 µL and at seven concentrations ranging from 0.02 to 0.4 mM (0.02, 0.03, 0.06, 0.12, 0.2, 0.3, 0.4 mM). The inflection point of the resulting breakthrough curves was determined by the apex of the first derivative of the chromatogram. The amount of 3-ABA that saturated the thrombin column at each concentration was calculated from the breakthrough time of 3-ABA subtracted with the breakthrough time of DMSO. A saturation binding curve was constructed from the number of moles of 3-ABA that bound to the column at each concentration versus the concentration of 3-ABA.
The number of specific binding sites (Btot) on column 1 and 2 was estimated by global nonlinear regression of the obtained saturation binding curve using Graph Pad Prism (v. 5.02; GraphPad Software, La Jolla, CA). The global fitting model of total one-site and nonspecific binding was used, which assumes that the total interaction is the sum of specific as well as nonspecific interactions in which the specific interaction follows the Langmuir equation 27 :
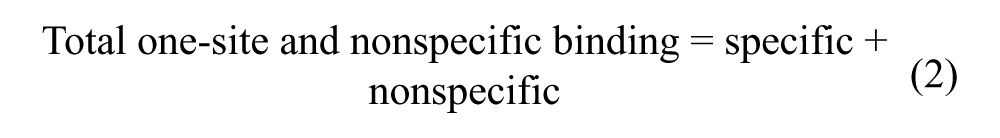
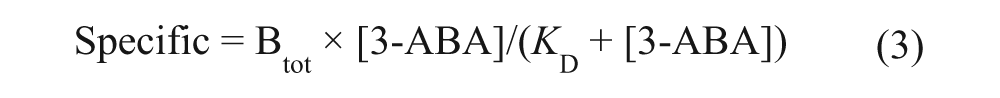
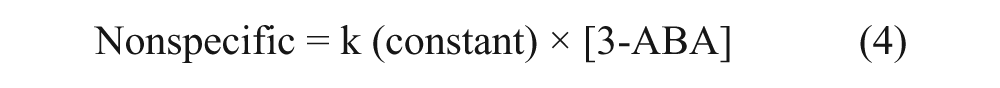
Frontal chromatography of 3-ABA was also performed on PPACK-inactivated column 2 employing identical conditions.
Pose Prediction by Docking
The confirmed hit (ST036785) and benzamidine (BZA) were docked into a thrombin crystal structure using the computational software package Schrödinger Suite 2010. The crystal structure of human thrombin (PDB ID: 1K22) 28 with resolution 1.93 Å was downloaded from the Protein Data Bank (www.rcsb.org) and employed to predict the binding pose of the fragments. All computational parameters were used as default, if not otherwise stated.
The fragments were first treated by LigPrep (v.2.4) and then docked into the active site of processed thrombin structure using Glide (v.5.6) in extra precision mode with the use of an expanded sampling scheme. When there were more than one tautomer or conformation generated from the LigPrep treatment, the binding pose that possesses the most negative Glide Emodel value was considered as most relevant.
Results
Overview of Fragment Screening
The screening procedure is illustrated in Figure 1 , and an overview of the results from the primary screening is shown in Figure 2 . In this step, 590 fragments, divided into 11 mixtures, were analyzed on column 1. Mixtures consisted of 35 to 65 compounds at individual concentrations ranging from 0.023 to 0.043 mM. The data collection time was 20 min per injection, and 90% (n = 530) of the fragments were detected in this time frame. This speed allowed a complete primary analysis of the library of 590 compounds in less than 4 h. The reanalyses of single solutions of fragments, which were not detected in the first run, needed 40 min for each fragment, and 6% more fragments (n = 36) were detected in this way. The thrombin consumption per capillary column was 0.87 mg, and the fragment consumption was in the range of pmol (9–17 pmol, or 0.7–4.9 ng). The retention time of the internal controls (BZA, 3-ABA, and 4-ABA in PBS) on column 2 was stable after more than 150 injections during 1 wk.
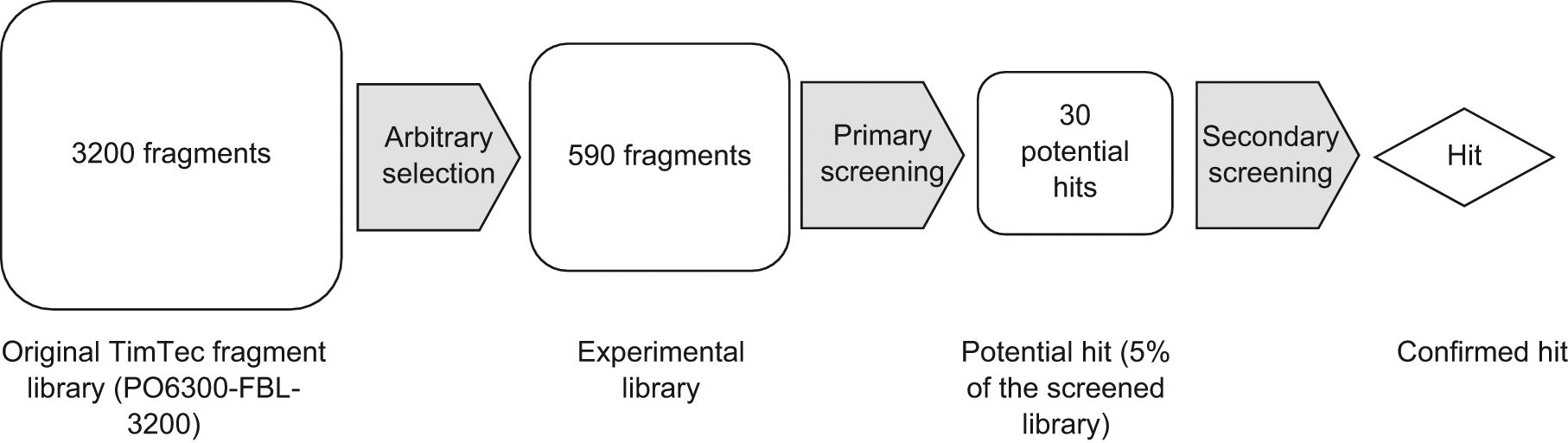
Procedure to screen the fragment library to select a hit.
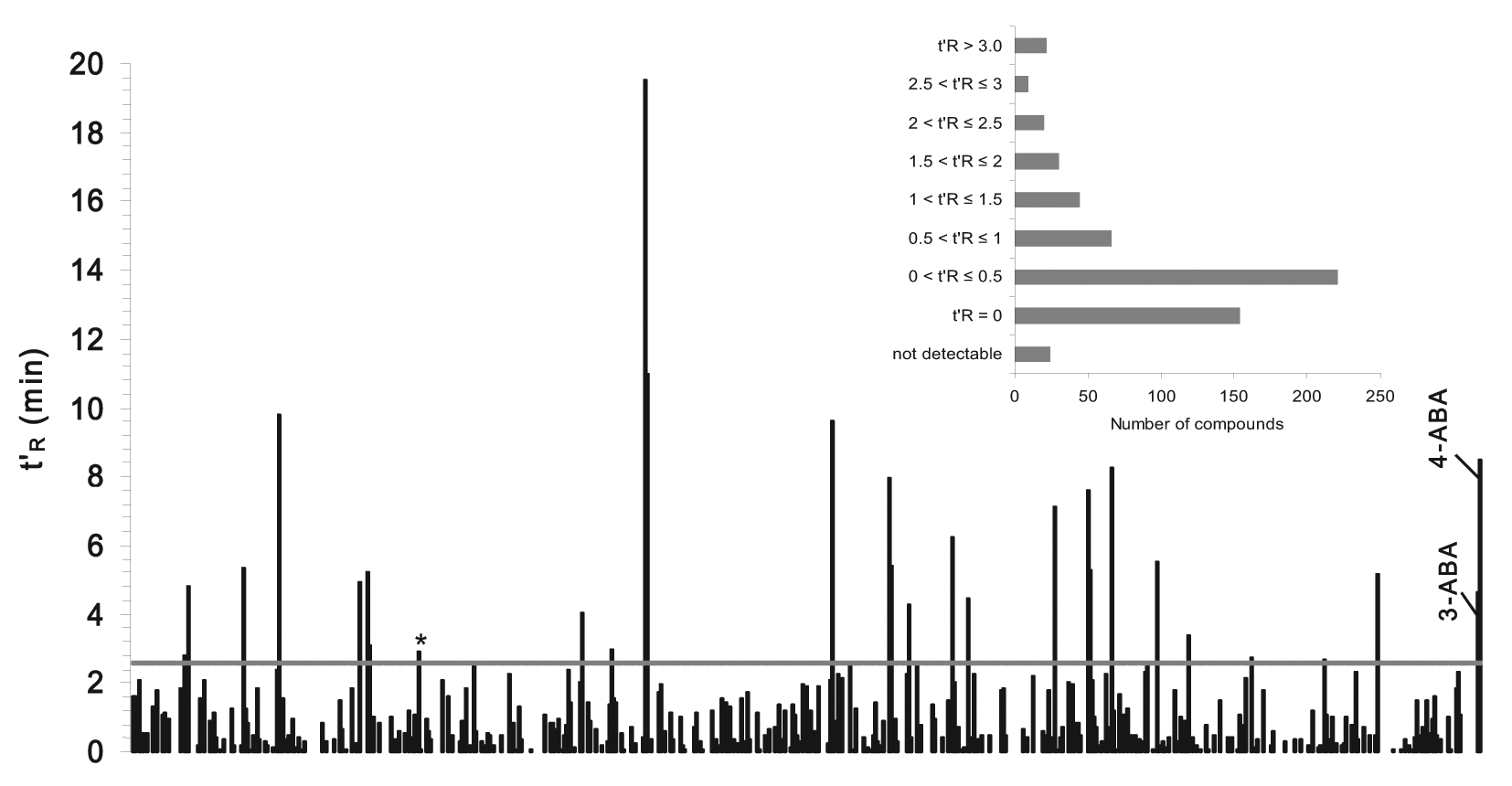
Detail of t′R of 566 compounds and two reference binders. The horizontal line indicates the cutoff for potential hits (t′R = 2.54 min). The compound marked with a star was the actual hit after confirmation. A more quantitative description of the library is depicted in the inset.
As depicted in Figure 2 , a significant number (n = 154; 26%) of the fragments eluted in the front with t′R = 0, and 221 compounds (37% of all fragments) had a t′R of less than 0.5 min. The number of fragments with longer retention times reduced gradually, and there was no natural border to separate hits from nonretained fragments. The expected hit rate in a fragment-screening campaign is about 5%; therefore, the cutoff limit in our study was set to include the 5% of most retained fragments. This cutoff produced 30 compounds, with a lowest adjusted retention time (t′R) of 2.54 min and highest of 19.51 min. These potential hits were included in the secondary screening.
The secondary screening was done with individual analyses of the 30 potential hits on thrombin column 2. Identical samples were analyzed on the active column and then reanalyzed when thrombin in the column had been inactivated by PPACK. Figure 3 shows chromatograms of 3-ABA in AmAc on column 2 before and after thrombin was inactivated. The known thrombin binders, BZA, 3-ABA, and 4-ABA, were analyzed in the same series with the potential hits and showed significant reduction in retention (tspec contributed to >80% of t′R in both AmAc and PBS mobile phases) when analyzed on the PPACK-inhibited column. Among the selected 30 potential hits, only one fragment (ST036785) showed a significant reduction of retention time on the PPACK-inhibited column in both PBS and AmAc (tspec contributed of 78% and 48% of t′R in PBS and in AmAc, respectively). The remaining 29 potential hits did not show enough reduction in retention time (<30% of its t′R) after PPACK inhibition of thrombin and were consequently considered as promiscuous binders. Estimated dissociation constant (KD) of ST036785 in PBS was calculated by equation 1 to be 124 µM, whereas the obtained KD values of the controls were 258 µM for benzamidine, 168 µM for 3-ABA, and 55 µM for 4-ABA, all in PBS. Böhm and others 29 reported inhibition constant (Ki) values by enzyme functional assay for BZA, 3-ABA, and 4-ABA toward human thrombin to be 250, 170, and 34 µM, respectively.
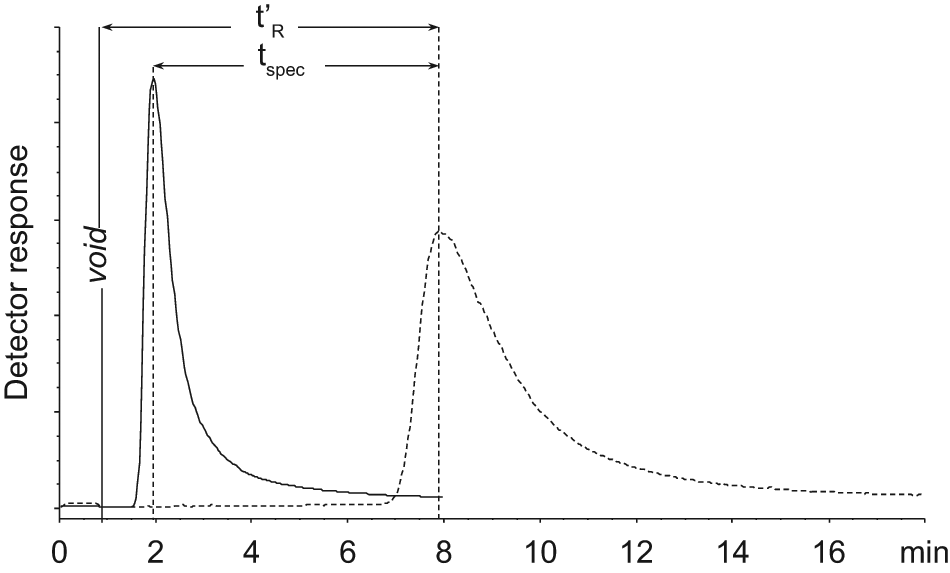
Total ion current of 3-ABA on active (broken line) and PPACK-inhibited (solid line) affinity column 2 with ammonium acetate 20 mM, pH 7.0 as mobile phase.
MS Detection
The primary screening of the 590 fragments was performed by analyzing fragment mixtures applying the SIM mode of the mass spectrometer. Ions were collected in single or mixed-polarity phase (negative or positive, or both phases) corresponding to the combination of fragments in the different mixtures. Among the 590 screened fragments, 10% (n = 60) were not detected. When reanalyzing these compounds individually, while reducing the DMSO concentration to 1.5% to 2.8%, resetting the MS to alternating positive and negative phases and collecting only 2 ions [M+H]+ and [M-H]−, 36 more fragments were monitored. The final number of ions that was impossible to detect was 4% (n = 24). Those compounds were also not detectable on the PPACK-inhibited column.
DMSO affected the detection of fragments by introducing molecular ions in analyses performed in positive phase. Adducts such as [M+H]+, [2M+H]+ with m/z of 79 and 157, respectively, were found, as well as ions obtained by fragmentation having identical m/z values as some analyzed fragments. The phenomenon is illustrated in Figure 4 , where the ion with m/z 212 is present as two peaks. The earlier peak in the chromatogram was later confirmed as originating from DMSO. Among the 24 fragments that were impossible to detect, 9 compounds have predicted m/z values coinciding with masses produced by DMSO, and they were suspected to be covered by the DMSO signal. The DMSO interference did not affect compounds that produced ions in the negative phase because DMSO generated no negatively charged ions.
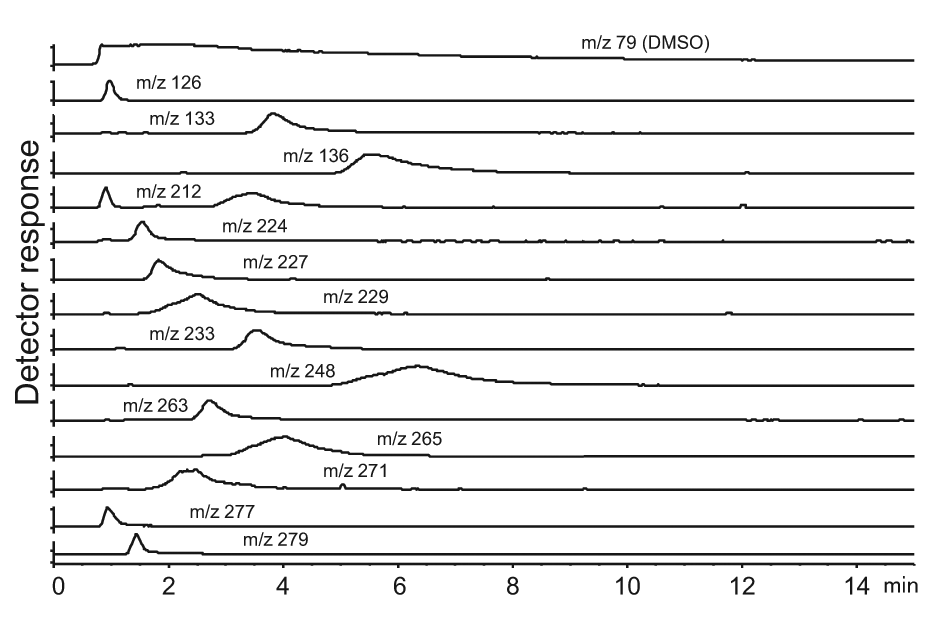
An example of 14 extractions of ion chromatograms. The ion with m/z 79 was the signal from DMSO.
Comparison of Fragments as Mixtures and as Singles
The chromatographic retention (t′R) of 30 fragments was determined in single solution and compared with the retention when analyzed as part of a mixture ( Fig. 5 ). Among the examined fragments, 24 had a longer retention time when analyzed as single solution. The highest variation was 1.78 min, equal to 29% of t′R of the fragment in single solution analysis. On the contrary, six fragments had similar or longer retention times when analyzed in mixture compared with single solution. The maximum fluctuation was in this case 0.63 min.
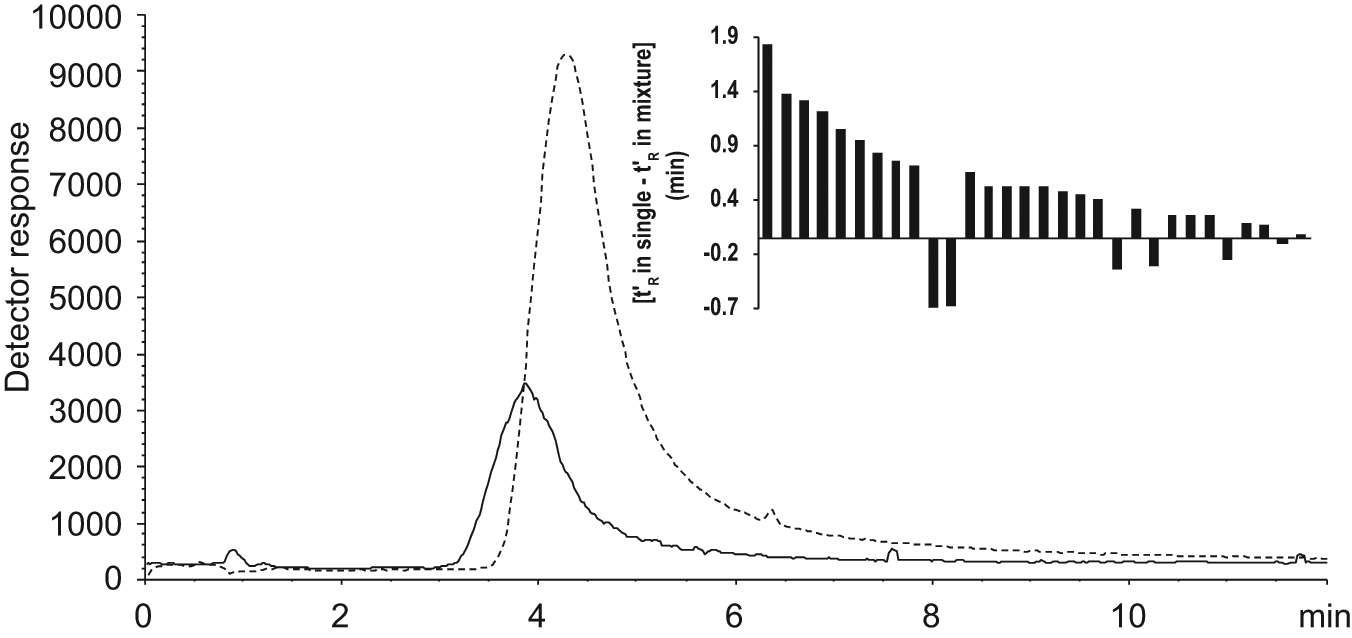
MS signals from compound ST036785 in mixture (solid line) and in single solution (broken line) on column 1 injected at the same concentration (0.023 mM). The inset shows the variation of retention of the examined 30 fragments.
Frontal Affinity Chromatography
Frontal affinity chromatography with 3-ABA in PBS was performed on column 1 and column 2 to estimate the amount of active thrombin (Btot) in the columns. The estimation of the nonspecific part of the interactions was determined experimentally by chromatography with the PPACK-inhibited thrombin column 2. Figure 6 depicts the fitting of saturation curves of 3-ABA to the model of one-site total and nonspecific binding (see the Materials and Methods section). When thrombin was inactivated by PPACK, the response curve turned into a straight line (R2 = 0.9997), which means the remaining interaction was only nonspecific and active sites were completely inhibited. Interestingly, the slope value (k) of the linear nonspecific interaction curve was 8.4 × 10−6 liter (inactivated column 2), which is very close to the estimated nonspecific binding from the global fitting on the active column that was 7.4 × 10−6 liter. Because of the different curve forms (saturation binding vs linear) of the specific and nonspecific interaction, the relative contribution from the nonspecific interaction increases with sample concentrations, as can be seen in Figure 6 . For example, when the 3-ABA concentration was 400 µM, the nonspecific interaction reached 49% of the total retention, whereas the nonspecific contribution was only 28% at 60 µM 3-ABA.
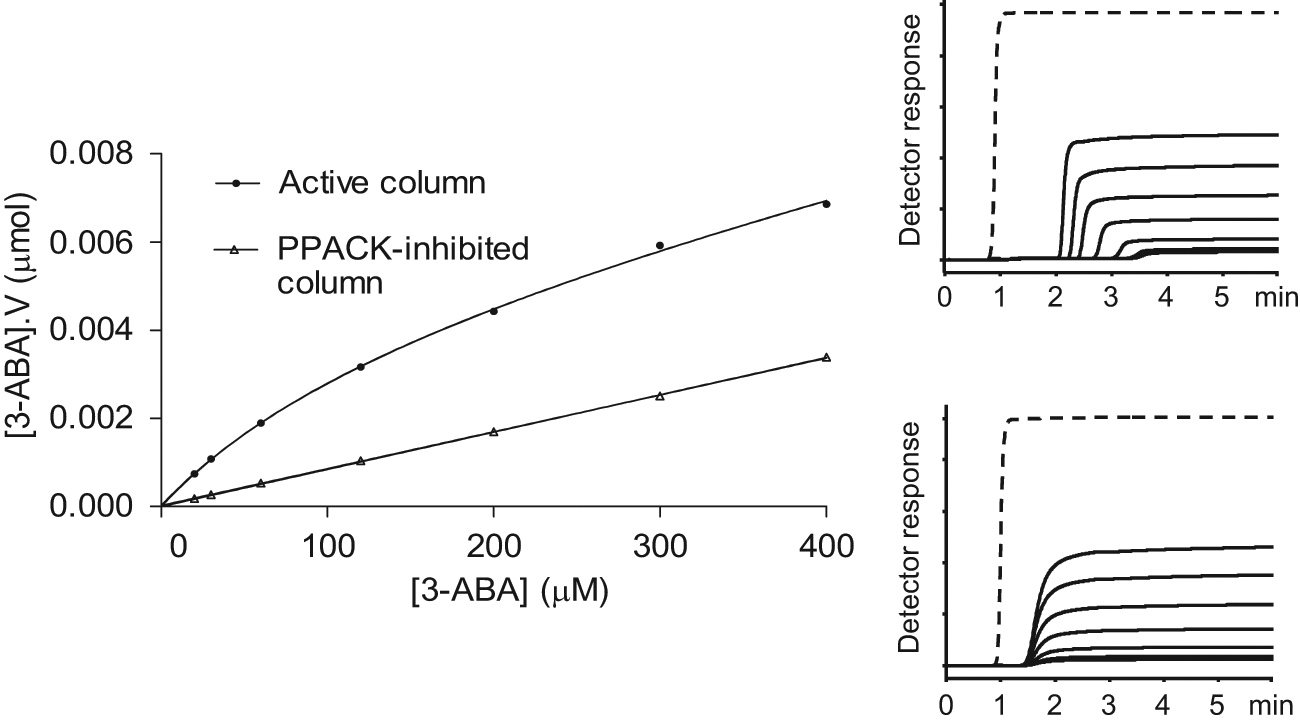
Frontal affinity chromatography data of 3-ABA in PBS from active and PPACK-inhibited thrombin capillary column 2. V is the adjusted retention volume and calculated by subtraction of the void volume from the observed volume. Figures on the right are corresponding frontal chromatograms on the active (upper) and PPACK-inhibited (lower) column. The broken lines in chromatograms are signals from the void marker DMSO.
Table 1 gives data on Btot, KD, and the nonspecific interaction of 3-ABA from frontal and zonal chromatography analyses. KD values of the active columns were extracted from the saturation binding curves using a global fitting procedure. The estimated KD values of 3-ABA from frontal chromatography on the two columns were 170 and 177 µM, respectively, which is close to the published value Ki of 170 µM. 29 By using the value of nonspecific binding from the PPACK inhibited column 2 in the fitting of the saturation binding curve, a KD value of 151 µM was obtained. The KD value of 3-ABA obtained from zonal chromatography on column 2 also agreed with the literature value. 29 It was calculated by equation 1 and based on Btot derived from global fitting including data from the PPACK-inhibited column.
Description of Functional Parameters for 3-ABA and Thrombin by Zonal Analysis and Frontal Analysis and Compared with Published Data a

Values are reported as mean ± SD (n = 7 concentrations), with SD obtained from the simulation. Frontal data from column 2 is given with and without PPACK adjustment. NA = nonapplicable.
Btot value from frontal analysis adjusted for PPACK was used to calculate KD.
Discussion
Selection Process: Primary and Secondary Fragment Screening
In the first round of fragment screening (primary), 30 hits were selected out of 590 fragments based on a threshold level of t′R = 2.54 min (5% of all 590 fragments), which corresponds to roughly three times the void time of the thrombin column and to a KD of approximately 0.14 mM. It is important to state that the figures on affinities should be regarded with caution, as no estimate of nonspecific binding is performed in the primary screening. It is further interesting to note that it is sufficient to run a single run of a fragment at one concentration to give directly an estimate of affinity and/or ranking of fragments.
Many fragments did not show any binding to target (26%), and most fragments (37%) showed an apparent affinity in the sub-mM range, close to the minimum affinity (approximately KD = 0.5 mM) detectable by the affinity LC/MS system. The window of affinity range that can be seen by the system is dependent on Btot and data collection time. In this study, the maximum affinity was in the range of 5 to 10 µM in KD. Analytes with even higher affinity will be severely retained and are typically not detected by the MS detector. This is generally no problem, as binders with higher affinities (KD < µM) are rarely present in a fragment library. The retention time in zonal affinity chromatography depends on the affinity of the analyte to target and can be adjusted by tuning the amount of immobilized active target (see equation 1). For example, to be able to see fragments typically binding in the mM to high µM range of KD, concentrations of active ligand should be in the mM range, as applied with column 1 and 2.
The 30 initial hits were evaluated in a secondary screening with a thrombin column (column 2) with higher Btot (46% higher than column 1), and total nonspecific interactions were estimated by blocking covalently the active sites of immobilized thrombin on column 2. The most selective hit was ST036785 (for structure, see Fig. 7 ), and the remaining 29 hits showed essentially no specific binding to the active site of thrombin.
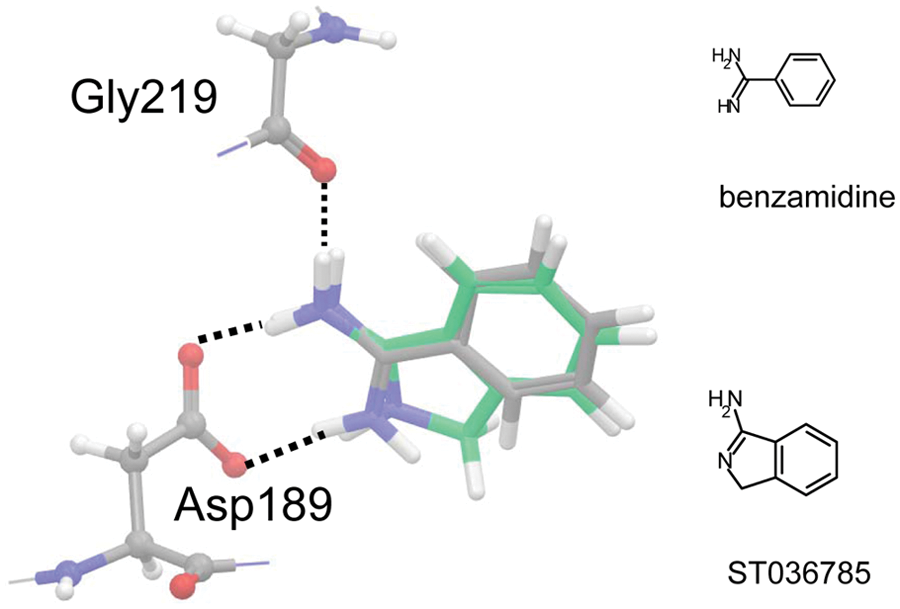
Predicted binding poses of ST036785 (green, large stick) and BZA (gray, large stick) into human thrombin (PDB ID 1K22). Amidino moieties were protonated in docked structures, and broken lines indicate positions of hydrogen bonds. The distance cutoff for hydrogen bond was 2.5 Å.
The well-known thrombin inhibitors BZA, 3-ABA, and 4-ABA were used as internal standards to monitor the condition of the thrombin columns during screening and storage. In addition, their specific binding to immobilized thrombin was measured during secondary screening, and they were well correlated with K i values obtained from enzymatic assays, 29 which further emphasizes the relevance in specific binding data with affinity LC/MS.
Mass Spectrometry Detection
Detection by MS offers the opportunity to follow the mass of each fragment, thereby allowing identification of single fragments even though chromatography peaks are not completely separated. This allows the use of mixtures where fragments with different MWs are run combined leading to drastically higher throughput (see the discussion below). In our study, we used a maximum of 65 fragments into one mixture that was analyzed as one sample. This maximum number depends on the sensitivity of MS, the concentration of each fragment, and the variety of different MWs of fragments in a library. Most fragments were detected (90% of 590) in this study when run in mixtures, but some (6% of 590) were detected only when run as individual compounds. Remaining substances (4% of 590) were not detected at all. The lack of signal from these compounds by MS may be due to difficulties in ionization in the presence of other fragment molecules and/or DMSO. It cannot be ruled out that some of these undetected fragments were retained by the thrombin column. A critical feature in the design of the mixtures is that different MWs should be distinguishable with at least a difference of 1 unit in m/z.
The commercial fragment library used in this study was composed of 3200 compounds, with MWs ranging from 75 to 289, and the most frequent MW was 221 (52 fragments). Consequently, in this case, the lowest possible number of mixtures for the whole library was 52, with 62 fragments in each mixture. Moreover, fragments that contain atoms with more than one stable isotope, such as Cl and Br, produce more than one m/z value in the MS detector and therefore further reduce the possible number of fragments to be included in a mixture.
DMSO is the preferred solvent of choice for molecular libraries because of its ability to dissolve a wide variety of compounds, its low toxicity, and its miscibility with water. 30 In our study, DMSO was also used as a void marker. However, DMSO can cause problems for MS detection by ion suppression and introducing ions obscuring detection of certain fragments as indicated previously. One way to reduce the effect of DMSO is to dilute samples with water to reduce the concentration. As the concentration of each fragment in the commercial library used in this study was rather low (1.5 mM in DMSO), mixtures of 35 to 65 compounds were used without further dilution to achieve a reasonable concentration of each fragment, and the DMSO concentration was 100% in sample mixtures. By diluting single samples with water down to a few percentage of DMSO, the detection of some fragments was facilitated. It is therefore advisable to have fragment libraries with as high concentration as possible, for example, in the 10 to 100 mM range. Another option is to find a solvent that is suitable for storage of the library, which does not produce a strong signal in the MS.
Regarding the effect of DMSO on binding, we assume that it has only a little influence on the ranking of fragment binders. In the chromatographic procedure, DMSO is not retained and eluted immediately at the front. Therefore, the retained fragment interacts with the immobilized protein mainly in the mobile phase environment. In our case, where DMSO was not diluted, there was still the presence of some solvent during an extended time of elution ( Fig. 4 ), which again emphasizes the need of a high-concentration compound library.
The extracted ion chromatogram (EIC) of a fragment mixture is necessary to detect single ions. Figure 4 shows a few examples of EICs of single ions as distinguishable peaks. As seen in Figure 4 , the DMSO peak is extremely large, and the influence on detection of some fragments is therefore difficult to avoid.
In comparison with AS-MS, especially size-exclusion procedures in homogeneous assays, affinity LC/MS offers several advantages: In fragment screening, weak binders are frequently studied, which are difficult to handle in size-exclusion chromatography because of very rapid off rates of fragments from protein target. Another key benefit is that we follow the binding of fragment directly by its retention, whereas in AS-MS it is a more indirect and complex process, leading to lower throughput, especially when considering the number of samples needed to receive affinity data. A disadvantage with affinity LC/MS is the possible ion suppression from DMSO. However, this effect is limited for retained fragment binders as they are moving out of the DMSO environment during chromatography.
Throughput of Operation
One of the unique features with affinity LC/MS, as a fragment-screening technique, is the ability to screen fragments in mixtures in each experiment and thereby drastically shorten the time of analysis of a fragment library. The throughput of screening depends on the number of fragments that can be analyzed in each experiment and the elution time of the fragments. Increasing the flow rate could be a simple option to enhance throughput, but it is limited by a high back-pressure and mass transfer limitations of the affinity column.
In this work, the entire 590-member fragment library was screened in less than 4 h. This corresponds to a throughput of 3000 to 4000 fragments per 24 h. The limiting factor is the processing of data and sample preparation, which will extend the total analysis time considerably. However, when automation is applicable to sample preparation and online data treatment is available, it can be predicted that the zonal affinity LC/MS could handle a normal fragment library of thousands of fragments in a matter of days. This speed is considerably faster than NMR 11 and ITC 13 and comparable with parallel SPR. 31
When using mixtures to increase throughput, there has been a concern about competition among the fragments for the binding sites. This can lead to the situation that individual fragments are not run under a strict linear adsorption isotherm causing reduced retention. Equation 1 is valid under the assumption of linear conditions with the criterion that KD >> [L], which means that deviations from linearity are more pronounced for tighter binders. As we noticed during screening ( Fig. 5 ), most fragments were more retained when introduced alone as compared with mixtures, which indicates competition for the same binding site. A few showed the reverse situation, probably caused by system variation. Therefore, affinity calculations from analyzing mixtures should be regarded as approximate. The affinity of the most retained compounds will consequently be slightly underestimated, but the screening will still indicate the ranking of evaluated fragments. This is usually sufficient for the first selection of interesting fragments.
Consumption of Target Protein and Fragments
The consumption of protein target should be kept as small as possible because it often is available in scarce amounts (<1–5 mg). By miniaturization into capillary columns, the amount of thrombin used in this study was in the sub-milligram range. Further down-sizing can decrease consumption even further, for example, by performing fragment screening in a chip format, which may reduce the required protein target to less than 0.1 mg. The consumption of target is also correlated to the lifetime of the immobilized column and how many experiments that can be conducted before the column needs to be replaced. The amount of consumed fragments in the study was low (nanogram range) because affinity is obtained from a few injections of fragments at low concentrations. When compared with other techniques, the amounts of target and fragments used by WAC are comparable with SPR and lower than for NMR.
Specificity of Operation
In screening of fragments, the specific binding to active sites on the target is usually in the mM to µM range in KD, which can be obscured by nonspecific interactions that are numerous but of very low affinity and therefore contribute significantly to retention in an affinity LC/MS system. This will result in a number of false-positives and may be the main cause of a low resulting hit rate (0.2%). The solution to the problem is either to include more hits to the secondary screening or to identify and eliminate from the library compounds with nonspecific interaction to the matrix before screening.
The cause of nonspecific binding is several-fold. Apart from binding to matrix and linkages between support surface and protein target, exposed areas of the protein outside active sites can offer binding that varies in binding strength. Matrices are designed to minimize nonspecific interactions (e.g., in the case of silica as a support, a diol layer is silanized on the support surface). However, apparent nonspecific binding to target surface protein can be of great interest, for example, when searching for allosteric drugs that bind to other areas other than the active sites or domains of the protein.
The extent of nonspecificity is measured by monitoring the retention in a control gel. Frequently, reference matrices with no or irrelevant immobilized ligand are used to estimate nonspecific binding. The major problem with this approach is that it may not be representative of the system under study. For example, the surface of the support in this study is covered by high amounts of thrombin creating an environment of its own. Therefore, an ideal control for nonspecific binding could be the immobilized target devoid of its active binding sites. When we study proteases, inhibitors are available that block covalently the active site of the protein. The inhibitors in themselves can therefore result in nonspecific binding, but these effects are considered to be negligible.
Of 30 fragments that showed the highest retention in the 590-member library (arbitrarily selected from the commercial library), only one (ST036785) showed a high selectivity in binding (78% in PBS). Many hits showed indiscriminate binding ( Fig. 2 ), which emphasizes the need for confirmation of specific binding. In our study, the blocking of the active site of thrombin was important to extract such data. We used the covalent blocker PPACK to inactivate the binding site of immobilized thrombin. PPACK mimics the binding pose of thrombin substrates and forms covalent bonds to residues Ser195 and His57, 25 permanently preventing analytes from reaching the binding pocket of thrombin by mainly sterical hindrance. The reference inhibitors (BZA, 3-ABA, and 4-ABA) all exhibited significant selectivity in binding (>80%), as is shown, for example, for 3-ABA in Figure 3 . However, some remaining binding is evident after PPACK inhibition, at least when compared with DMSO as a void volume control ( Fig. 3 ). The same picture is demonstrated when performing frontal analysis on 3-ABA, where the presence of nonspecific interactions is obvious on the PPACK-inhibited column ( Fig. 6 ). In the estimate of Btot by frontal analysis to achieve proper estimates of KD by zonal analysis, it is important to estimate the contribution from nonspecific binding. This was achieved by using the global fitting models for binding and by subtracting the nonspecific binding determined by PPACK-inhibited thrombin column (see the results in Table 1 ). As the relative contribution of nonspecific binding increases with fragment concentration, it is important to highlight that weak affinity LC/MS screening is performed at low fragment concentrations (<43 µM). This contrasts, for example, with SPR, in which high concentrations of fragments have to be used to detect the affinity of low-affinity binders.
Characteristics of Hit
The Glide docking program was employed to predict the binding pose of the confirmed hit (ST036785). The amidine moiety has long been considered a mimic of the arginine part of the substrate of thrombin, 32 and the presence of this functionality in a fragment facilitates specific binding. The found hit (ST036785) having its amidine functionality partly incorporated in a ring structure possessed an expected binding pose identical to BZA ( Fig. 7 ). As shown, both compounds formed bifurcated hydrogen bonds to the two carboxylate oxygen atoms of Asp189, and they further participated in another hydrogen bond to the oxygen atom of Gly219, which is a typical pose of the benzamidino group when binding into the S1 pocket of the thrombin active site. 28
Conclusions
Fragment screening complements high-throughput screening procedures and is now becoming a recognized strategy for drug discovery. The value of this approach comes from the fact that from a small library of simple fragments, a number of weak but rather specific binders can be selected and used as synthetic building blocks for new leads or drug candidates. The success of this technology is evidenced by the number of fragment-based substances that are currently in clinical trials 33 and the recent approval of the drug vemurafenib.7,8 Fragment screening faces a number of analytical challenges relating to its weak and often promiscuous binding of fragments to different sites on target. Therefore, several different biophysical methods, including NMR, SPR, thermal shift assay, and X-ray as well as computational docking procedures have been implemented to address these needs. This study introduces an addition to this arsenal of methods in which weak-affinity zonal chromatography, integrated with MS, was used for the first time to screen a protein target using a large fragment library. This affinity LC/MS platform shows a number of potential benefits, including high throughput, low consumption of target and fragments, ease and robustness of operation, and—last but not least—its ability to be performed on standard HPLC equipment. A unique feature of the affinity LC/MS approach to fragment screening is that it is based on separation of interacting molecules to a target. This will have important consequences; for example, mixtures of substances (including synthetic, natural, contaminants, and stereoisomers) can be monitored for binding. Affinity LC/MS promises to be a valuable and complementary tool for fragment screening, but major work still remains to be done in future studies to realize the full potential of the technology. This will include comparison with other methods, application to other important targets such as kinases and membrane proteins, and finally extraction of kinetic information from fragment binding to target.
Footnotes
Acknowledgements
The authors thank Göran Karlsson and Stefan Winge from Octapharma for providing thrombin. Linnaeus University and Agilent Technologies are acknowledged for providing financial and other support. We also thank Dr. Gerard Rozing for critically reviewing the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.