
Abstract
Microsomal prostaglandin E synthase-1 (mPGES-1) is the major enzyme catalyzing the isomerization of prostaglandin (PG) H2 to PGE2. Here we report the development of a robust and practical automated assay in a 384-well format for room temperature screening of mPGES-1 inhibitors with high precision and low reagent consumption. The assay should enable precise structure-activity relationship development. It uses acetonitrile as solvent for PGH2, FeCl2/citrate as stop reagent, and a short reaction time. Combined with high-precision liquid transfer and extensive mixing after addition of reactants, these properties let the assay reach Z′ > 0.7 and high reproducibility of inhibitor IC50 values. Thorough investigation of the quality of mixing in all liquid transfer steps proved crucial for reaching high-precision performance.
Abbreviations: mPGES-1 (microsomal prostaglandin E synthase-1); FRET (fluorescence resonance energy transfer); HTRF (homogeneous time-resolved fluorescence); PGH2 (prostaglandin H2); PGE2 (prostaglandin E2); SAR (structure-activity relationship); COX-2 (cyclooxygenase-2); GSH (glutathione); ALP (automated labware positioner)
Keywords
Introduction
Prostaglandin E2 (PGE2) is an important lipid metabolite with a wide range of physiological functions in mammals as well as pathophysiological roles (e.g., in cancer, inflammation, and pain).1,2 The biosynthesis of PGE2 from arachidonic acid involves two enzymatic steps: the rate-limiting and first step is cyclooxygenase (COX)–producing prostaglandin H2 (PGH2). The major inducible enzyme isomerizing PGH2 to PGE2 is microsomal prostaglandin E2 synthase-1 (mPGES-1). 1 Often upregulated in concert with COX-2, it is a potent catalyst with a KM for PGE2 in the low micromolar range. 3 In recent years, treatment with COX-2 selective inhibitors has been associated with increased cardiovascular risk. A number of studies in mouse models of hypertension and myocardial damage have indicated that inhibition of mPGES-1 could potentially confer lower cardiovascular side effects than inhibition of COX-2.1,4 This has spurred interest in mPGES-1 as a potential target for pharmaceutical intervention in several areas of disease 1 and highlighted need for a high-precision protocol for use in developing inhibitors of mPGES-1.
In vitro, PGH2 spontaneously and rapidly isomerizes to mainly PGE2 in parallel to its enzymatic conversion, 5 which in assay development demands a very short reaction time to keep background down. Massé and coworkers 6 addressed this, in an automated assay protocol for screening in 96-well microplates, by running the reaction at 0 °C and using the water-miscible organic solvent acetone to keep PGH2 stable until the start of the enzymatic reaction. This 13-step protocol, which used an enzyme immunoassay (EIA) for PGE2 quantification, was later simplified to fewer steps in a protocol that used homogeneous FRET-based PGE2 detection (HTRF). 7 These protocols stopped the reaction by converting remaining PGH2 to PGF2α. For the utility of this assay for high-precision SAR–driving inhibitor optimization, it was desired to improve aspects such as precision, robustness, and feasibility.
We report here the development and optimization of a robust automated assay in 384-well format for high-quality screening of mPGES-1 inhibitors at room temperature. The solvent for PGH2 and the reagent for stopping the reaction were changed compared with the earlier protocols. This change, together with meticulous optimization of liquid transfer and innovative labware handling, enabled high Z′, low interexperimental variation in IC50 for pharmacological mPGES-1 inhibition, and low reagent consumption. Specific attention to the liquid transfer revealed that careful investigation of the precision of mixing was crucial for assay quality, which should also be of general interest.
Materials and Methods
Materials
All chemicals were of pro analysi or of similar high quality. PGH2 (Larodan Fine Chemicals [Malmö, Sweden], shipped as 2 mg/mL in acetone) was diluted in acetonitrile (Fluka [Buchs, Switzerlnad] 00709, puris, over mol. sieve, ≤0.001% H2O), using dried glassware and equipment, and aliquoted in 15-mL argon- or nitrogen-filled polypropylene tubes (352096; Falcon Fischer Scientific, Gothenburg, Sweden), which were frozen on dry ice and stored at −80 °C for later use. Microsome preparations containing high mPGES-1 activity (~10 mg total microsomal protein/mL, 10 mM potassium phosphate [pH 6.8], 0.1 mM EDTA, 10% [v/v] glycerol, Complete Protease inhibitor [EDTA free; Roche, Basel, Switzerland, 1 836 153, 1 tablet/10 mL]) were produced according to a standard microsome preparation procedure 8 from SF9 cells expressing the human mPGES-1 gene (GenBank BC008280) by use of the baculo expression system (BaculoDirect; Invitrogen, Carlsbad, CA). mPGES-1 microsomes used as blank-rate controls in the determination of KM for PGH2 were heat inactivated by incubation at 80 °C for at least 20 min and subsequently stored at −80 °C before use. Compound AZ1 was found within an in-house research program and has been published previously. 9 Compound MK-886 (P1833aa) was from Affiniti Research Products Ltd (Mamhead, UK).
Final prostaglandin E synthase assay
A small volume (0.4 µL) of test compound in DMSO was added (Axygen tips, cat. no. FX-384R; Axygen, Union City, CA) to a 60-µL microsome preparation with human mPGES-1 in potassium phosphate buffer (50 mM), pH 6.8, with 2.5 mM glutathione (GSH) in the sample wells of a 384-well polypropylene microplate (781280; Greiner Bio-One, Monroe, NC) and preincubated for 15 min. Corresponding solutions without test compound were used in 16 positive-control wells, and corresponding solutions without test compound and without microsomes were used in 16 negative-control wells. The enzymatic reaction was then started by addition of 5 µL of 140 µM PGH2 in dry acetonitrile. The reaction was stopped after 1 min by addition of 20 µL of an acidic and degassed solution of ferric chloride and citrate, pH 1.9 (to reach final concentrations of 7 mM and 47 mM, respectively, pH to 3–4), which reduced remaining PGH2 to lipids—mainly, 12-hydroxy-heptadecatrieneoic acid (12-HHT). 10 12-HHT is not detected by the subsequent PGE2 detection step. The resulting solution was then pH neutralized by addition of 20 µL potassium phosphate buffer (1.2 M, pH 7.5), prior to two sequential 37-fold dilutions (in total 1350-fold dilution) in a weak potassium phosphate buffer (50 mM, pH 6.8) containing 0.2% bovine serum albumin (BSA; w/v, A-3294, Fraction V, protease free; Sigma, St. Louis, MO). The PGE2 formed was quantified by use of a commercial HTRF-based kit (cat. 62PG2PEC or 62P2APEC) from Cisbio International (Cisbio International, Bagnols-sur-Cèze, France): 6 µL of the sample solution was transferred to a Corning 3676 plate (Corning, Inc., Corning, NY) and, after subsequent addition of 3 µL PGE2-d2 and 3 µL anti-PGE2-cryptate from the HTRF kit, this plate was incubated overnight at 4 °C. Labcyte’s MicroClime Environmental Lid (LL-0310; Labcyte, Sunnyvale, CA) filled with water was used to cover the detection plate during the incubation. The large dilution prior to quantification was needed due to the highest concentration of PGE2 quantifiable by the kit being more than 1000-fold lower than the concentration of PGE2 formed (a very low ratio of substrate concentration over KM,PGH2 was not desired for pharmacological reasons). After incubation, the plate was read in an EnVision reader (PerkinElmer, Waltham, MA) according to Goedken et al. 7 and kit instructions.
The whole assay procedure described above was performed on a Biomek FX system (Beckman Coulter, Brea, CA) equipped with one 384-head and one 96-head. The latter was used as an alternative gripper for parallel transportation of plates and lids. This parallel processing was a prerequisite for enabling automation of a precise assay with a reaction time of only 60 s. Liquid transfers and mixing are described in
The typical reaction condition of the enzymatic reaction was thus:
Test compound: ranging from 60 μM to 0.002 μM (diluted in half-logarithmic steps in DMSO), or zero in positive and negative controls; Potassium phosphate buffer pH 6.8: 50 mM; GSH: 2.5 mM; mPGES-1-containing microsomes: 2 µg/mL (total protein concentration, sample and positive controls) or 0 µg/mL (negative control); PGH2 (diluted in dry acetonitrile): 10.8 µM; Acetonitrile: 7.7 % (v/v); DMSO: 0.6% (v/v).
Temperature in cooled assay microplate
The temperature in individual wells of assay microplates was determined with a digital thermometer (ELFA CIE mod. 305 with thermoelement, Thermo-Electra 8010 [type K]; Thermo Electra BV, Pijnacker, the Netherlands).
Determination of KM for PGH2
Determination of KM for PGH2 was performed according to the mPGES-1 assay protocol with the following deviations. The PGH2 solutions in the PGH2 reservoir plate were diluted in dry acetonitrile to various concentrations so that final assay concentrations varied from 2 to 115 µM both in wells used for enzyme reactions as well as in blank-rate wells (triplicates, 50 mM potassium phosphate, 2.5 mM GSH). After subtraction of PGE2 levels in blank-rate wells from their cognate enzyme wells, equation (1) was fitted to the resulting net PGE2 levels. The experiment was repeated once with a lower microsome concentration (1 µg/mL) with a similar result.
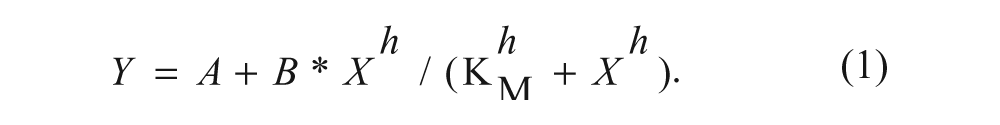
Determination of KD for GSH
Determination of KD for GSH was performed similarly to the determination of KM above. Assay solutions with (enzyme reactions) and without (blank-rate controls) mPGES-1–containing microsomes (2 µg/mL) were dispensed into wells of an assay plate, which were subjected to the mPGES-1 assay protocol as described above. Equation (1), with the variable h fixed to unity, was fitted to the net PGE2 produced during a 1-min reaction.
Calculation of Z′
Z′ values were calculated according to the equation of Zhang and coworkers. 11
Calculation of IC50
Concentration-response curves were fitted according to equation (3), using a four-parameter logistic equation where x is concentration (M); y is effect (% inhibition); A and B are the bottom and top plateau, respectively, of the curve; C is IC50; and D is the slope (Hill) coefficient. The effect y is sample (s) signal normalized to positive (p) and negative (n) control well medians of (16 wells each) according to equation (2).
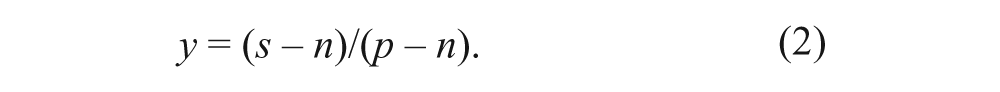
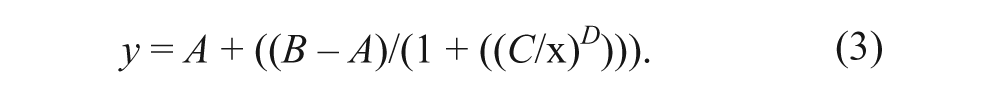
Calculation of HTRF ratio
Calculation was performed according to equation (4), where F665 and F620 are the fluorescence values for the 665-nm and 620-nm channels, respectively.
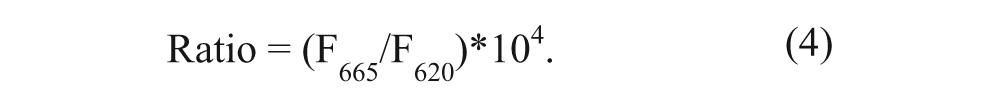
Determination of adequate mix
The importance of adequate mixing of reagents in microplates was determined according to Poulsen et al. Poulsen, E.; Smith, R.; Norman, M.; Leijon, J.; Otroka, M., (manuscript in preparation). Briefly, to measure whether a mixture of two different solutions (e.g., DMSO and aqueous buffer) is homogeneous, tartrazine was dissolved in the liquid about to be transferred. After the transfer and the following mix, three to five layers (depending on the volume in the plate) of the mixture were transferred to Greiner 781101 microplates, and absorbance was read with a Molecular Devices (Sunnyvale, CA) SpectraMax 340 (at 426 nm). If the different layers were differing less than 5%, then the mixture was deemed homogeneous. To quickly achieve acceptable mixing, the maximum tip volume, minus some volume reserved for washing, was used.
Accuracy and precision of liquid transfers
Accuracy and precision of dry and wet transfers were performed essentially according to Taylor and coworkers 12 with the following exceptions. Absorbance of tartrazine (Sigma) was used instead of rhodamine green fluorescence. Dilution after transfer, when required, was done in water.
Results and Discussion
Room Temperature Automated Assay in 384-Well Format with PGH2 in Acetonitrile
We sought to set up a practical high-capacity assay of high precision for use in robust SAR-driving IC50 determination. The starting point for assay development was a commercially available HTRF PGE2 detection kit 7 and microsome preparation from mPGES-1 overproducing cells rather than the purified enzyme used in the previous assays by Massé et al. 6 and Goedken et al. 7 The reaction catalyzed is challenging for assay development because, in parallel to the mPGES-1–catalyzed reaction, PGH2 spontaneously isomerizes to PGE2 at a significant rate in aqueous solution at ambient temperature. To minimize background and simplify the assay, we chose FeCl2/citrate to quench PGE2 production, instead of SnCl2/HCl, which converts PGH2 to PGF2α. PGF2α cross-reacts weakly with PGE2 in the detection kit, which could potentially contribute to background and thus reduce signal window and Z′. The FeCl2/citrate reaction reduces PGH2 to mainly 12-HHT, 10 which does not cross-react with the PGE2 on the HTRF antibody.
Several available mPGES-1 assay protocols keep PGH2 in a water-miscible organic solvent until the start of the reaction to let blank-rate hydrolysis to PGE2 start simultaneously with the enzymatic reaction. However, the choice of solvent was a concern for us: The vapor pressure of the solvents used (e.g., acetone or isopropanol3,7) is high, with elevated risk for evaporation-related issues (liquid transfer precision, work environment, number of plates per batch). Here, we instead opted to use dry acetonitrile, which has fairly low vapor pressure, as a solvent for PGH2. We confirmed that this solvent evaporated at a very low rate from the 384-well microplate that we would use as a reservoir (data not shown) and that loss of PGH2 to the wall of propylene tubes when stored at −80 °C were negligible at stock concentrations of 140 µM and above. The 140-µM PGH2 preparation contained little PGE2 and was fairly stable in dry acetonitrile (
Our first attempt at a robotized protocol, which involved keeping the aqueous reaction components cooled locally on the robotic system by using a cooled ALP and a dito cabinet (4 °C), resulted in large difficulties with plate edge effects: The temperature in the wells of the assay plate differed by several degrees. To avoid these effects, we instead ran the assay at room temperature with the short enzyme reaction time of 60 s. This protocol (see Material and Methods, Final Prostaglandin E Synthase Assay) worked well in several aspects. First, it displayed low blank formation of PGE2 from spontaneous PGH2 hydrolysis (0.3 µM), which allowed for a signal window of ~7 for the uninhibited reaction. Second, the total PGE2 production over time from addition of PGH2 until termination was approximately linear (
Fig. 1
). The graph indicates that 1 min of assay reaction with 2 µg microsomes/mL lies within the stable and fairly linear domain of the assay reaction with respect to enzyme concentration and time: Assay components seem to be stable up to 3 min in the reaction. Third, further characterization of the assay system showed that PGE2 detection by HTRF was precise and without plate-border effects. PGE2 was stable in the intermediate steps of the reaction (
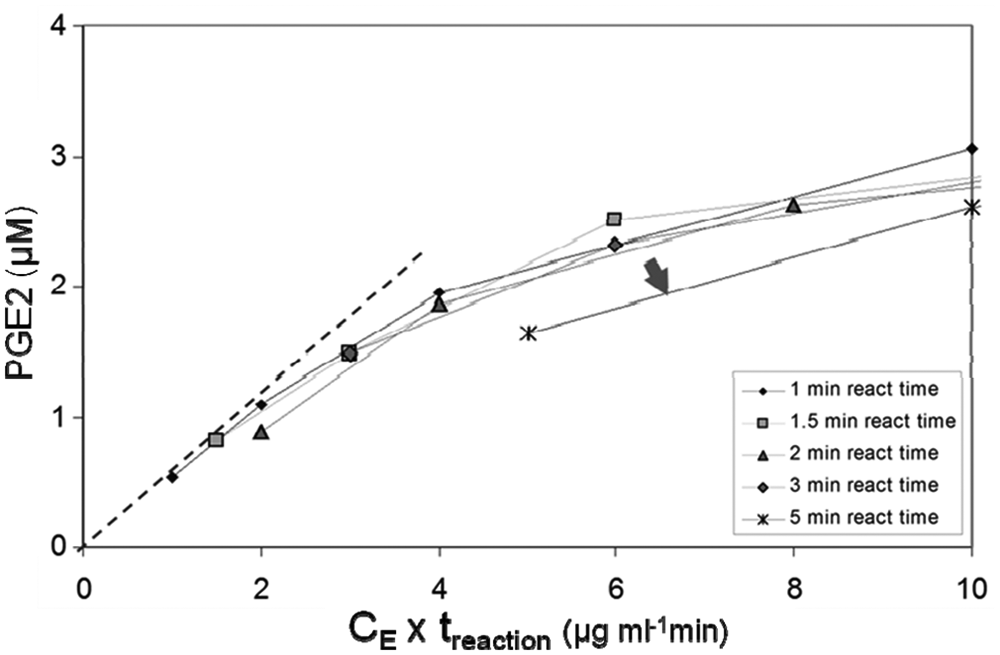
Analysis of time-dependent prostaglandin E synthase (PGES) activity of microsomal prostaglandin E synthase-1 (mPGES-1) microsomes in the automated assay. Plotting according to Selwyn 14 of the PGE2 formation versus the product of enzyme concentration and reaction time is useful for identifying time-dependent instabilities of assay components and products. Here, connecting the data points of equal reaction time clearly indicates (arrow) that the curve of 5 min assay-reaction time displays a lower activity than the other curves. Microsome concentrations were 0.5, 1, 2, 4, and 10 µg/mL (total microsome protein concentration). The dotted line is an approximate tangent to the curves of the 1- and 1.5-min assay reaction time.
Basic Assay Characterization and Enzyme-Kinetic Properties
It has previously been shown with purified mPGES-1 that GSH is not consumed during the mPGES-1–catalyzed reaction and thus is a true cofactor.
3
With the microsome preparation used here, we obtained an apparent KM for PGH2 isomerization of 22 ± 5 µM at 2.5 mM GSH and a KD for GSH of 0.6 ± 0.1 mM in accordance with earlier estimates.
3
The enzyme-catalyzed reaction rate in response to variation in PGH2 concentration was slightly sigmoid with a slope coefficient of 1.8 ± 0.2 (
Pharmacological validation confirmed IC50 for MK-886 to 7 ± 3 µM ( Fig. 2 ), which is similar to a published value of 3 µM for which direct determination of PGE2 was used. 3 The slightly higher value obtained here likely derives from the fact that we calculated IC50s from HTRF ratios: The HTRF ratio relates to PGE2 concentration in a nonlinear manner. We therefore compared IC50 calculated from HTRF ratios and from concentration of product formed, respectively, for four mPGES-1 inhibitors and found that IC50 was offset by a constant factor of around 2 in favor of the former over a potency range from 3 to 400 nM (data not shown). This thus explains the twofold difference to the published MK-886 value.
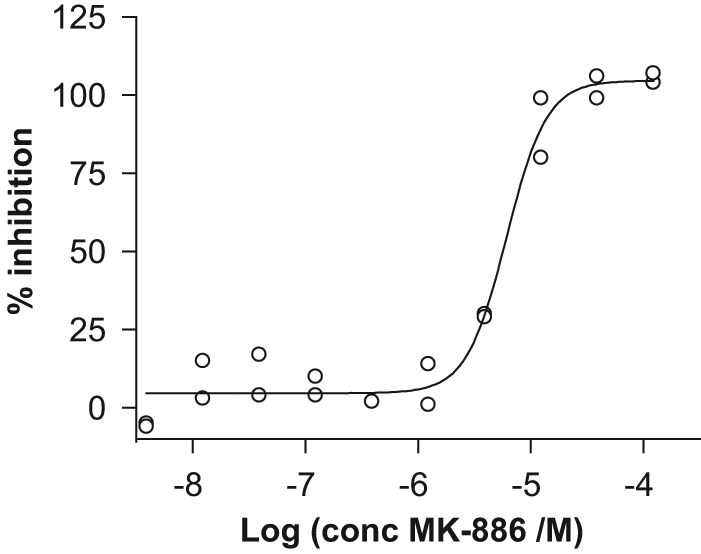
Inhibition of microsomal prostaglandin E synthase-1 (mPGES-1) by compound MK-886. The curve was fitted to 20% inhibition values calculated from homogeneous time-resolved fluorescence ratios from 10 inhibitor concentrations in duplicate on the microplate. The slope coefficient of MK-886 concentration-response curves was generally higher (mean 2.2 ± 1.0) than for other tested compounds. No correlation between slope coefficient and IC50 for this compound was observed.
Investigation of Mixing Enabled Very High Precision and Reproducibility
All liquid transfers were validated for accuracy and precision. The result from this work indicated that precision of the assay needed further optimization ( Fig. 3 , circles; Fig. 4a ) and that suboptimal mixing rather than calibration of transfer volumes needed to be addressed. The relatively high volumes in the plates combined with the narrow wells of a 384-well plate were expected to lead to an inhomogeneous mixture if not mixed thoroughly, which would lead to less precision and reproducibility with respect to Z′ and IC50. To address this, we endeavored to validate all liquid transfers for homogeneity.
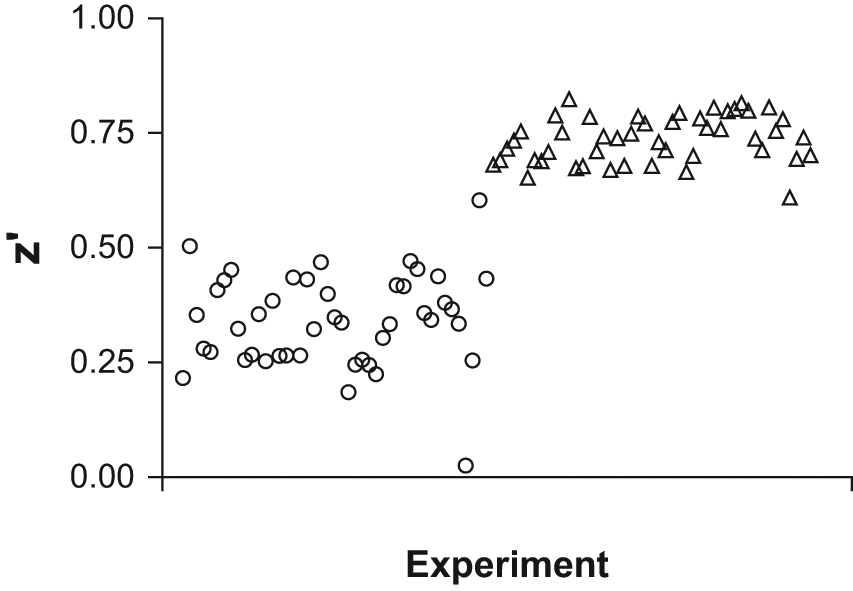
Signal to noise of positive and negative controls: Z′ before and after optimization of mixing. Circles: experiments before optimization of mixing. Triangles: experiments after optimization of mixing. Z′ values were calculated from homogeneous time-resolved fluorescence ratios.
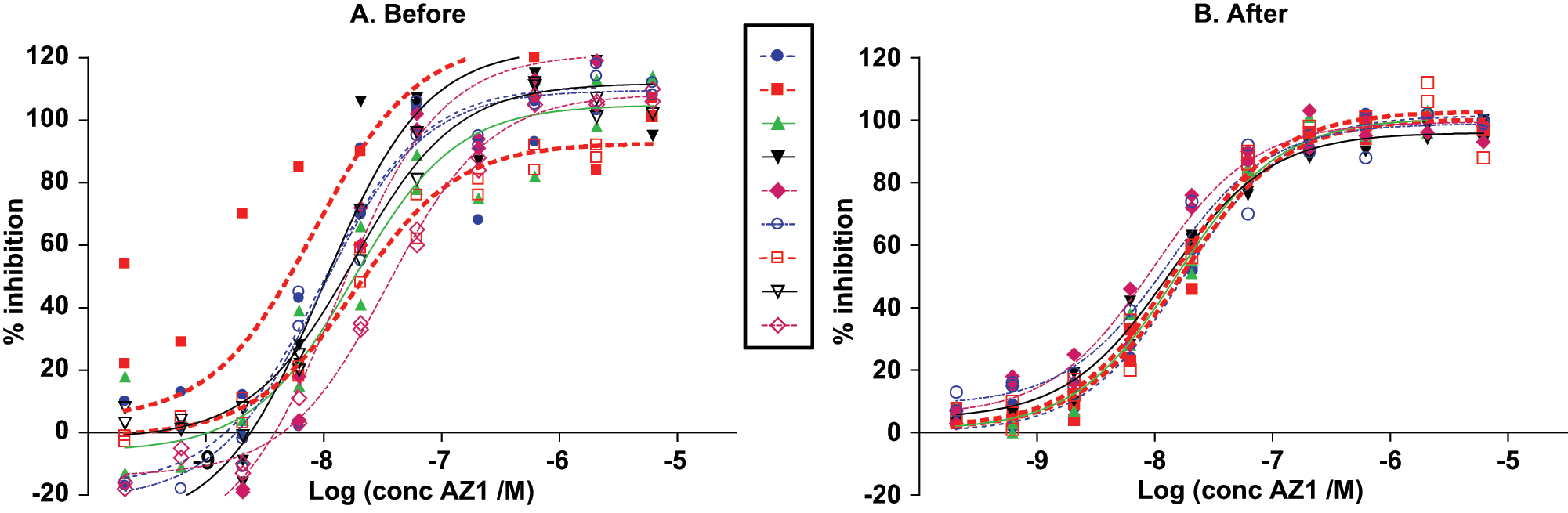
Microsomal prostaglandin E synthase-1 (mPGES-1) assay IC50s of compound AZ1 (
The homogeneity of mixing was investigated thoroughly using a novel method developed at our lab (Poulsen, E.; Smith, R.; Norman, M.; Leijon, J.; Otroka, M., manuscript in preparation). Our investigation showed that the mixing indeed could be drastically improved—an increase of the number of mixes from 3 up to 15 in some steps (
A critical parameter in pharmacological inhibitor screening is cost. By using adequate pipette tips and an appropriate liquid-handling protocol, very small volumes (0.4 µL) of test compound in DMSO could be transferred, enabling low final DMSO concentration and a small final reaction volume. Washing combined with centrifugation enabled reuse of pipette tips at least 20 times without diminishing quality of data. To prevent sticky compounds to be carried over between plates, however, it was necessary to exchange some tips regardless of extensive washing. Reducing volumes in the HTRF detection step by 40% down from the manufacturer’s recommendation, together with a significant reduction of dead volume to 4 µL by introduction of a low-volume source plate (Greiner 784201), reduced detection reagent cost significantly. This reduction did not affect CV of the assay controls (below).
Improvement of mixing of all steps in the assay led to a large decrease in variation in the pIC50 of the reference compound AZ1: CV dropped from 3% to 1% ( Fig. 4 ). This improved CV meant that ±3 standard deviations around pIC50 covers as little as 0.5 units on the pIC50 scale, which guarantees good separation between compounds during the building of the SAR for a chemical series. The final assay displayed a Z′ ≥ 0.7, as well as a CV of <5% and <3% for positive-control wells and negative-control wells, respectively (average of two plates; data not shown), as measured by the HTRF ratio. Z′ above 0.5 could be reached with even lower (fourfold) enzyme activity (data not shown). Whether the assay protocol could be amenable for greater capacity for use in high-throughput screening was not investigated here. The capacity of the present assay, however, was six 384-well microplates per batch of reagent plates, which is adequate for compound optimization and characterization in medical-chemistry efforts. Thorough investigation of the quality of mixing in all liquid transfer steps thus proved crucial for reaching the high-precision performance of the present assay—an observation that may be of general relevance for assay development in 384-well microplates, which cannot be properly mixed by shaking.
We have presented here, to our knowledge, the first screening assay in a 384-well format for mPGES-1 inhibitors that uses acetonitrile as a dry solvent for PGH2 and is devoid of cooled steps during the enzyme reaction. With these properties, together with its high-precision pIC50 determination, the presented assay should be of utility in generating SARs of high quality and in driving optimization of mPGES-1 inhibitory compounds. Also, given the recent crystallographic progress in mPGES-1 research, 13 the reaction catalyzed by mPGES-1 demands further examination of its biochemistry in screening assays as well as in vivo. The present protocol should be of utility in detailed investigation of the enzyme-kinetic characteristics of mPGES-1 in vitro.
In the work of developing the present assay, it was learned that thorough investigation of mixing is a prerequisite for reaching very high precision in an assay for determining IC50s of mPGES-1 inhibitors, at least with the type of protocol used here. It is our belief that the challenge of investigating the quality of mixing may have been underestimated in pharmaceutical assay development and that setup of future screening assays may generally benefit from increased focus on this important aspect of assay development.
Footnotes
Acknowledgements
We thank Rolf Johansson for critically reviewing the manuscript. During the submission of the present manuscript, Leveridge and coworkers published a COX-2–coupled high-throughput screening assay for mPGES-1 inhibitors that also addresses the challenge of PGH2 instability but in a quite different manner. 15 The assay presented here and the protocol of Leveridge, which incidentally also uses FeCl2 for stopping PGE2 formation, complement each other and considerably strengthen the molecular pharmacologist’s toolbox for high-capacity generation and high-precision optimization of mPGES-1 inhibitors as well as investigation of kinetic properties and inhibitor mode of action.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.