
Abstract
The development of assays in single-addition mode is of great interest for screening purposes given the multiple advantages of minimizing the number of intervention steps. Binding assays seem to be more prone to this attractive format because no functional biological activity is taking place but instead a biophysical process, whose dynamics seem easier to control without introducing significant alterations, is happening. Therefore, single-addition assays based on the displacement of prebound labeled ligands can be conceived, but careful kinetic considerations must still be taken to maximize the sensitivity of the assay and to avoid jeopardizing the identification of compounds with slow-binding kinetics. This article shows the development of a single-addition, displacement-based binding assay intended to identify modulators that act by binding to the gabapentin site of the ion channel regulatory protein α2δ1. After studying the kinetics of gabapentin binding and the influence they might have on the assay sensitivity, the best conditions were identified, and the sensitivity was compared with that of the more classical two-additions competition-based assay. Although the present study focuses on α2δ1 and its interaction with gabapentin, the rationale and the methodology followed are of broad purpose and can be applied to virtually every binding assay.
Introduction
Nonfunctional binding assays, based on the detection of the interaction of a small-molecule ligand with its biological target, are frequently used in screening programs. They represent a common strategy in the search for putative modulators of all classes of therapeutic targets, and their utility is remarkable when functional assays are not available either because the biological role of the target is unknown or because its function cannot be reproduced in vitro. Among the different binding assay options acceptable for high-throughput screening (HTS), the scintillation proximity assay (SPA) technology is probably one of the most used. SPA was one of the first homogeneous technologies introduced in the screening arena more than two decades ago, 1 and its popularity grew quite rapidly among the drug discovery community because it enabled the use of radioactive assays in a high-throughput mode because of the absence of separation and filtration steps. Although many other nonradioactive alternatives for binding assays have emerged throughout these years, 2 SPA is still widely used in HTS. The main reason is that it uses the original ligand with a radioactive isotope replacing one of the original atoms, a subtle modification compared with the introduction of large groups (e.g., a bulky fluorescent moiety), which may affect notably the affinity for the target. This makes the use of SPA almost universal for every target as long as a suitable ligand is known and available in its radioactive form.
Nonetheless, SPA is affected by some drawbacks. In addition to the environmental and safety concerns inherent to the use of radioactive isotopes, other inconveniences related to the use of beads may become critical and may eventually preclude the attainability of an optimal HTS assay. 3 The insoluble nature of the beads can cause variations in the way they settle in the well, and this may affect adversely the final readout due to optical interferences. Such insolubility is also detrimental for the liquid-handling steps, leading to high variability and poor Z′ values. Whereas the optical issues are surmountable because of the availability of modern luminescence readers with improved functions,4,5 the problems related to pipetting are harder to solve. For this reason, keeping the number of pipetting steps to a minimum can be highly beneficial for the performance of the assay.
SPA binding assays are usually configured in a competition- based mode in which both the radioactive ligand and the interrogated sample are incubated simultaneously with the biological target that has usually been coupled to the SPA bead. 6 A competition is then established between the probe and the compound to bind to the target. This configuration demands two dispensing steps onto the wells containing the tested samples: one for the target and another one for the radioligand. However, an alternative displacement-based format can be conceived to reduce the number of pipetting steps. In such configuration, the SPA-bound target is mixed with the radioligand, and this mixture is then dispensed to the wells in a single and unique step; then, the tested sample will have to displace the prebound probe during the course of the assay. Because the effect yielded by the sample will depend on its ability to displace the prebound ligand, it follows that the duration of the assay will be critical for its sensitivity: A reasonable amount of time must be given to ensure that every active sample has been able to displace the probe, and the length of this time will be dictated by the kinetics of binding (association and dissociation) of both the probe and the sample. Therefore, reasonable kinetic studies must be performed to ensure that the sensitivity of the single-step, displacement-based assay is not hampered with respect to the two-steps, competition-based one.
We have performed such studies when configuring a binding assay intended to find modulators of the ion channel regulator α2δ1. This is a regulatory protein forming part of voltage-dependent calcium channels, which is composed of two distinct subunits (α2 and δ) covalently bound by disulfide bridges. 7 It has been shown that α2δ1 is an essential protein complex for excitation-contraction coupling in skeletal muscle, 8 and it modulates the calcium traffic across the channel. 9 α2δ1 is involved in several disorders such as epilepsy 10 and neuropathic pain 11 ; indeed, the two most used anticonvulsants, gabapentin and pregabalin, bind to α2δ1, thereby reducing the synaptic transmission of overstimulated neurons and returning neuronal firing to basal levels. 12 The clinical success of both drugs has generated a notable activity in the search of new molecules acting through similar mechanisms, and for this reason, a screening campaign was intended for the identification of molecules binding to the gabapentin site of α2δ1, with the final purpose of using such molecules for the treatment of severe pain. The current article describes the kinetic considerations undertaken for the development of a suitable single-step assay monitoring the binding to α2δ1 through the displacement of prebound gabapentin, in which the reduction in the number of pipetting steps contributed to a significant improvement in assay quality.
Materials and Methods
Materials
Wheat germ agglutinin-polystyrene (WGA-PS) beads were from Perkin Elmer (Waltham, MA), and [3H]-gabapentin was from American Radiolabeled Chemicals (St. Louis, MO). All other reagents were of the highest purity available from Sigma (St. Louis, MO) unless otherwise stated. Nonlinear regression fitting of the experimental data to the corresponding equations described below was done with Grafit 5.0.8 (Erithacus Software Limited).
Preparation of Cell Membranes Expressing α2δ1
PrecisION hCav2.2 (α1B/β3/αδ1)-HEK recombinant cell line was obtained from Millipore (Billerica, MA) and cultured according to the instructions from the supplier. Cells were harvested after reaching 80% to 90% confluence and washed twice with Hanks balanced salt solution containing 0.6 mM EDTA. Then, lysis buffer (50 mM Hepes pH 7.5, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], 0.1 mM leupeptin, 2 µM pepstatin A, and 25 µg/mL bacitracin) was added in a volume equal to 12 times that of the pellet, and the mixture was homogenized in a waring blender for 2 cycles of 15 s. The resulting suspension was clarified by centrifugation at 500 × g for 20 min at 4 °C, and the supernatant was centrifuged at 48 000 × g for 36 min at 4 °C. The pellet was resuspended in the same buffer as above without PMSF and pepstatin A, and the resulting material was forced to pass through a 0.6-mm needle, made up to 4 times the original cell pellet volume with the latter buffer and frozen at −80 °C until its use. The membrane preparation used in these studies was titrated with [3H]-gabapentin using a classical filtration method with GF/C membranes (GE Healthcare, Waukesha, WI) and resulted in a binding site titer (B max ) of 3000 fmol/mg.
Kinetic Determinations
SPA assays were performed at 25 °C in 384-well polystyrene white plates (Greiner, Gloucestershire, UK) in 20 µL final volume including α2δ1-expressing membranes at a total protein concentration of 150 µg/mL (450 pM binding sites concentration according to the B max value), with 3 mg/mL WGA-PS beads and 50 nM (110 kBq/mL) [3H]-gabapentin in 10 mM Hepes, pH 7.4. The concentration of radiolabeled gabapentin was selected to be in the range of its reported KD value according to the bibliography12,13 and to our previous data. Nonspecific binding was determined with 100 µM unlabeled gabapentin. To investigate the kinetics of gabapentin binding, varying concentrations of radioligand were mixed at different times with membranes and beads in the corresponding wells and incubated at 25 °C with shaking. At a given time the reaction was stopped simultaneously in the whole plate by centrifugation at 700 × g for 5 min, and final readout was performed in a ViewLux ultraHTS Microplate Imager (Perkin Elmer, Boston, MA). Nonspecific binding was discounted from the readouts, and the resulting values for each gabapentin concentration were fitted by nonlinear regression to the following exponential equation:
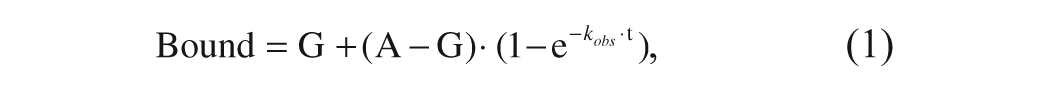
where A is the maximum amount of binding obtained at equilibrium for each gabapentin concentration, G is the background binding level detected at t = 0, and k obs is the kinetic constant to approach the equilibrium. Assuming that the binding of gabapentin to α2δ1 follows a one-step process, such constant is related to the association and dissociation rates through the expression 14
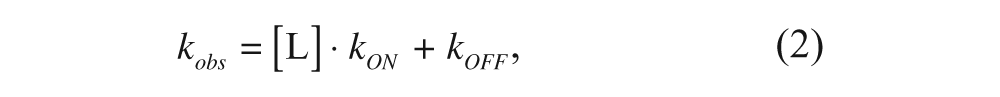
where [L] is the free radioligand concentration and kON and kOFF are the kinetic constants for association and dissociation, respectively. Both eq 1 and 2 assume that the concentration of free radioligand remains unaltered because it is in large excess over the binding sites, as detailed below. Therefore, plotting the resulting k obs values versus the concentration of gabapentin will deliver a straight line, enabling the calculation of k ON and k OFF from the slope and the intercept, respectively.
To investigate the kinetics of gabapentin dissociation, membranes, beads, and 50 nM (110 kBq/mL) [3H]- gabapentin were mixed in a conical tube and incubated for at least 4 h at 25 °C under gentle shaking on a rocking platform. Then the mixture was dispensed at different times in the wells of a plate prefilled with unlabeled gabapentin to reach a 100 µM final concentration of the latter, and the mixture was further incubated in the plate until it was processed as above. The time elapsed between the addition to the plate and the centrifugation step was the time considered for the dissociation time course. The readouts obtained at each time were fitted by nonlinear regression to the following exponential decay equation:
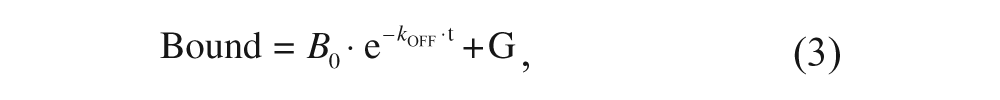
where B 0 corresponds to the initial levels of bound gabapentin and G corresponds to the background observed at infinite time.
pEC50 Determinations
Compounds were evaluated for their ability to reduce the amount of [3H]-gabapentin binding (either in a competition- or displacement-based assay) at different concentrations in 1% DMSO. The assay was performed under the same conditions as above in 384-well polystyrene white plates. Controls for total binding (including 1% DMSO) and nonspecific binding (including 100 µM gabapentin in 1% DMSO) were also included in the corresponding wells, so that the effect of each compound was quantified as
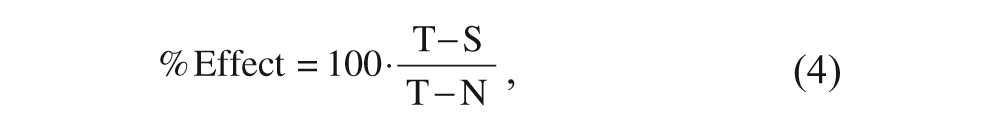
where T is the signal corresponding to total binding, N that corresponding to nonspecific binding, and S that of the tested sample. The resulting values obtained for each compound concentration were then fitted by nonlinear regression to eq 5:
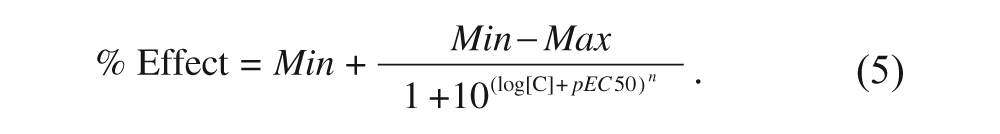
Equation 5 is a modified version of the classic isotherm equation where Min and Max are the lower and higher asymptotes of the resulting sigmoid curve, respectively; [C] is the concentration of the tested compound in molar units; n is the Hill coefficient; and pEC50 is the negative logarithm of the EC50. The use of this equation is preferred to the classical isotherm equation because the experiments were carried out by performing serial dilutions, and therefore the range of concentrations was evenly distributed in a logarithmic rather than a linear scale. Hence, it is more appropriate to take the logarithm of the concentration as the independent variable instead of the plain concentration.
Resolution of Differential Equations
Numerical solutions to the differential equations describing the kinetics of binding equilibria described in eq 6 to 8 (see below) were approximated by an iterative procedure following the Runge-Kutta method using the software Scientist 3.0 (Micromath, St. Louis, MO). The equations were introduced into the system using the appropriate syntax as recommended by the manufacturer, and the individual values for the parameters (detailed in the
Results
When developing a new assay for HTS purposes, it is mandatory to ensure that the sensitivity to possible modulators of the target is not jeopardized because of the singular features of the assay that may lead to missing interesting compounds. In the particular case of a single addition, displacement-based, radioligand binding assay, this point is particularly critical because such sensitivity might be compromised with respect to the classical two-additions, competition-based format. Indeed, because the assay is now based on the displacement of a prebound radioligand by a test compound, the rates of radioligand dissociation and compound association are crucial because they will dictate how easily the latter will displace the former from the target binding site. It is therefore necessary to explore the kinetics of the process to understand whether such displacement-based assay format is feasible and what are the best conditions for it.
The scheme displayed in Figure 1 shows the two processes being considered. Although both will eventually lead to the same final equilibrium, each one is initiated from different starting points: free protein “P” for the competition-based assay and the preformed equilibrium between “P” and “PL” for the displacement-based one. Consequently, the time course of the evolution of the individual species is different in each case, and it is ruled by the individual kinetic constants as well as by the initial conditions. Because PL is the species being measured in the assay, the differences in its concentration at a given time between the two formats will inform on the differences in sensitivity among them.
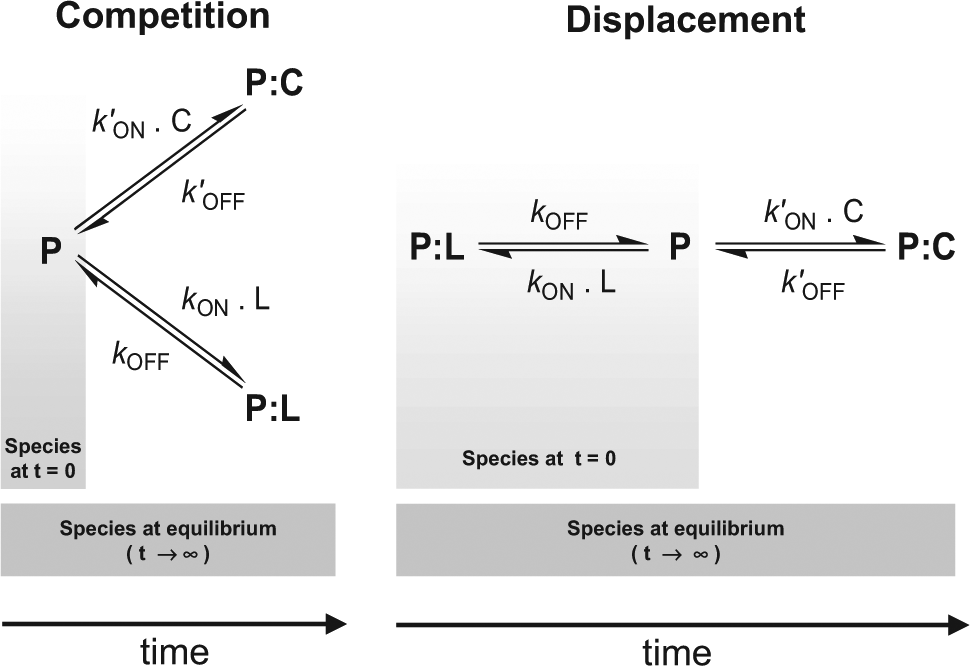
Schematic representations of the way the equilibrium is reached for the two additions, competition-based assay format and the single-addition, displacement-based one. The final equilibrium is identical in both cases, but the initial conditions are different and so are the kinetics to reach the equilibrium, as discussed in the text. P denotes protein (α2δ1 for the case considered in this article), L denotes the radioligand used ([3H]-gabapentin in this case), and C is the compound being tested.
The kinetics governing the evolution of the three species are described by the following equations:
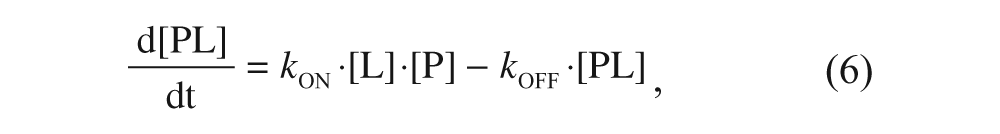
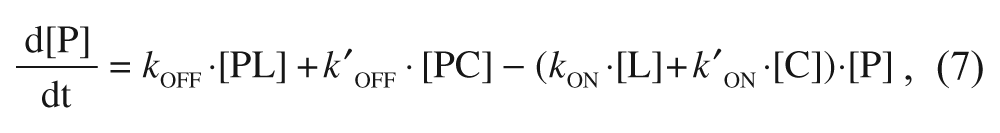
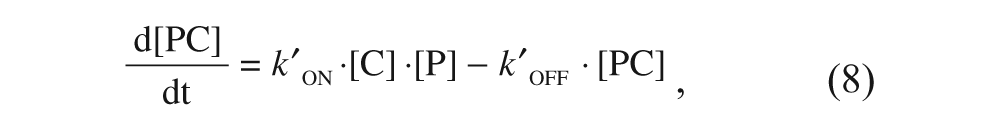
where P is the target protein (i.e., α2δ1 in the particular case being considered in this article), L is the free radioligand (i.e. [3H]-gabapentin), and C is the tested compound. kON and kOFF are the association and dissociation rate constants for gabapentin, respectively, whereas k′ON and k′OFF are the corresponding constants for the compound. The explicit solutions to these equations have already been described only for the competition case. 15 To cover both the competition and displacement cases at once, an alternative approach has been undertaken based on a numerical simulation of these differential equations with the help of a suitable software (Micromath Scientist), fixing the values of all the involved parameters in a reasonable manner and expressing the concentrations of the three species as fractional amounts. To that end, a few premises have been established. First, because the compounds are usually screened at a concentration of 10 µM, which is far above that of the protein target (in the case being considered, this latter value is that of the binding sites concentration, which is in the sub-nM range), it follows that changes in [C] are insignificant compared with changes in [P], and therefore the product k′ON·[C] can be considered as a constant, and the same reasoning applies to [L] provided that it is far above [P]. Second, because [3H]-gabapentin is present at a concentration corresponding to its KD and given that this value equals the quotient between kOFF and kON, it follows that under these conditions, kON·[L] = kOFF. Third, the initial concentrations of each species are different for the competition and the displacement cases: In the competition case, where radioligand and compound are mixed with free protein, such concentrations (in fractional amounts) are [P]0 = 1; [PL]0 = [PC]0 = 0, whereas in the displacement case (where the binding equilibrium has previously been reached at a total concentration of radioligand corresponding to its KD), such values are [P]0 = [PL]0 = 0.5; [PC]0 = 0. Finally, because of the mass conservation law, it is obvious that for both cases, the sum of the concentrations of three species must always be 1 throughout the entire process.
With these premises, the system of differential equations can be numerically resolved just by entering the values of kOFF, k′ON and k′OFF. The two latter correspond to the unknown compounds and therefore could be simulated according to the different cases considered, as explained below. The value of kOFF must be determined experimentally for [3H]-gabapentin. With that aim, two experimental strategies have been followed measuring the kinetics of gabapentin binding as well as of gabapentin dissociation. In the first case, the time course of radioligand binding was monitored ( Fig. 2A ) and fitted to an exponential equation from which the kinetic constant to approach the binding equilibrium, kOBS, at every gabapentin concentration could be obtained. Bearing in mind that the concentration of gabapentin is far above that of binding sites (50 nM in the lowest case for the former versus 450 pM for the latter), its depletion due to binding can be considered negligible, and therefore the concentration of free gabapentin remains essentially unaltered, hence allowing the use of eq 1 and 2 to describe the kinetics of binding. The secondary plot obtained from these values ( Fig. 2B ) delivered a straight line that enabled the determination of the off-rate from the intercept. Such value was 3.62 ± 0.08 × 10−4 s−1, a small figure that suggests that gabapentin is a slow-dissociation ligand and therefore its displacement by a competing molecule may be challenging. To confirm this conclusion, the kinetics of gabapentin dissociation were measured by monitoring the time course of the displacement of prebound radiolabeled ligand with a large excess of the unlabeled molecule. Because of this large excess, the binding of the displaced radioligand is fully prevented; hence, the rate of disappearance of the protein-radioligand complex obeys to a single exponential decay, 14 and therefore, a kOFF value of 2.78 ± 0.74 × 10−4 s−1 could be determined ( Fig. 3 ), confirming the range for the value obtained above. Thus, a value of 3 × 10−4 s−1 will be used for the off-rate constant in the simulations.
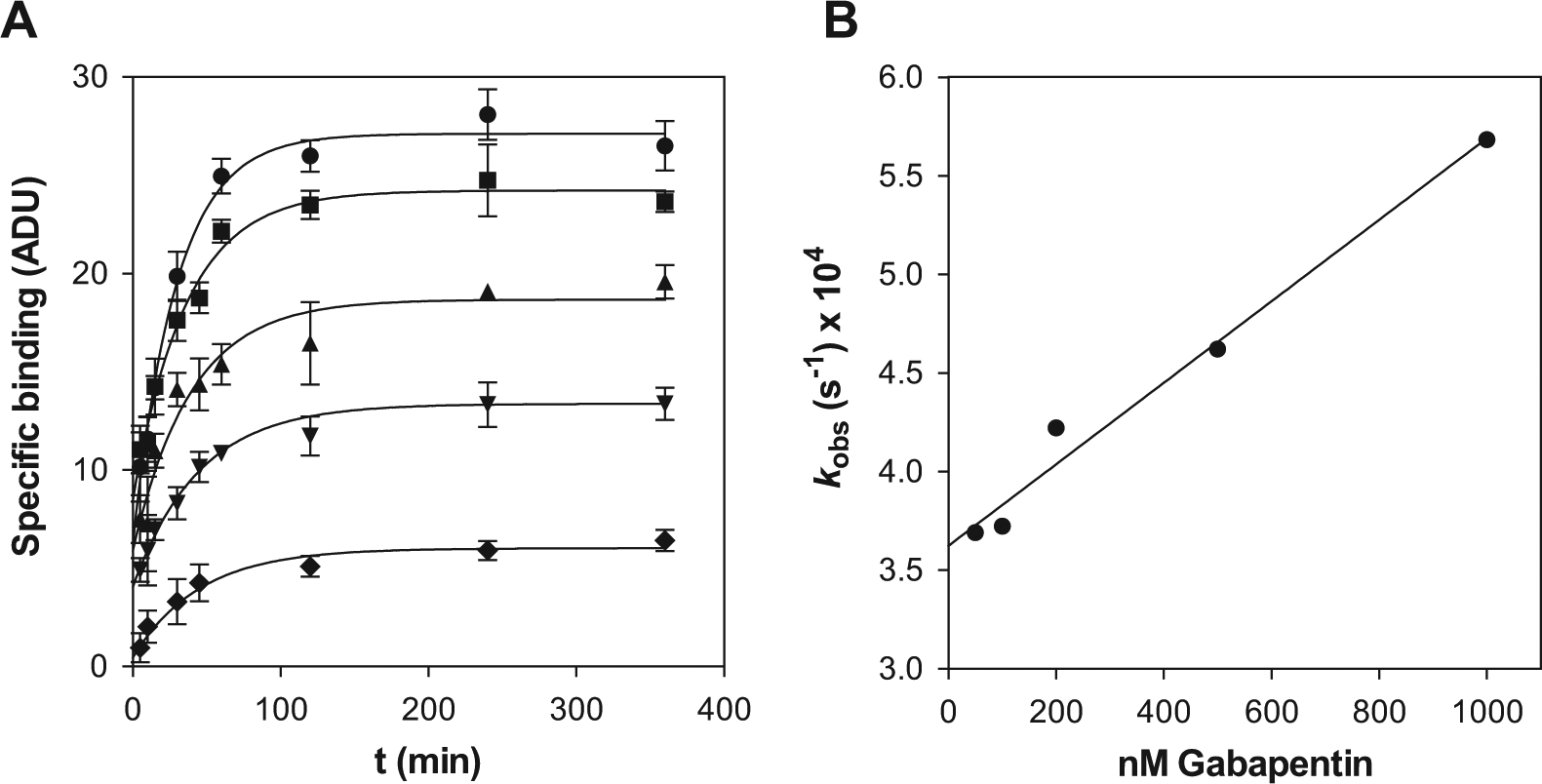
Kinetics of [3H]-gabapentin association to α2δ1. (
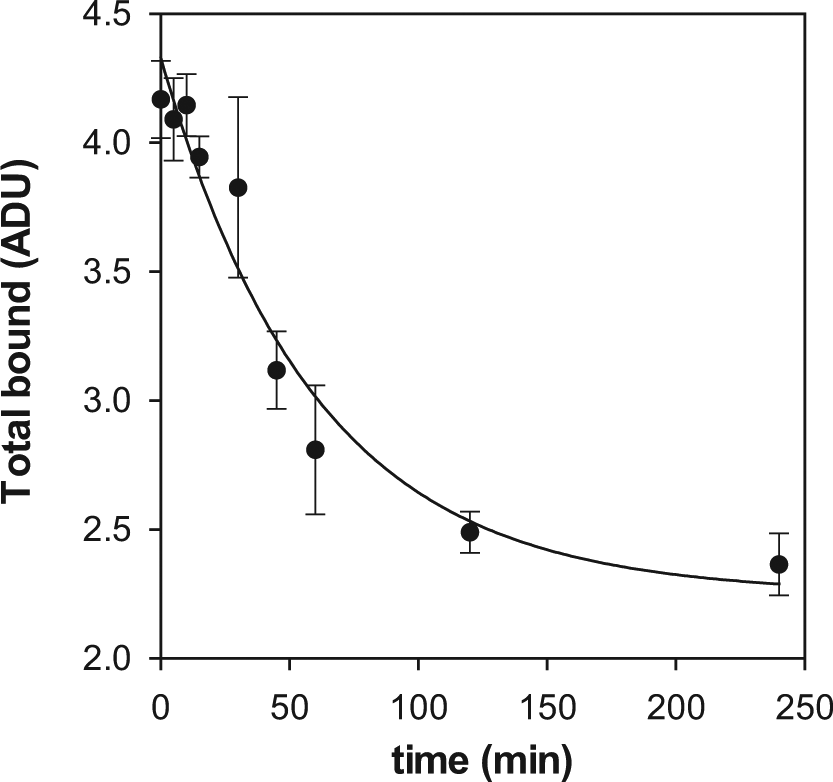
Kinetics of [3H]-gabapentin dissociation from α2δ1. The experiment was performed as described in the text, and the experimental points were fitted to eq 3 by nonlinear regression. Each determination was done in triplicate. The points denote the average value, whereas the error bars correspond to the SD.
Once kOFF was available, a number of cases with different k′ON and k′OFF values were considered covering all the possible type of ligands in terms of both affinity and binding kinetics, with the aim of understanding how their idiosyncrasy would affect the outcome of the assay. Sixteen possible cases were contemplated (their detailed features are summarized in a
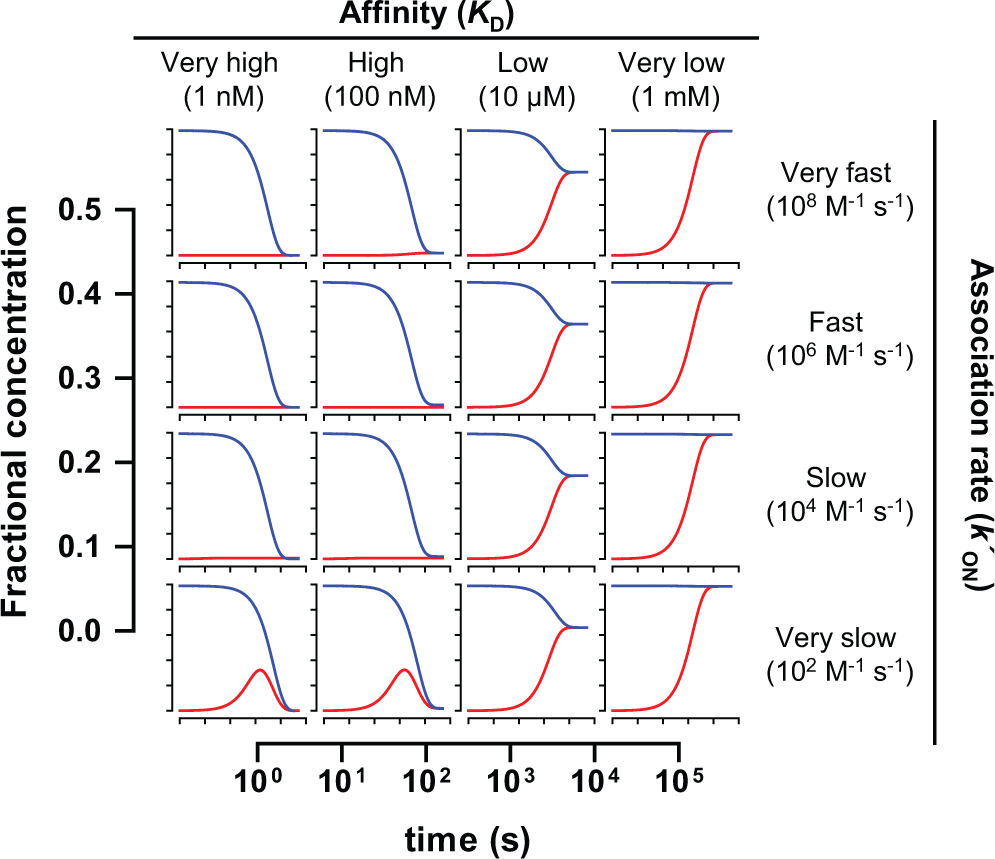
Time course of the evolution of the α2δ1:[3H]-gabapentin complex (denoted as “PL” in eqs 6 to 8 and in Fig. 1 ) for each of the 16 possible cases of compounds. The values were obtained by approaching the numerical solutions to eqs 6 to 8 as described in the text. The red line shows the evolution of the complex in the competition-based format, whereas the blue line shows the evolution in the displacement-based one. All graphs share the same scale displayed in the two axes represented in the margins.
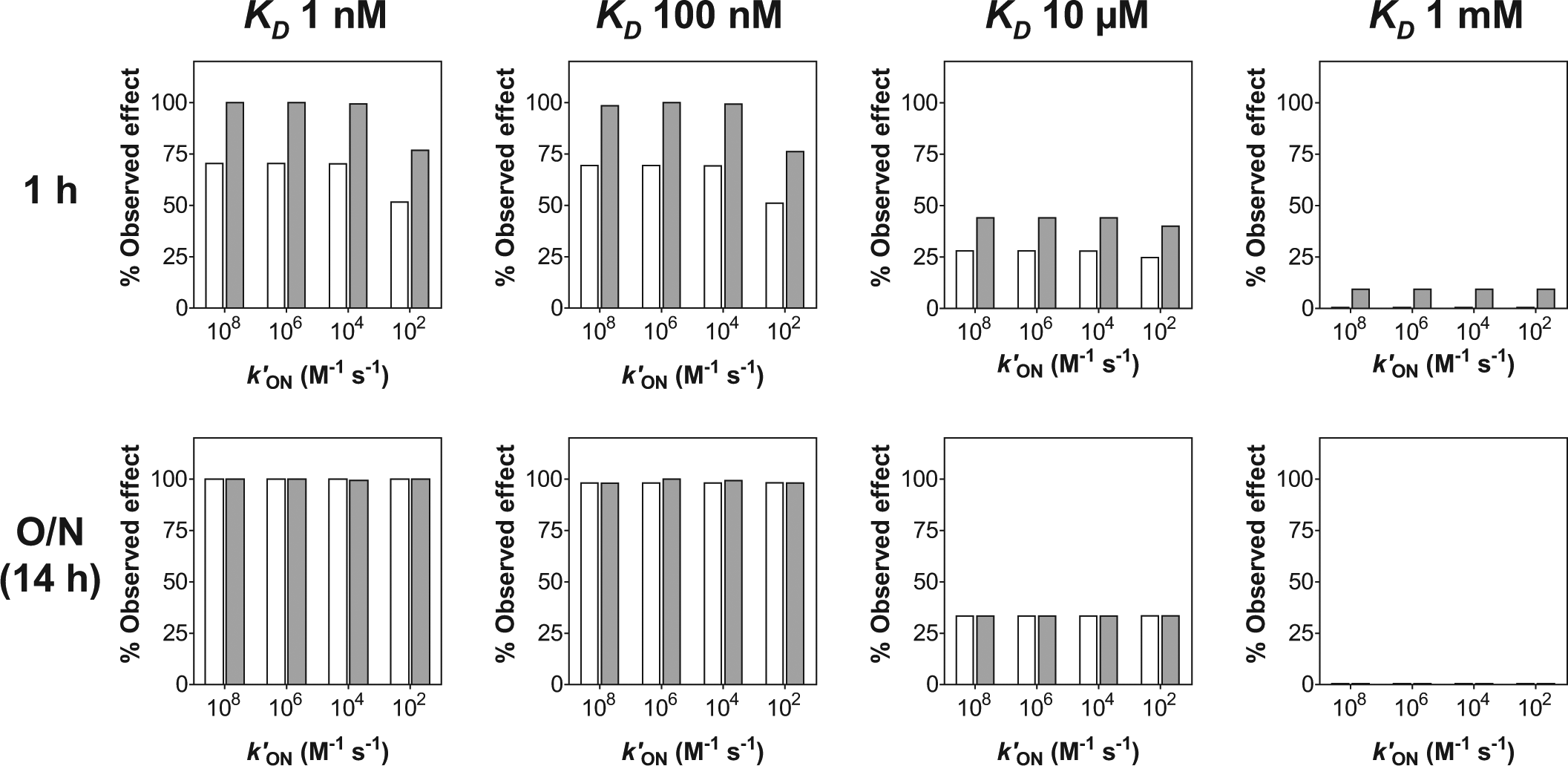
Effect caused by each of the 16 cases considered after 1 h and 14 h (overnight, O/N) depending on whether the assay followed a displacement-based format (white bars) or a competition-based one (gray bars). Such effect was measured as the percentage of [3H]-gabapentin bound with respect to the control in the absence of compound and was determined from the values of fractional binding obtained at 1 h and 14 h for the complex PL in the numerical solution to eqs 6 to 8, assuming that such value for the control in the absence of compound was 0.5.
According to the prediction above, the sensitivity of the single-addition format may be compromised if the duration of the assay is kept short (1 h) but is preserved if such duration is extended overnight. To experimentally confirm this conclusion, the effect of seven compounds belonging to several chemical series known to interact with α2δ1 at the gabapentin-binding site was evaluated, and their potencies (measured as pEC50) were determined in both assay formats including an overnight incubation. These compounds were gamma-aminoacids (analogues of gabapentin), pyrrolopyridazines (described as α2δ1 ligands 16 ), acridines, 17 quinolines, 18 and alpha-aminoacids 19 (some of the structures are displayed as supplemental Data). Figure 6 shows a good correlation among the pEC50 values obtained in both assay formats when the incubation was extended overnight, with a concordance correlation coefficient 20 value of 0.955 which, according to some authors, 21 is indicative of identity between the two sets of data being compared.
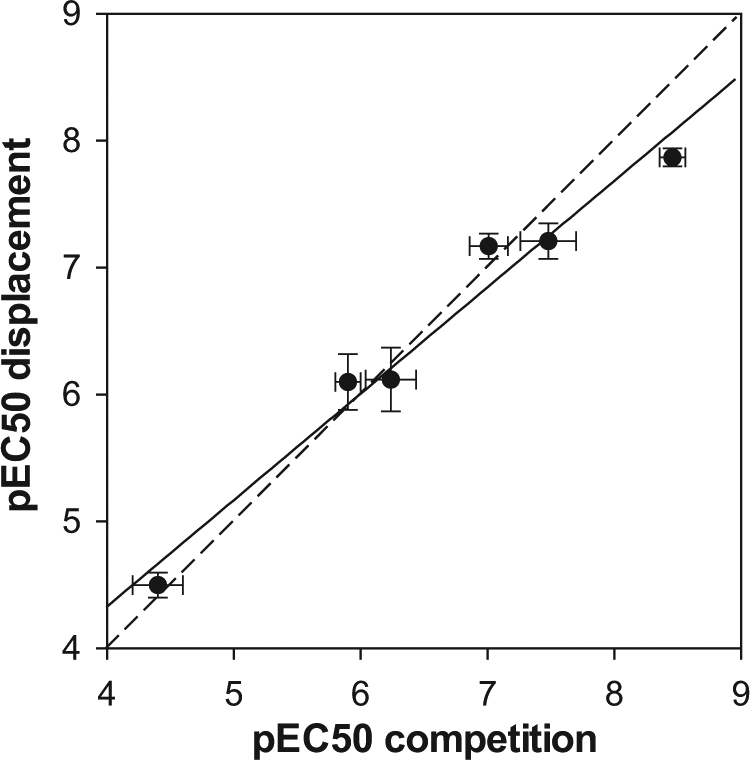
Correlation among the pEC50 values obtained in both assay formats for six selected compounds belonging to several chemical series. The solid line corresponds to the linear regression fitting (R2 = 0.971), whereas the dashed line denoting identity (y = x) is presented for visual reference. The determinations were done in triplicate. Each point corresponds to the average value, and the error bars denote SD.
This pharmacologic equivalence of the displacement-based assay with respect to the competition-based one enabled its use for screening purposes. Before doing that, the improvements in assay performance provided by the single addition of reagents were demonstrated by evaluating the Z′ factor 22 in a series of plates: 11 plates were tested in two consecutive days, including in each plate 20 wells for measuring total binding and 20 wells for nonspecific binding as “high” and “low” controls, respectively. The evaluation was performed using both assay formats. As observed in Figure 7 , the Z′ values obtained for the single-addition, displacement-based assay were always above those for the competition-based one, and indeed the average Z′ value obtained was almost 0.2 units higher (0.78 ± 0.05 versus 0.61 ± 0.04), demonstrating the benefits delivered by the simpler format. The availability of this assay allowed the execution of a full screening campaign in which 1 200 000 compounds were evaluated in 3 weeks with less than 10% plate failures and an average Z′ of 0.7, which led to the identification of about 20 000 hits and the initiation of chemical programs on some of them.
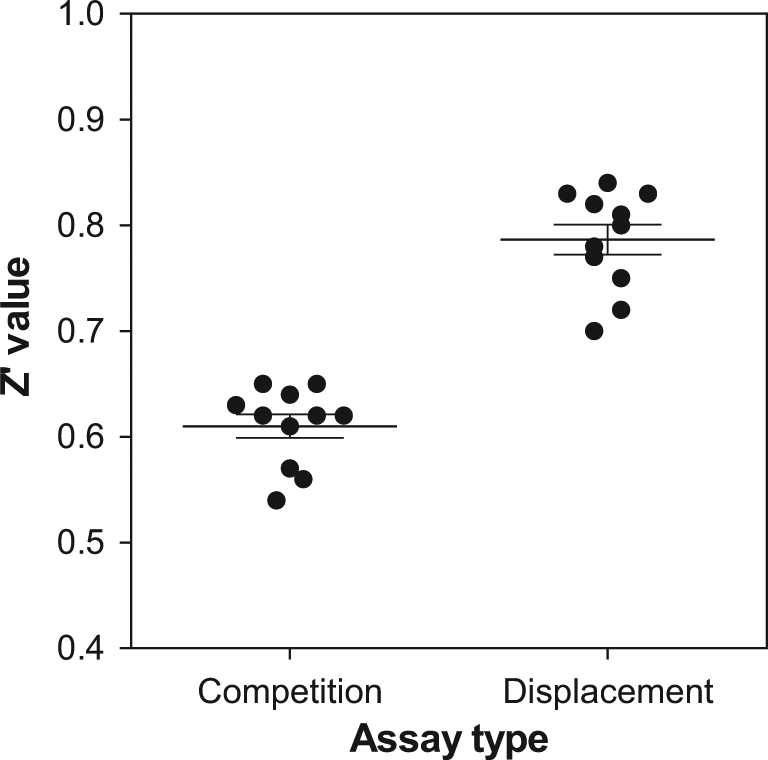
Z′ values obtained in the two-additions, competition-based format and in the single-addition, displacement-based one. The lines correspond to the average values of 11 determinations as described in the text, whereas error bars denote SD.
Discussion
Despite the tremendous benefits offered by SPA technology, which two decades ago revolutionized the performance of radioactivity-based assays in HTS environments, the issues associated with handling insoluble heterogeneous material such as the scintillation beads are still a major drawback, which is challenging the complete adaptation of this technology to modern ultra-high-throughput screening strategies. The reduction of liquid-handling steps derived from the availability of single-addition assays would help to circumvent those issues, minimizing their impact on assay quality as demonstrated by the improvement in Z′ factor for the α2δ1 assay described in this article. However, the implementation of such an assay format is by no means an elementary process, and serious considerations must be undertaken to avoid jeopardizing assay sensitivity. As for every binding assay with an endpoint readout, it is always necessary to ensure that the system has reached equilibrium before the readout is recorded to allow the tested compound to fully display its effect. The kinetics to approach this global equilibrium are not only governed by the binding of the tested compounds but also by that of the radioligand used, and such influences are more critical in a displacement-based assay, especially if the radioligand shows low dissociation rates and therefore its displacement is intrinsically slow. This is the case of gabapentin, whose low off-rate (in the range of 10−4 s−1) hinders its removal from the binding site. Then, compounds with low values for their on-rate constant will need more time to evacuate the radioligand, hence to display their full effect, and therefore their potency may be underestimated if the readout is performed prematurely. This fact is clearly illustrated by Figure 4 : In none of the 16 cases considered does the displacement-based format reach the equilibrium earlier than the competition-based one. Therefore, for some compound cases, the effect observed in the former version will be lower than in the latter one if the readout is performed after only 1 h of incubation, as observed in Figure 5 . It is particularly noticeable that, for the K D 10 µM case, the effect is artificially higher in the competition case after 1 h incubation (comparing the height of the gray bars in the 1 h and overnight cases) because the global equilibrium has not been reached yet for that particular case. This highlights the importance of ensuring enough time to allow the equilibrium to be fully established. Because equilibrium has been reached in all cases after 4 × 104 s (i.e., more than 11 h), it was decided to leave the incubation overnight and this has contributed to the pharmacologic equivalence between both formats, as demonstrated in Figure 6 .
The rationale presented here has been applied to the particular case of α2δ1 but can be extended to any binding assay regardless on the nature of the probe (radioligand or fluorescent ligand), as long as the concentration of free ligand exceeds that of binding sites, hence, free ligand depletion can be considered negligible. In the particular case of an SPA-based assay, the reduction in liquid-handling steps was translated into an improvement in the Z′ factor, which might eventually convert an intractable assay into a feasible one. For any other type of binding assays, even though the reproducibility of reagent dispensing may not be so critical, the reduction in the number of dispensing steps will provide other advantages such as an increase in throughput and a reduction in expenses due to the decrease in cumulative dead volumes in robotic reservoirs. This underlines how a better knowledge of the kinetics of ligand binding may contribute to an improvement in the efficiency of HTS strategies.
Footnotes
Acknowledgements
The authors thank Jim Coote, Andy Powell, and Morven Allen for the preparation of cell membranes. They also thank Paloma Pérez and Alberto Alaminos for the generation of the HTS data.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The research presented in this article corresponds to work done in the course of employment by GlaxoSmithKline. There was no financial support above and beyond normal compensation for employees.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.