
Abstract
Breast cancers expressing hormone receptors for estrogen (ER) and progesterone (PR) represent ~70% of all cases and are treated with both ER-targeted and chemotherapies, with near 40% becoming resistant. We have previously described that in some ER+ tumors, the resistant cells express cytokeratin 5 (CK5), a putative marker of breast stem and progenitor cells. CK5+ cells have lost expression of ER and PR, express the tumor-initiating cell surface marker CD44, and are relatively quiescent. In addition, progestins, which increase breast cancer incidence, expand the CK5+ subpopulation in ER+PR+ breast cancer cell lines. We have developed models to induce and quantitate CK5+ER−PR− cells, using CK5 promoter-driven luciferase (Fluc) or green fluorescent protein (GFP) reporters stably transduced into T47D breast cancer cells (CK5Pro-GFP or CK5Pro-Luc). We validated the CK5Pro-GFP-T47D model for high-content screening in 96-well microplates and performed a pilot screen using a focused library of 280 compounds from the National Institutes of Health clinical collection. Four hits were obtained that significantly abrogated the progestin-induced CK5+ cell population, three of which were members of the retinoid family. Hence, this approach will be useful in discovering small molecules that could potentially be developed as combination therapies, preventing the acquisition of a drug-resistant subpopulation.
Keywords
Introduction
Human breast cancers can be subdivided into five subtypes based on gene expression profiling. 1 These include two subtypes termed luminal that are estrogen receptor (ER) and usually progesterone receptor (PR) positive, a Her2 amplified group, and two “triple-negative” (TN) subtypes devoid of ER, PR, and Her2. 1 ER+ breast cancers constitute 70% of all cases, and in these tumors, estrogens and ER-regulated pathways drive cancer growth. Treatment of ER+ breast cancers includes ER-targeted therapies such as tamoxifen and aromatase inhibitors as well as conventional chemotherapies. 2 In general, ER+ breast tumors have a better prognosis than Her2 ER− or TN breast tumors, which lack ER and PR. 3 Despite this, drug resistance and tumor recurrence are a critical problem for ER+ disease, and >5 years out from initial diagnosis, ER+ tumors account for a similar number of cancer-related deaths as ER− tumors. 4 One plausible explanation for this is the existence of tumor cells with a drug-resistant phenotype termed cancer stem cells (CSCs). However, CD44+CD24− and aldehyde dehydrogenase (ALDH)+ markers used to identify CSCs are more prevalent in TN tumors and are low and sometimes absent in luminal ER+ breast cancers.5,6 Thus, there is a need to better understand and define markers of prospective luminal breast CSCs and to discover agents that inhibit them.
We previously identified a subpopulation of cells within ER+PR+ breast tumors that is cytokeratin 5 (CK5) positive. 7 CK5 is a characteristic marker of a group of poor prognostic basal-like or metaplastic TN breast cancers and a marker of luminal stem and progenitor cells in the normal breast.8,9 We demonstrated that tumor CK5+ cells are inherently negative for steroid receptors (ER−PR−); express stem-like genes, including the tumor-initiating marker CD44 and multidrug resistant pump ABCG2; and have increased expression of self-renewal genes. 7 CK5+ cells display resistance to endocrine therapies and conventional chemotherapies compared with CK5− cells, likely due to ER negativity and quiescence, respectively. 10 Moreover, these cells increase in number in patients’ tumors following neoadjuvant endocrine therapy. 10 Up to half of all luminal breast cancers contain small (~1%–10%) subpopulations of CK5+ cells, with increasing CK5+ cell content correlating with worse prognosis.10,11 Thus, small-molecule modulators that suppress or revert the CK5+ phenotype may prove to be effective for the prevention or treatment of breast cancer.
Progestins play a pivotal role in regulating the number of stem and progenitor cells in the normal mammary gland and breast cancer. In the normal murine mammary gland and human breast, progesterone is the critical hormone controlling the number of mammary epithelial stem and progenitor cells. 12 Progestins in combination with estrogens increase breast cancer risk 13 by stimulating proliferation and metastatic potential, as well as through a mechanism postulated to involve amplification of quiescent stem cells. 14 Progestins also expand the number of CK5+ cells in breast cancer cell lines and xenograft tumors. 7 Notably, two articles recently described the conversion of non-CSCs to a CSC state, suggesting that tumor cells can acquire this undesirable phenotype from internal or external stimuli.15,16 Thus, it is critical to develop methods to both prevent and revert the CSC phenotype during therapeutic intervention.
We have previously developed a model to track dynamic expression of the biomarker vimentin in mesenchymal TN human MDA-MB-231 breast cancer cells. 17 Using this model, we identified small molecules that revert epithelial-mesenchymal transition (EMT) and prevent invasion. These data demonstrated that surrogate reporters of well-characterized biomarker expression offer an effective methodology, suitable for high-throughput drug discovery, to identify small molecules that can revert or suppress the malignant phenotype to a relatively more benign state.
In an effort to discover small-molecule agents that specifically modulate the CSC phenotype in luminal subtype ER+PR+ breast cancers, we developed a reporter-based assay using the putative stem cell biomarker CK5. This model uses a CK5 promoter-driven enhanced green fluorescent protein (GFP) reporter stably transduced into T47D human luminal breast cancer cells (CK5Pro-GFP-T47D). GFP functions as a biomarker readout to identify small-molecule modulators of this CSC phenotype. Herein, we have characterized the CK5 promoter activity as a faithful reporter of CK5 protein expression using a CK5Pro-Fluc (firefly luciferase) reporter and a CK5Pro-GFP reporter. In this model system, addition of progesterone boosts the <2% de novo baseline CK5+ cell population up to 20% of the total cell population, a number that is suitable for high-content screening (HCS). We validated the CK5Pro-GFP-T47D model for HCS in 96-well microplates and subsequently performed a pilot screen using a focused library of 280 compounds obtained from the National Institutes of Health (NIH) clinical collection number 2 that yielded four effective compounds.
Material and Methods
Cell Culture and Reagents
Monoclonal antibodies NCL-L-CK5 (1:500 dilution; Leica Microsystem, Buffalo Grove, IL), MAB3580 (1:2500 dilution; Millipore, Billerica, MA), and AC-15 (1:5000 dilution; Sigma Aldrich, St. Louis, MO) were used to detect CK5, GFP, and β-actin, respectively. Infrared secondary antibody IRDye 800CW (catalog # 926-32210, 1:10000 dilution; Li-Cor, Lincoln, NE) was used for detection following incubation with primary antibodies. Alexa Fluor 594 goat antimouse was purchased from Invitrogen (Carlsbad, CA), and protease inhibitors were purchased from Sigma Aldrich. ONE-Glo Luciferase Assay Reagent and CellTiter-Glo reagent were purchased from Promega (Madison, WI). T47D (ATCC) and CK5Pro–(Fluc or GFP)–T47D breast carcinoma cells were maintained (37 °C, 5% CO2) in cell culture using Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS), 200 ng/mL puromycin (for stably transduced cell lines), and 1 nM insulin. For human HEK 293T (ATCC, Manassas, VA) cells and experiments with T47D cells (including reporter cell lines), the cell culture medium was limited to DMEM plus 10% FBS unless otherwise stated.
CK5 Promoter Reporter Cloning and Stable Cell Line Engineering
CK5Pro-Fluc and CK5Pro-GFP cloning
A 6034-bp fragment (–6002 to +32) of the proximal promoter for the human KRT5 gene 18 was amplified by PCR and cloned into the pCDH1 lentiviral vector in place of the cytomegalovirus (CMV) promoter using ClaI/EcoRI sites. Reportable markers luciferase ((luc)2p fragment; Promega) and enhanced GFP were placed downstream of the promoter using Swa1/Not1 sites (luc2P) and EcoRI/NotI sites (GFP) within the multiple cloning site of the pCDH1 lentiviral vector. All PCR products and plasmids were confirmed by sequencing.
Stable transduction of CK5Pro-Fluc-T47D and CK5Pro-GFP-T47D cell lines
The 7.5 × 106 HEK 293T cells were seeded in T-75 flasks 24 h prior to transfection (to be 70% confluent at the time of transfection). Plasmids pCMV-VSV-G, pHR-8.2 ΔR, and pCDH1–CK5–(Fluc or GFP)–EF1–puro were transfected using liposomal LT 1 transfection reagent as described by the manufacturer (Mirus, Pittsburgh, PA). Transfected cells were incubated for 48 h. The virus-containing medium was collected and filtered through a 0.45-µm filter. The supernatant containing viral particles was supplemented with polybrene (8 µg/mL), added to T47D cells (50% confluent) in 100-mm culture dishes, and incubated for 24 h. The medium was removed and then replaced with fresh medium. After 3 days, transduced T47D cells were selected with 1 µg/mL puromycin for 7 days.
CK5Pro-Fluc-T47D Cell Luciferase Assay
CK5Pro-Fluc-T47D cells were seeded at 1 × 104 cells per well in white clear-bottom 96-well plates and incubated overnight. Cells were treated with either 100 nM progesterone alone or 100 nM progesterone plus 1 µM RU486 for 48 h, and luminescence was measured using the ONE-Glo assay (Promega). A cell viability assay using the CellTiter-Glo luminescent assay (Promega) was performed in parallel to normalize CK5 promoter activity. For both assays, the medium was replaced with 50 µL of fresh medium plus 50 µL of ONE-Glo or CellTiter-Glo Luciferase Assay Reagent. After a 10-min incubation at room temperature, CK5Pro-Fluc activity was measured and luminescence was recorded with an integration time of 1 s/well using a Clarity Luminescence Microplate Reader (Bio-Tek Instruments, Winooski, VT).
CK5Pro-GFP-T47D Cell Fluorescence Assay
CK5Pro-GFP-T47D cells were seeded at 4 × 103 cells per well in black clear-bottom 96-well plates (Greiner Bio-One, Monroe, NC) in phenol red–free DMEM plus 10% FBS and incubated overnight. Cells were treated with either 100 nM progesterone or 100 nM progesterone plus 1 µM RU486. The final concentration of DMSO, which was the solvent for both progesterone and RU486, was 0.5%. After a 72-h incubation, cells were stained with 10 µM Hoechst 33342 for 10 min and washed with phosphate-buffered saline (PBS). Cells in 15 fields per well were analyzed using a 10× objective and the Operetta (PerkinElmer, Waltham, MA) high-content imaging system. The fraction of GFP-positive cells was determined using Harmony software (PerkinElmer) with the following settings: 10× long working distance, nonconfocal mode, 50% excitation, 0% transmission, 400 ms exposure for GFP, 100 ms exposure for Hoechst 33342, method B for nuclei, and method C for cytoplasm.
Western Blot Analysis
CK5Pro-GFP-T47D cells were seeded into 6-well dishes at 3 × 105 cells/well. After 24 h, cells were treated with 100 nM progesterone or DMSO (vehicle control) for 24 h, rinsed with cold PBS, and then scraped in cold PBS containing protease inhibitors and centrifuged at 1500 g for 5 min at 4 °C. Total cellular extracts were prepared by resuspending cell pellets in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 50 mM Tris [pH 7.5]) containing protease inhibitors. Lysates were incubated on ice for 10 min and centrifuged at 10 000 g for 15 min. The supernatants were collected and protein concentrations were determined using the Bradford assay. Then, 100 µg of lysate from each sample was separated on 4% to 12% Bis-Tris NuPAGE gels (Invitrogen) and transferred to PVDF membranes. Membranes were blocked with 5% bovine serum albumin in phosphate buffered saline with Tween20 (BSA/PBST) for 1 h followed by overnight (CK5, GFP) or 1-h (β-actin) incubation with primary antibodies in 3% BSA/PBST. Membranes were incubated with secondary antibodies for 1 h in 3% BSA/PBST. Signals were detected using the Li-Cor Odyssey infrared imaging system, and proteins were quantified using ImageJ (NIH, Bethesda, MD) software normalized to the β-actin loading control.
Immunofluorescence
CK5Pro-GFP-T47D (7 × 103) cells per well were plated on black clear-bottom 96-well plates and cultured overnight. Cells were treated with either 100 nM progesterone or 100 nM progesterone plus 1 µM RU486. After a 48-h incubation, cells were washed with PBS and fixed with 100 µL of 4% paraformaldehyde in PBS for 15 min at room temperature. Cells were washed twice with PBS, incubated with blocking buffer (1 × PBS, 1% BSA, 0.3% Triton X-100) for 1 h at room temperature, washed with PBS 3×, and incubated with anti-CK5 antibody (1:250 dilution) overnight at 4 °C. Cells were then washed with PBS (3×), incubated with Alexa Fluor 594 secondary antibody (1:250 dilution) for 1 h at room temperature, and washed 3× with PBS. Images were taken using the Operetta high-content imaging system (PerkinElmer) and analyzed using Harmony Software (PerkinElmer).
NIH Clinical Collection 2 Pilot High-Content Screen
For the pilot screen, column 1 of each 96-well plate contained negative control (progesterone) and column 12 contained positive control (progesterone and RU486). All control lanes included 0.5% DMSO. Cells were dosed from 384-well plates containing 2-mM stock solutions in DMSO of the NIH Clinical Collection 2 (provided through the NIH Molecular Libraries Roadmap Initiative). Then, 0.5 µL of compound or controls was added using a Janus liquid-handling robot (PerkinElmer) with a 96-tip MDT dispensing head, corresponding to a final compound concentration of 10 µM and 0.5% DMSO, incubated at 37 °C in 5% CO2 for 72 h, and stained as above prior to imaging on the Operetta.
Statistical Analysis
Data were subjected to an unpaired 1-tailed t-test statistical analysis using Prism (GraphPad Software, San Diego, CA), and p-values ≤0.01 (**) or ≤0.001 (***) were considered statistically significant. The NIH Clinical Collection 2 pilot screen was validated using the Z′ factor statistical parameter. 19
Results
Validating the CK5Pro-Reporter in T47D Cells
Assays were performed to determine if CK5 promoter-driven Fluc and GFP serve as adequate markers of endogenous CK5+ cells. To measure progesterone responsiveness, CK5Pro-Fluc-T47D cells were treated with vehicle, 100 nM progesterone, or 100 nM progesterone plus the PR-specific antagonist RU486 20 (1 µM) for 48 h and luciferase assays performed. Progesterone treatment led to a statistically significant sixfold increase in Fluc activity compared with cells treated with vehicle only ( Fig. 1A ). RU486, which competes with progesterone for ligand binding to PR, effectively antagonized progesterone-driven Fluc expression, suggesting this is a PR-mediated mechanism. Twenty percent of progesterone-treated CK5Pro-GFP-T47D cells expressed GFP, consistent with our published studies of progesterone-induced CK5 expression.7,10 Progesterone-induced CK5-Pro-GFP activity was blocked by RU486 ≥94% ( Fig. 1B , D ). Appropriate and simultaneous induction of GFP and CK5 was confirmed by Western blot analysis ( Fig. 1C ). In the presence of progesterone, we observed 22-fold and 25-fold increases in CK5 and GFP protein expression, respectively. Finally, we observed that CK5Pro-induced GFP overlapped CK5 protein expression in CK5Pro-GFP-T47D cells stimulated with progesterone as assayed using immunocytochemistry ( Fig. 1E ). In whole, these data verify that Fluc and GFP readouts appropriately mark the CK5+ population in T47D CK5Pro-Fluc and CK5Pro-GFP cells and that proper hormone control was retained. Thus, we concluded that the CK5-promoter GFP-reporter in T47D cells could be used as a biomarker readout to identify small-molecule modulators of this particular breast CK5+ CSC phenotype.
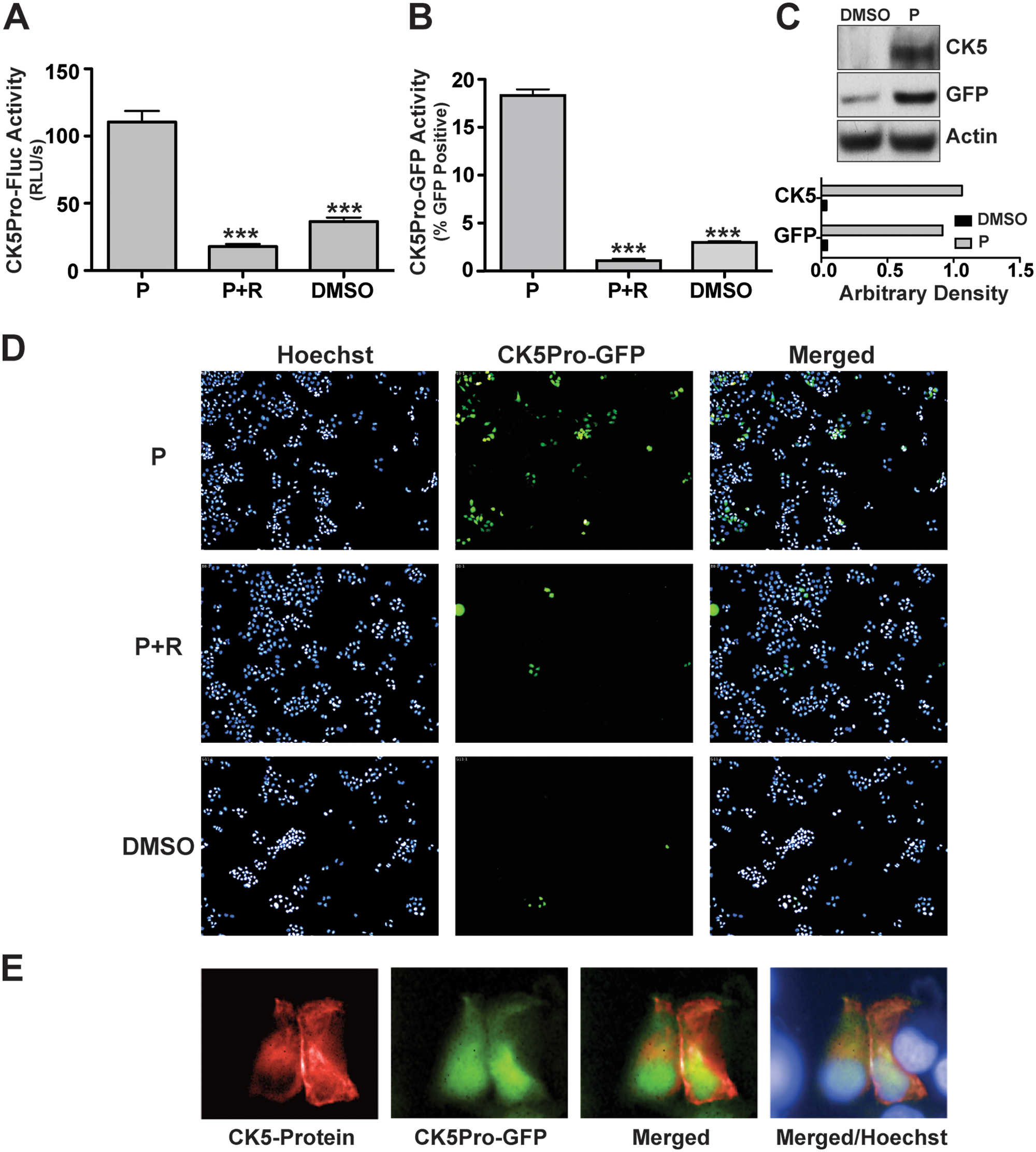
CK5 expression in response to progesterone in T47D cells. CK5 promoter activity was assessed by (
Validating the CK5Pro-GFP-T47D Model for High-Content Screening
Prior to screening for small-molecule modulators of CK5 expression, the quality of the assay was assessed by determining the Z′ factor.
19
Cells were stimulated with progesterone, and the vehicle (DMSO) was used as the negative control, whereas progesterone plus RU486 was used as the positive control. DMSO was held constant at 0.5% v/v in all wells throughout validation. Z′ factor was calculated from the user-defined field “fraction GFP-positive cells” using Harmony software. The fraction of GFP-positive cells was determined by dividing the number of cells that express GFP by the total number of cells, which were determined by counting nuclei stained with Hoechst 33342. To account for variability caused by different locations of control wells, we determined the Z′ factor using data obtained from various locations of controls from three different 96-well plates (see
NIH Clinical Collection 2 Pilot High-Content Screen
To identify small-molecule modulators of CK5 expression and further validate our assay model, we performed pilot screening with 280 compounds from the NIH Clinical Collection 2. Using 3 standard deviations away from the mean of the negative control wells as our hit limit, we identified four compounds that downregulate CK5Pro-GFP activity at 10 µM, including miconazole, retinoic acid, istoretinoin, and acitretin ( Figs. 2 and 3 ). Three of the four 96-well plates had Z′ factors ≥0.5 ( Fig. 2B ). The assay for the plate that failed the Z′ factor cutoff was repeated, and the average Z′ factor between the two runs was 0.3. Z′ factors between 0 and 0.5 are considered suitable for screening in duplicate. 19 One of the four hits (acitretin) came from this plate and exhibited a similar degree of CK5Pro-GFP suppression on both duplicates of the plate. Importantly, all four compounds were shown to be noncytotoxic based on the CellTiter-Glo cell viability assay as compared with untreated controls ( Fig. 2C ). Interestingly, three of the hits were structurally similar retinoids, indicating that they potentially share a common target. Using the same assay conditions as the HCS, dose-response curves were generated for miconazole and retinoic acid, and the IC50 values were found to be 4 µM and 800 pM, respectively ( Fig. 4 ).
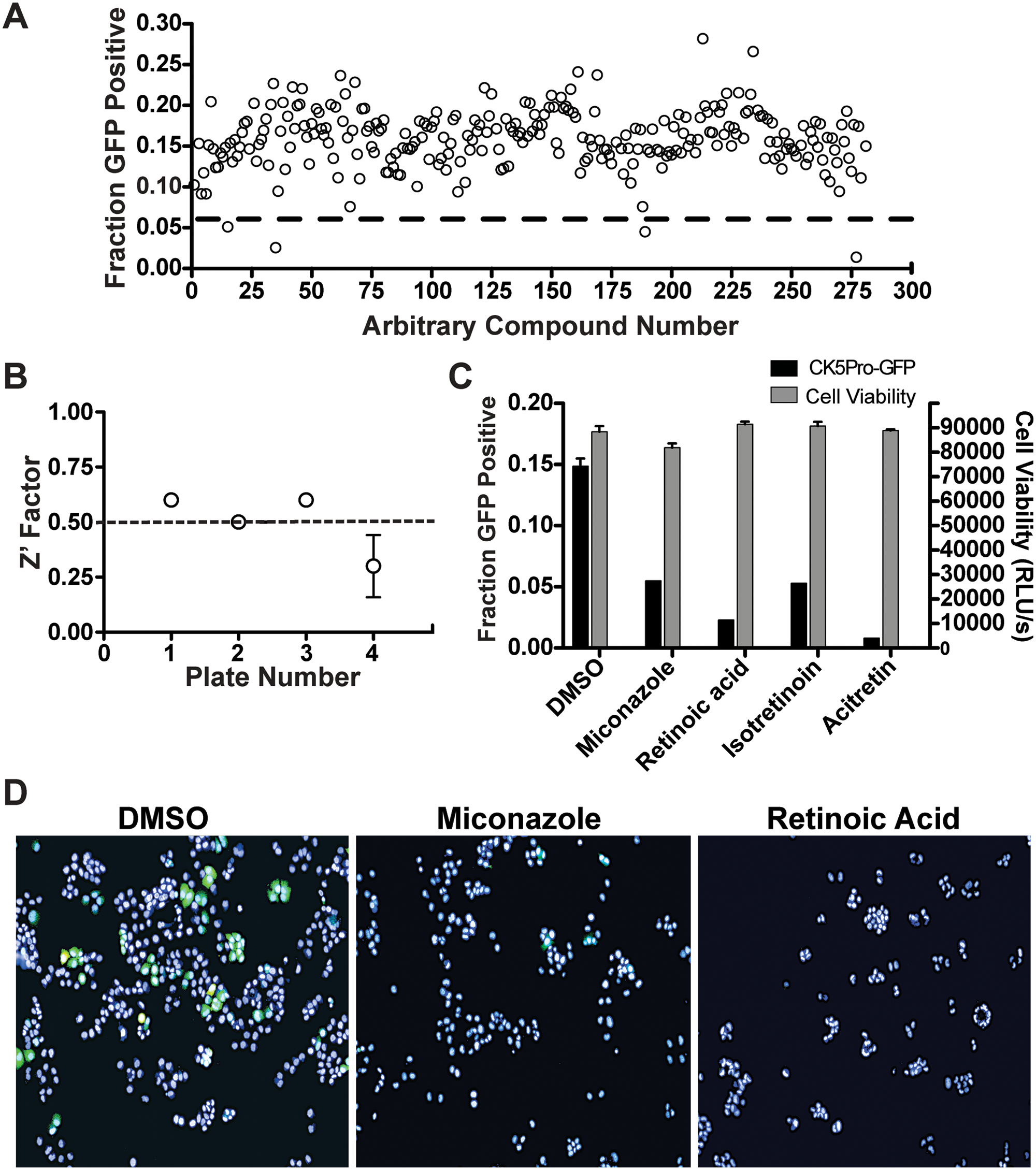
National Institutes of Health (NIH) Clinical Collection 2 high-content pilot screen. (
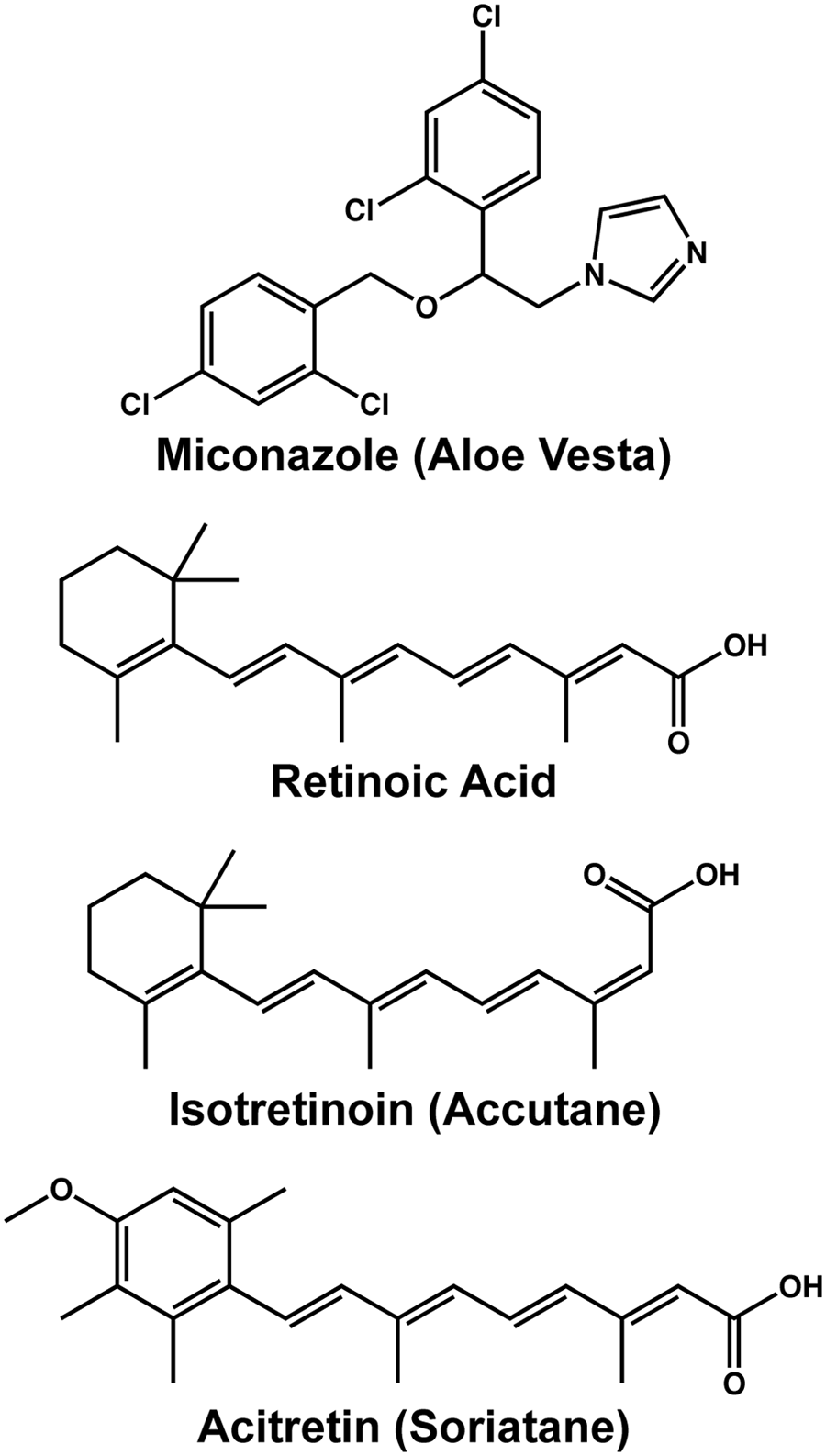
The chemical structures of four hits identified from a high-content pilot screening.
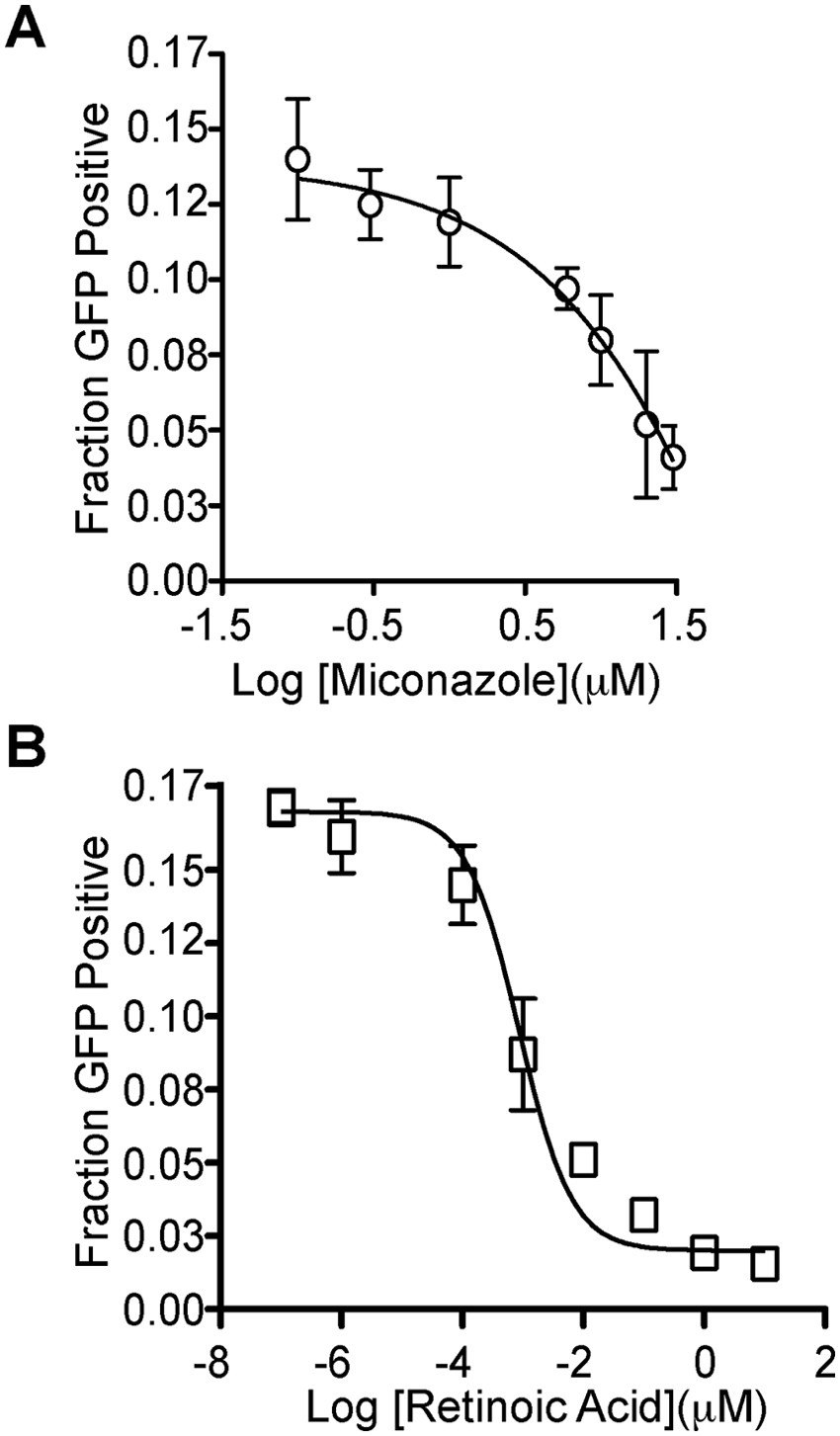
Dose-response curve for the downregulation of CK5Pro-GFP activity by (
Discussion
Breast cancer is a disease of complex heterogeneous tissues, including immune cells, fibroblasts, endothelial cells, extracellular matrices, and proliferating cancer cells that vary by phenotype. It is well accepted that breast tumors harbor a biologically distinct and relatively rare population of “tumor-initiating cells,” defined as CSCs because they have the ability to self-renew, develop into any cell in the overall tumor population, and have the capacity to proliferate and metastasize. 21 Based on these criteria, CSCs may be responsible for the malignant phenotype of breast carcinomas, including drug resistance and high metastatic potential. Treatment regimens such as immunotherapy, chemotherapy, radiation, and surgery cannot guarantee eradication of the CSC population, without which frequent relapse occurs with overall poor prognosis.21,22 Therefore, therapies that specifically eradicate CSCs could offer a more effective strategy for the treatment of malignant breast cancer with reduced risk of relapse and metastasis.21,22 In the most common type of breast cancer, luminal ER+ disease, a subpopulation of stem-like cells is marked by CK5. 7 Therefore, we hypothesized that small molecules that downregulate CK5 expression, independent of cell death, would offer an effective strategy to eradicate this CSC population by sensitizing this population to conventional endocrine or chemotherapy, preventing recurrence and metastasis.
Progesterone induces expansion of the CK5+ population in luminal ER+ breast cancer cells and thus was used as a tool to measure induction and blockade of the CK5 CSC phenotype ( Fig. 1A , B , D ). To characterize the responsiveness of the CK5 reporters, we treated both stable T47D-reporter cell lines with progesterone and progesterone plus RU486. As expected, progesterone induced CK5, whereas the addition of RU486 prevented the progesterone-mediated expansion of CK5+ cells. Importantly, we observed increased expression of GFP in CK5Pro-GFP-T47D cells treated with progesterone, and CK5Pro-GFP activity co-localized with CK5 protein expression ( Fig. 1C , E ).
We postulated that CK5Pro-GFP-T47D cells, developed for high-content screening, would offer an efficient and informative assay to identify small-molecule modulators of the CK5-expressing CSC subpopulation. Therefore, we optimized the assay in 96-well plates, using RU486 as a positive control and DMSO as a negative control (see
In the epidermis, expression of several cytokeratin genes, including CK5, is regulated by keratin-response elements (KREs) via binding of nuclear receptors, including glucocorticoid (GR), thyroid hormone receptor, and retinoic acid receptors (RARs). 23 Binding of four monomers of ligand-bound GR to KREs leads to transcriptional repression of epithelial keratins, including CK5. 24 Interestingly, miconazole can act as a GR ligand and thus could be regulating CK5 expression through GR in breast cancer cells. 25 The fact that PR, a steroid receptor closely related to GR and that binds to similar hormone response elements (HREs), activates rather than inhibits CK5 expression suggests cell type and receptor specificity of the CK5 promoter. Retinoids can inhibit expression of CK5 by several plausible mechanisms. First, retinoic acid can antagonize PR-mediated transcription, likely by reducing PR protein levels. 26 Retinoids can be negatively regulating CK5 expression through direct transcriptional repression by RAR/retinoid X receptor (RXR) heterodimer binding to KREs. 24 A third mechanism involving suppression of nuclear factor–κB (NF-κB) is further discussed below. Therefore, it appears that the hits we have identified all potentially target the expression of CK5 through nuclear receptor–mediated mechanisms of action.
Miconazole is also a ligand for pregnane X receptor (PXR), another member of the nuclear receptor family. PXR binds to its DNA response elements as a heterodimer with RXR.27,28 Recently, Papi et al 29 reported that agonists for RXR, RAR, and peroxisome proliferator-activated receptor (PPAR-γ) reduce the inflammation-dependent survival of breast CSCs through hampering the activity of proinflammatory NF-κB, which downregulates genes that regulate the CSC phenotype, notably including the notch pathway and potentially CK5 expression. Moreover, NF-κB and downstream target genes are part of the “invasiveness gene signature” (IGS) of breast cancer, associated with high metastatic potential and poor prognosis. 30 Papi et al 29 also showed that inhibiting NF-κB activity leads to upregulation of CK18 and ERα, both absent in CK5+ cells, indicating a loss of the CSC phenotype. Therefore, it is plausible that miconazole and the retinoids may act by inhibiting NF-κB activation, leading to reduced inflammation-dependent survival of the CSC population in T47D breast cancer cells.
In conclusion, we have developed a novel fluorescence-based biomarker model to measure the modulation of the CK+ CSC phenotype in luminal ER+ breast cancer. We have validated this model through pilot screening using a focused library of clinically used drugs, demonstrating the assays’ ability to identify potential new uses for existing clinically used drugs. Future studies will focus on expanded HCS to identify other potential lead compounds and to test our hypothesis that small molecules that downregulate CK5 expression can sensitize the drug-resistant and malignant CK5+ CSC population in ER+ breast cancer.
Footnotes
Acknowledgements
The authors thank Professor Elaine Fuchs, Rockefeller University, for providing us with the 6-kb fragment of the human CK5 5′ promoter region. The authors also thank the High Throughput and High Content Screening and DNA sequencing core facilities at the University of Colorado Anschutz Medical Campus for their contributions to this manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded in part by grants from the Cancer League of Colorado (to C.A.S. [principal investigator (PI)] and D.V.L. [co–PI]), Cancer League of Colorado (S.D.A.), the National Institutes of Health (NCI R01 CA140985; C.A.S. [PI], and D.V.L. [co-PI]), and startup funds provided by the Skaggs School of Pharmacy and Pharmaceutical Sciences (D.V.L.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.