
Abstract
Apolipoprotein A-I (ApoA-I), a primary protein component of high-density lipoprotein (HDL), plays an important role in cholesterol metabolism mediating the formation of HDL and the efflux of cellular cholesterol from macrophage foam cells in arterial walls. Lipidation of ApoA-I is mediated by adenosine triphosphate (ATP) binding cassette A1 (ABCA1). Insufficient ABCA1 activity may lead to increased risk of atherosclerosis due to reduced HDL formation and cholesterol efflux. The standard radioactive assay for measuring cholesterol transport to ApoA-I has low throughput and poor dynamic range, and it fails to measure phospholipid transfer. We describe the development of two sensitive, nonradioactive high-throughput assays that report on the lipidation of ApoA-I: a homogeneous assay based on time-resolved fluorescence resonance energy transfer (TR-FRET) and a discontinuous assay that uses the label-free Epic platform. The TR-FRET assay employs HiLyte Fluor 647–labeled ApoA-I with N-terminal biotin bound to streptavidin-terbium. When fluorescent ApoA-I was incorporated into HDL, TR-FRET decreased proportionally to the increase in the ratio of lipids to ApoA-I, demonstrating that the assay was sensitive to the amount of lipid bound to ApoA-I. In the Epic assay, biotinylated ApoA-I was captured on a streptavidin-coated biosensor. Measured resonant wavelength shift was proportional to the amount of lipids associated with ApoA-I, indicating that the assay senses ApoA-I lipidation.
Introduction
Adenosine triphosphate (ATP) binding cassette transporter A1 (ABCA1) is the critical transporter of phospholipid and cholesterol to lipid-free and lipid-poor apolipoprotein A-I (ApoA-I) and is responsible for the formation of nascent high-density lipoprotein (HDL).1–3 ABCA1 mediates cholesterol efflux from peripheral tissues.1,4 In humans, low HDL is a risk factor for the development of cardiovascular disease (CVD).5,6 Numerous studies using knockout and transgenic mouse models, studies of patients with Tangiers disease (natural genetic ABCA1 knockout), and clinical association studies have validated ABCA1 as a critical transporter in the formation of HDL and a protein that is responsible for HDL levels and resistance to the development of atherosclerosis.1,3,7
Based on these studies, ABCA1 is a logical target for the development of drugs aimed at enhancing HDL formation, improving HDL function, and facilitating cholesterol removal from peripheral tissues. Several ABCA1-targeted strategies to increase transport of phospholipids and cholesterol have been employed, including screening for compounds that would increase ABCA1 mRNA expression, decrease ABCA1 turnover, enhance ABCA1 function, and increase the efflux of cholesterol with the use of synthetic ApoA-I mimetic peptides. Large-scale screening campaigns to identify molecules that upregulate ABCA1 gene expression have identified mainly liver X receptor (LXR) ligands with unacceptable side effect profiles. 8 ApoA-I peptide mimetics show promise as antiatherogenic agents, increasing ABCA1 protein level and enhancing its function. 7 However, issues of cost, delivery, and potential antigenicity are yet unresolved. In the past few years, it has become increasingly evident that a major component of ABCA1 regulation occurs at the posttranslation level through regulation of protein turnover and lipid transport function.9,10 For these reasons, novel assays are required for large-scale screening efforts to identify compounds that increase ABCA1 protein expression and/or function. The current gold-standard assay for ABCA1 function is a radioactive assay that measures the efflux of cholesterol to lipid-poor ApoA-I. 1 Unfortunately, this assay is not well suited for high-throughput screening (HTS) as it employs tritiated cholesterol, has narrow dynamic range, ignores phospholipid transport by ABCA1, and is not easily adaptable to measuring hepatic HDL formation. For these reasons, new tools are required to facilitate the development of sensitive assays with large dynamic range and flexibility allowing for automation. Here, we describe the development and characterization of two novel assays: a homogeneous binding assay that uses a fluorescent ApoA-I variant with the capacity to report changes in lipidation state (lipid mass) in a sensitive and specific application of time-resolved fluorescence resonance energy transfer (TR-FRET) and an orthogonal ApoA-I lipidation assay that employs label-free biosensor technology and is amenable to high-throughput applications.
Materials and Methods
Buffers
Buffers were made with reagent grade chemicals and distilled water that was further deionized by treatment with a Milli-Q water purification system (Millipore, Bedford, MA). Spectrophotometric grade glycerol was from Sigma-Aldrich (St. Louis, MO). Phosphate-buffered saline (PBS) buffer consisted of 8 mM Na2HPO4, 2 mM KH2PO4, 2.7 mM KCl, and 137 mM NaCl (pH 7.4) at 25 °C. Tris-buffered saline (TBS)/homogeneous time-resolved fluorescence (HTRF) buffer consisted of 25 mM Tris, 150 mM NaCl, 5% glycerol, and 0.5 mM tris(2-carboxyethyl)phosphine (TCEP) (pH 7.4) at 25 °C.
Cloning, Expression, and Purification of Mature N8H-Avi-ApoA-I and N8H-Avi-ApoA-I(E136C)
Full-length human ApoA-I 11 was obtained from Genecopoeia (Rockville, MD) as an N-terminal Avi-tagged construct optimized for insect cell expression. To obtain a sequence encoding only the mature ApoA-I, a pair of forward and reverse primers was designed (GATGAACCCCCCCAGAGCCCCT and AAGCTTTTC-TTCGTGCCATTCGATTTTC, respectively) to remove the propeptide sequence by site-directed mutagenesis using Phusion Site-Directed Mutagenesis kit (NEB, Ipswich, MA). A sequence of the gene encoding the mature (with propeptide sequence removed) N-terminal Avi-tagged ApoA-I was confirmed by sequencing and then PCR-amplified with a pair of PCR primers carrying an NdeI site and a HindIII site at the 5′ and 3′ end, respectively. The PCR product was digested by NdeI and HindIII, gel purified, and cloned into a modified pET28a vector (Novagen, Darmstadt, Germany) with an engineered rTEV-cleavable N-terminal 8-His tag. For the generation of E136C mutant, a pair of oligonucleotides—E136Cplus (CGCCAGAA-GCTGCACTGTCTGCAAGAGAAGCT) and E136Cminus (AGCTTCTCTTGCAGACAGTGCAGCTTCTGGCG)—was used to mutate the glutamic acid residue at position 136 to cysteine using the Strategene QuikchangeII XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) with the wild-type construct as template. N8H-Avi-ApoA-I(E136C) expression construct was cotransformed with pACYC-BirA plasmid into BL21DE3 cells for protein expression. An overnight culture of the mutant ApoA-I expression strain was used to inoculate fresh LB broth containing kanamycin and chloramphenicol at an initial density of 0.05 OD600/mL. The culture was grown at 37 °C and 250 rpm for 3 to 4 h until the OD600/mL reached 0.8 to 1.0. The culture was cooled to 30 °C, and protein expression was induced by the addition of isopropyl-β-D-thio-galactoside (IPTG) to a final concentration of 1 mM. At the time of induction, L-biotin was also added to a final concentration of 50 µM. After 4 h of induction at 30 °C, cells were harvested by centrifugation and stored at −80 °C.
An 8-g cell pellet was resuspended in 60 mL of lysis buffer (50 mM Tris [pH 8.0], 200 mM NaCl, 2 mM CHAPS, 10% glycerol, 3 mM 2-mercaptoethanol, 2 mM PMSF) and the resulting suspension passed through a microfluidizer two times at 18 000 psi. The lysate was centrifuged for 45 min at 4 °C at 22 680 g, and the pellet was discarded. The clarified lysate was adjusted to 20 mM imidazole and loaded onto a His-Trap FF column (GE Healthcare Life Sciences, Piscataway, NJ) preequilibrated with buffer A (50 mM Tris [pH 8.0], 200 mM NaCl, 20 mM imidazole, 10% glycerol, 3 mM 2-mercaptoethanol) using an AKTA FPLC at 3 mL/min flow rate. Proteins were eluted using a linear gradient from 0% to 100% buffer B (50 mM Tris [pH 8.0], 200 mM NaCl, 500 mM imidazole, 10% glycerol, 3 mM 2-mercaptoethanol) over 20 column volumes. The fractions were analyzed by Caliper LabChip (Caliper Life Sciences, Hopkinton, MA), and fractions containing the target protein were pooled, diluted 1:5 in PBS buffer (pH 7.0), and loaded onto a streptavidin resin preequilibrated with 10 column volumes of PBS buffer (pH 7.0). Bound proteins were eluted with 10 mL of PBS buffer (pH 7.0) containing 8 M urea. After analysis on a 4% to 12% Bis-Tris NuPAGE gel (Invitrogen, Carlsbad, CA) with MES-SDS running buffer (Invitrogen), the biotin-containing protein fractions were pooled and dialyzed against the storage buffer (50 mM Tris [pH 7.0], 150 mM NaCl).
Concentration of N8H-Avi-ApoA-I(E136C) was determined spectrophotometrically from absorbance at 280 nm using an extinction coefficient of 3.942 × 104 M−1cm−1 calculated from an amino acid sequence with the program Sedenterp. 12 The presence of a biotin moiety incorporated into the Avi-tag of the protein during expression in Escherichia coli cells and the intact molecular mass of recombinant proteins were confirmed by mass spectrometry on an Agilent 6210 time-of-flight mass spectrometer with an Agilent 1200 Rapid Resolution HPLC. The samples were run on an Agilent Zorbax 300 Extend C18 Rapid Resolution column at 70 °C, using reverse-phase chromatography with a gradient from 20% to 90% of acetonitrile containing 0.1% formic acid. Data were processed via Agilent Masshunter B.03 Qualitative Analysis, with the Bioconfirm upgrade, allowing for protein deconvolution.
Labeling of N8H-Avi-ApoA-I(E136C) with HiLyte Fluor 647 C2 Maleimide
Labeling of N8H-Avi-ApoA-I (E136C) was performed as described previously with the following modifications. 13 N8H-Avi-ApoA-I(E136C) at 2.03 mg/mL was dialyzed overnight against buffer containing PBS and 1.5 mM TCEP. Dialyzed ApoA-I was denatured by addition of 8 M guanidine-HCl in PBS buffer to the final concentration of 5 M guanidine-HCl. The denatured protein (1.7 mg) was loaded onto a column packed with 1.2 mL Ni2+-NTA resin equilibrated with 5 M guanidine-HCl in PBS buffer. The column was washed with 15 column volumes of 5 M guanidine-HCl in PBS to remove TCEP from the protein sample. N8H-Avi-ApoA-I(E136C) bound to the Ni2+-NTA column was labeled with HiLyte Fluor 647 C2 maleimide (Anaspec, Fremont, CA) by the addition of 1.5 mL of a solution containing 0.43 mM fluorophore in 5 M guanidine-HCl and PBS (the molar excess of HiLyte Fluor 647 over the protein in the labeling reaction was 12-fold). The labeling reaction was allowed to proceed for 2 h at room temperature in the dark with slow end-to-end mixing. Unincorporated fluorophore was removed by washing the column with 20 volumes of a buffer containing 5 M guanidine-HCl in PBS. N8H-Avi-ApoA-I(E136C)–HiLyte 647 was refolded on the column by washing with 20 volumes of TBS/HTRF buffer. Subsequently, the protein was eluted by addition of TBS/HTRF buffer with 0.5 M imidazole while collecting ~0.5-mL fractions. The fractions were analyzed by UV-Vis spectroscopy and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 4% to 12% Bis-Tris NuPAGE gel with MES-SDS running buffer. Fluorescently labeled proteins were detected by scanning the unstained gel with a Typhoon Fluoroimager (Molecular Devices, Sunnyvale, CA), and then proteins were visualized by Coomassie blue staining. Fractions containing N8H-Avi-ApoA-I(E136C)–HiLyte 647 were pooled and dialyzed against 2 L of TBS/HTRF buffer overnight at 4 °C. Aliquots of N8H-Avi-ApoA-I(E136C)–HiLyte 647 were flash-frozen using liquid nitrogen and stored at −80 °C. The concentration of N8H-Avi-ApoA-I(E136C)–HiLyte 647 was determined spectrophotometrically from the absorbance at 280 nm, corrected for the contribution from the absorbance of HiLyte 647 fluorophore at this wavelength (11% of absorbance of HiLyte Fluor 647 at 652 nm; extinction coefficient of HiLyte Fluor 647 at 280 nm in TBS/HTRF buffer was determined to be 22135 ± 2224 M−1cm−1) and using an extinction coefficient calculated from an amino acid sequence (3.942 × 104 M−1cm−1) with the program Sedenterp. 12 The protein concentration was also determined using the Bradford assay (Bio-Rad, Hercules, CA) with bovine serum albumin (BSA) as a standard. Both determinations of the concentration of N8H-Avi-ApoA-I(E136C)–HiLyte 647 were within experimental error. The concentration of the HiLyte Fluor 647 in protein samples was determined spectrophotometrically from absorbance at 652 nm in TBS/HTRF buffer using an extinction coefficient of ϵ652 = 17881 ± 1499 M−1cm−1 calculated from the dilutions of known concentrations of fluorophore stock as described previously. 14
Homogeneous TR-FRET Lipid-Sensing ApoA-I Assay
A lipid-sensing ApoA-I assay based on homogeneous TR-FRET technology was performed using terbium cryptate as a donor of fluorescence and HiLyte Fluor 647 as an acceptor of fluorescence. HiLyte Fluor 647 was covalently attached to the cysteine residue engineered at the 136 position of mature ApoA-I. Terbium cryptate–labeled streptavidin was purchased from Cisbio (Bedford, MA). The assay was performed in a final volume of 50 µL in TBS/HTRF buffer containing 0.1 mg/mL BSA, 0.667 nM streptavidin–terbium cryptate conjugate, and up to 700 nM N8H-Avi-ApoA-I(E136C)–HiLyte 647 or recombinant fluorescent HDL particles prepared as described below. Plates were read after a 1-h incubation at room temperature on an EnVision 2102 Multilabel Reader (PerkinElmer, Waltham, MA) using a UV2 (TRF) 320-nm excitation filter and 590-nm and 665-nm emission filters with dichroic mirror D400. Raw counts per second (cps) at 665 nm and 620 nm were collected, and the signal was expressed as the ratio of (cps at 665 nm/cps at 620 nm) × 1000.
Preparation of Recombinant High-Density Lipoprotein Particles
Recombinant HDL (rHDL) were prepared using either recombinant N8H-Avi-ApoA-I(E136C)–HiLyte 647 or unlabeled N8H-Avi-ApoA-I(E136C), as previously described for native ApoA-I. 15 Briefly, free cholesterol and phospholipid 1-palmitoyl-2-oleoyl-sn-glycero-3- phosphocholine (POPC) (Avanti Polar Lipids, Alabaster, AL) solutions were extensively dried in a Speedvac at 37 °C. Recombinant N8H-Avi-ApoA-I(E136C) was added to yield the desired molar ratio of phospholipids to free cholesterol to recombinant protein (PL:FC:N8H-Avi-ApoA-I(E136C)). The solution was emulsified, and the lipids were dispersed with the addition of solution of 300 mg/mL sodium cholate (Sigma-Aldrich) to a final molar ratio of sodium cholate:phospholipid of 6:1. For some rHDL preparations, the solution remained opaque and required addition of extra quantities of sodium cholate (10 µL at a time) until it become clear. Then, 10× TBS was added to give a final concentration of 50 mM Tris HCl, 150 mM NaCl (pH 7.4). The solution was then incubated at 37 °C in a shaking water bath for 30 min prior to extensive dialysis, using 6 × 4 L of TBS containing 0.02% NaN3 over 24 h using 10 000 MWCO dialysis tubing. rHDL were characterized by native PAGE using 4% to 20% Tris-Glycine gels (Invitrogen). Gels were run at 4 °C for 1400 Vh and visualized either by red channel fluorescence or after staining with SimplyBlue Safestain (Invitrogen) using a Versadoc MP4000 (Bio-Rad). The Stokes diameters for the rHDL particles were calculated from high molecular weight (HMW) protein standards with known Stokes diameters (GE Healthcare).
Cholesterol Efflux from Differentiated THP1 Cells in the Presence of Recombinant ApoA-I
THP1 cells (ATCC, Manassas, VA) were cultured in RPMI medium (Cellgro, Manassas, VA) containing 10% fetal bovine serum (FBS), 0.05 mM 2-mercaptoethanol, and antibiotic solution (Cellgro; 100 U/mL penicillin, 100 µg/mL streptomycin) at 37 °C in a humidified atmosphere consisting of 95% air and 5% carbon dioxide. THP1 cells were seeded in 12-well plates at a density of 1.2 million cells/well and differentiated into macrophages by incubation in complete medium containing 100 ng/mL phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) for 3 days. For the cholesterol efflux assay, cells were labeled and cholesterol loaded overnight in 1 mL/well RPMI containing 1% FBS, 100 ng/mL PMA, 100 µg/mL ac-LDL (Intracel, Frederick, MD), and 2 µCi/mL 3H-cholesterol (PerkinElmer). Prior to medium preparation, the ac-LDL and 3H-cholesterol were mixed and incubated at 37 °C for 30 min. After overnight labeling and cholesterol loading, the cells were washed twice with PBS containing 2 mg/mL fatty acid–free albumin (FAFA; Sigma-Aldrich) and incubated overnight in serum-free RPMI containing 2 mg/mL FAFA and 100 ng/mL PMA. This additional overnight incubation allowed for equilibration of the labeled and native cholesterol. Immediately prior to initiation of the efflux, the cells were washed twice with RPMI/FAFA. Cholesterol efflux was initiated with the addition of 0.6 mL RPMI, 2 mg/mL FAFA, 100 ng/mL PMA, and 5 nM LXR agonist T0901317, with or without 15 µg/mL of ApoA-I variant. After 6 h of efflux, 100 µL of the medium was removed and counted in 5 mL scintillation fluid. Human apolipoproteins used in the cholesterol efflux experiments were purchased (Sigma-Aldrich), whereas recombinant biotinylated ApoA-I proteins were prepared as described above. Normalized cholesterol efflux was calculated by subtracting the cholesterol efflux detected in samples that did not contain ApoA-I (background) and normalizing the background- subtracted values to the radioactivity of the samples containing human plasma ApoA-I after 6 h of incubation, which was set to 1.0.
Label-Free ApoA-I Capture Assay
The label-free ApoA-I capture assay was performed on the Corning Epic System (Corning, Lowell, MA). Recombinant HDL particles containing N8H-Avi-ApoA-I(E136C) were made as described earlier, and streptavidin was purchased from Sigma-Aldrich. Biochemical Epic plates (Corning) were activated by adding 15 µL/well of 200 mM 1-ethyl-3(3-dimethylaminopropyl)carbodiimide (EDC) and 50 mM Sulfo-NHS (Pierce Thermo Scientific, Rockford, IL) and incubated for 60 min at room temperature. Plates were washed three times with MilliQ water and dried by centrifugation (1 min at 400 rpm). Streptavidin and BSA immobilization was accomplished by resuspending the proteins at a concentration of 40 to 50 µg/mL in 20 mM sodium acetate (pH 5.1) and adding 10 µL per well. The resulting Epic plates were incubated overnight at 4 °C and subsequently blocked with 150 mM borate buffer (pH 9.2) supplemented with 200 mM ethanolamine (Sigma-Aldrich) for 15 min and washed three times with assay buffer containing 1× TBS (Bio-Rad) and 0.02% sodium azide. Assay buffer (15 µL/well) was added, and after a 2-h incubation, a baseline read was taken to determine the amount of streptavidin or BSA immobilized. A new baseline read was determined before 15 µL/well of rHDL containing N8H-Avi-ApoA-I(E136C) was added and incubated for 2 h. Epic plates were washed with assay buffer three times to remove any loosely bound or unbound rHDL with incorporated N8H-Avi-ApoA-I(E136C), and the plates were read to determine the amount of ApoA-I captured.
Results
In the current study, we describe the development and characterization of two orthogonal high-throughput assays that sense the amount of lipids that are associated with ApoA-I. These assays have the potential to be implemented in large-scale screening campaigns aiming to identify new molecules that prevent and treat CVD by targeting ABCA1.
Design of Lipid-Sensing Fluorescent ApoA-I Variant
Design of the fluorescent ApoA-I used in the TR-FRET16,17 lipidation assay was based on several available pieces of information—a published crystal structure of lipid-free ApoA-I 18 ( Fig. 1A ) and the double superhelix model of ApoA-I in rHDL particles derived from small-angle neutron scattering data 19 ( Fig. 1B )—and was inspired by the kinetic studies of ApoA-I transitions between lipid-associated and lipid-free states that was recently published by Cavigiolio et al. 20 Moreover, we used a structural model of lipid-free ApoA-I in solution described in literature. This model was constructed based on a sundry of biophysical studies such as electron paramagnetic spectroscopy of site-directed spin labels in the N-terminal domain of ApoA-I and hydrogen-deuterium exchange studies coupled to mass spectrometry.21,22 These structural data suggest that E136, chosen by Cavigiolio et al. 20 for fluorophore attachment, is located in a flexible, unstructured segment of the protein. Therefore, substitution of a glutamate residue in the 136 position or introduction of a fluorescent probe should have only minimal impact on ApoA-I structure and its lipid-binding activity. In view of that, the lipid-free fluorescent variant of ApoA-I described by Cavigiolio et al. 20 was able to promote rapid exchange of ApoA-I between rHDL-associated and lipid-free states in a manner similar to the unmodified protein. Moreover, the ApoA-I exchange rates were dependent on the relative lipid affinity, the extent of cross-linking, and the extent of MPO-mediated oxidation of lipid-fee ApoA-I, further showing that the fluorescent ApoA-I was functionally active. 20
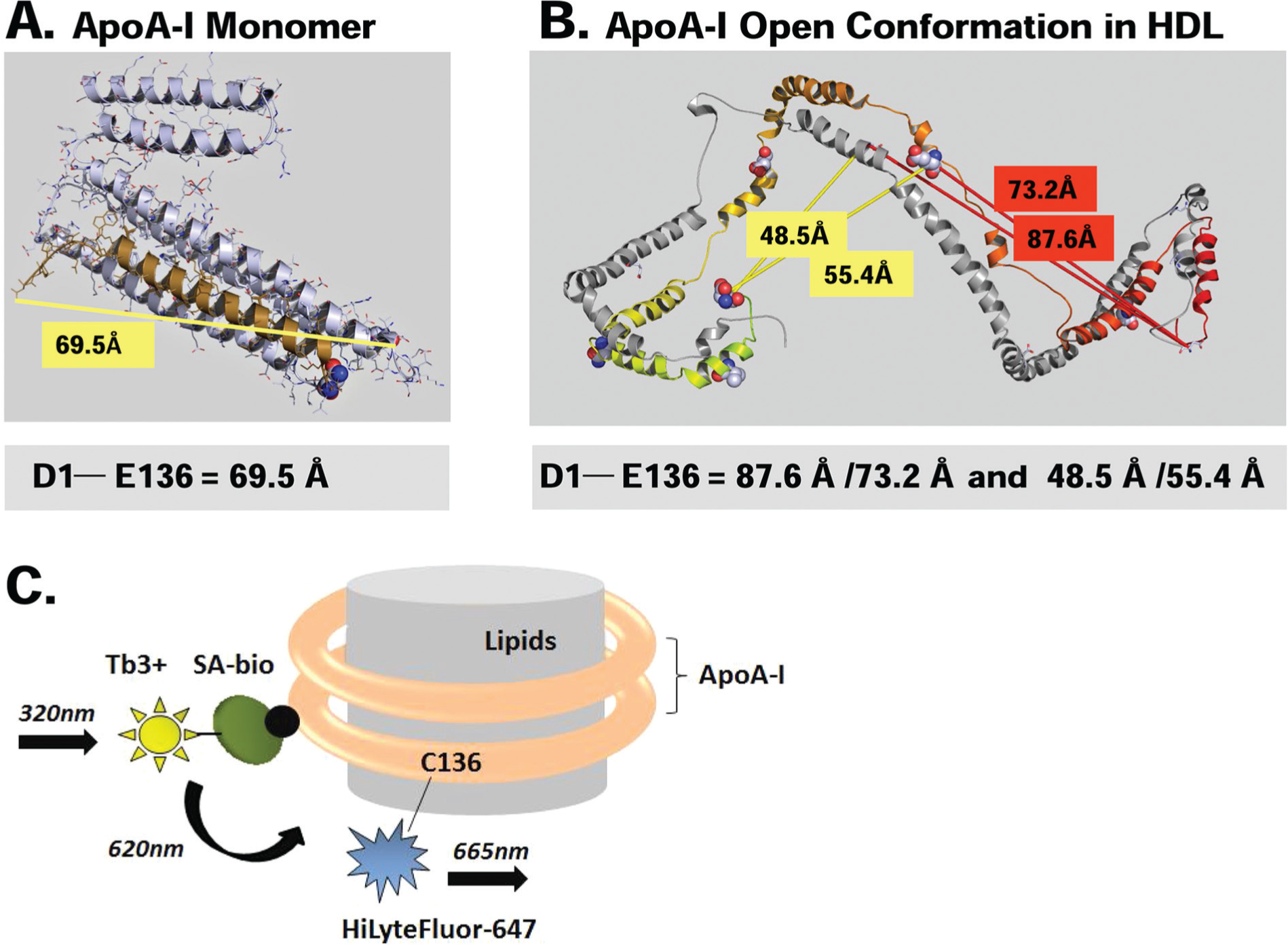
Design of fluorescent apolipoprotein A-I (ApoA-I) and homogeneous time-resolved fluorescence resonance energy transfer (TR-FRET) ApoA-I lipidation assay. (
The fluorescent ApoA-I probe developed in our studies did not require extensive protein engineering to remove endogenous tryptophan residues as it was in the case of protein employed by Cavigiolio et al. 20 In our recombinant ApoA-I construct, a single cysteine residue was introduced at position 136 of the human ApoA-I sequence, replacing a glutamate residue. Recombinant ApoA-I was expressed in E. coli as a fusion with an rTEV-cleavable N-terminal 8-histidine tag followed by an Avi-tag sequence that can be biotinylated in vivo by coexpressed BirA biotin ligase. Cysteine 136 was used to specifically label ApoA-I with FRET acceptor—a HiLyte Fluor 647 using maleimide chemistry. The N-terminal biotin moiety of ApoA-I was bound to the streptavidin labeled with terbium-cryptate conjugate, a cage-like entity that confers increased fluorophore stability.16,23 This moiety serves as the fluorescence donor in the homogeneous TR-FRET assay ( Fig. 1C ). Terbium, similar to other lanthanides, has an extremely long half-life (up to 1 ms) when compared with more traditional fluorophores.16,17,23 The use of long-lived fluorophores, combined with the time-resolved detection (a physical delay between an excitation and the measurement of fluorescence emission), minimizes the interferences from screening compounds and proteins potentially present in cell culture media or biological fluids, the fluorescence of which decays nearly to zero during the delay period. 17 The Förster distance of the terbium and HiLyte Fluor 647 donor-acceptor pair is approximately 70 Å, which is comparable to the distance between Asp1 and Glu136 in monomeric ApoA-I ( Fig. 1A ). The spatial separation between Asp1 and Glu136 in rHDL should be within the range where efficiency of FRET between this fluorophore pair is sensitive to the physical distance between them ( Fig. 1B ). In an assay, terbium-cryptate can be excited at 320 nm. A portion of the energy captured by cryptate during the excitation is emitted at 620 nm, whereas the remaining energy is transferred to the proximally located HiLyte Fluor 647 that emits at 665 nm ( Fig. 1C ).
Characterization of Fluorescent ApoA-I Variant
Recombinant N8H-Avi-ApoA-I(E136C) was purified by Ni-NTA affinity chromatography, and the biotin- conjugated protein fraction was enriched using streptavidin affinity resin. Cys136 of N8H-Avi-ApoA-I(E136C) was labeled with HiLyte Fluor 647 C2 maleimide. UV-Vis spectra and mass spectrometry of N8H-Avi-ApoA-I(E136C) before and after labeling are shown in
N8H-Avi-ApoA-I(E136C)–HiLyte 647 displayed a fluorescence excitation and emission spectra characteristic of the HiLyte Fluor 647 fluorophore with an excitation maximum at 648 nm and emission maximum at 658 nm ( Fig. 2A ). TR-FRET was measured for increasing concentrations of N8H-Avi-ApoA-I(E136C)–HiLyte 647 in the presence of streptavidin–terbium cryptate conjugate ( Fig. 2B ). TR-FRET signal was calculated from the ratio of acceptor fluorescence emission at 665 nm to donor fluorescence emission at 620 nm and corrected for the background fluorescence of TBS/HTRF buffer. In the absence of streptavidin–terbium cryptate (donor), there was no appreciable TR-FRET signal. For a particular amount of streptavidin-terbium, the TR-FRET signal increased initially with an increase in the concentration of N8H-Avi-ApoA-I(E136C)–HiLyte 647 (acceptor) before reaching a plateau ( Fig. 2B ), as a result of the streptavidin-terbium conjugate binding to the biotinylated Avi-tag of fluorescent ApoA-I and formation of a FRET pair ( Fig. 1C ). As the concentration of streptavidin–terbium cryptate increased, the amount of N8H-Avi-ApoA-I(E136C)–HiLyte 647 required to reach the half-maximum TR-FRET signal also increased, reflecting again donor/acceptor complex formation ( Fig. 2B ). TR-FRET obtained for the ApoA-I variant was very robust, with a high signal-to-background ratio of around 35 to 38.
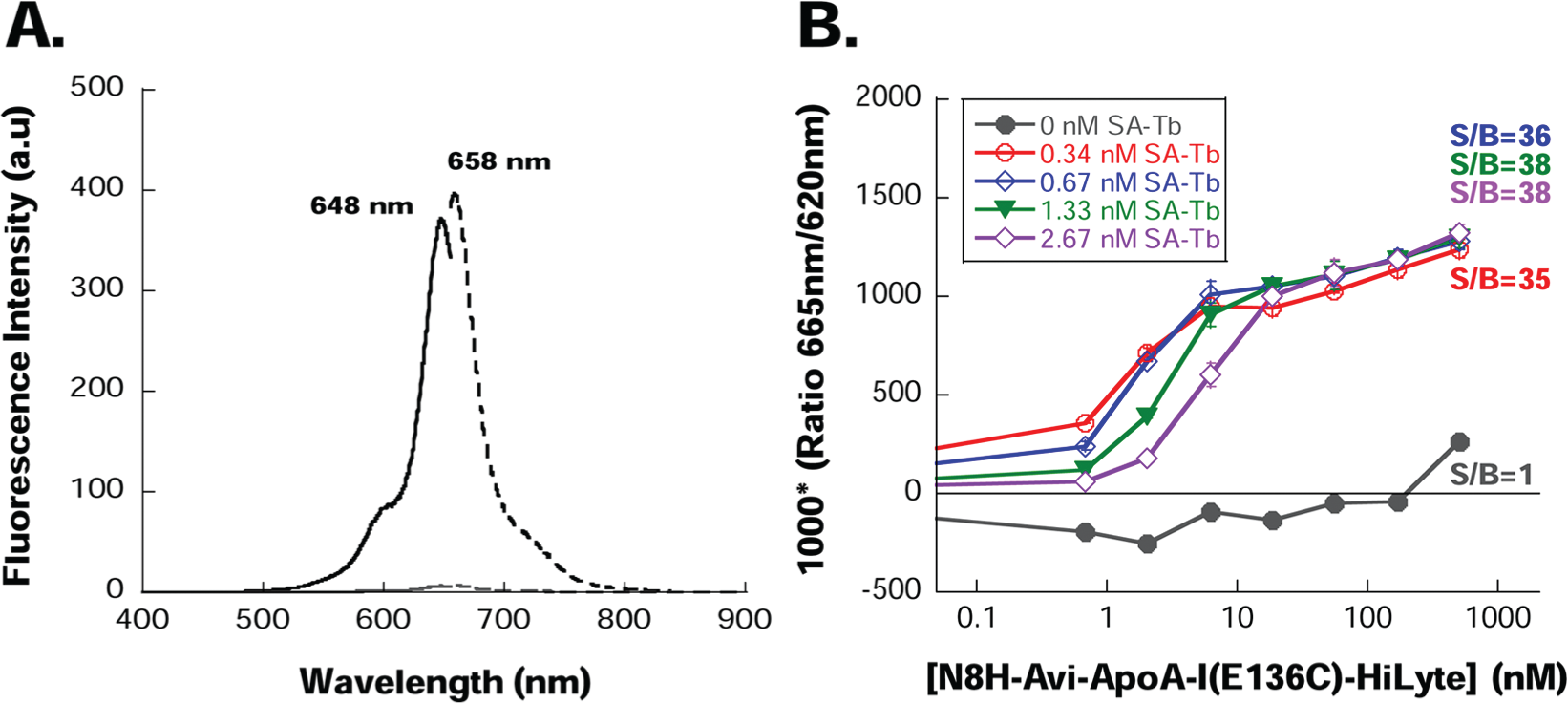
Characterization of fluorescent properties of the lipid-sensing apolipoprotein A-I (ApoA-I). (
The biological activity of unlabeled and HiLyte Fluor 647–labeled N8H-Avi-ApoA-I(E136C) was measured in comparison to a native ApoA-I purified from human plasma in a radioactive cholesterol efflux assay as described in Materials and Methods. Figure 3 depicts normalized cholesterol efflux from differentiated THP1 cells with various ApoA-I constructs after a 6-h incubation. Both unlabeled N8H-Avi-ApoA-I(E136C) and its fluorescent variant were able to participate in cholesterol efflux from differentiated THP1 cells with efficiencies of ~52% to 56% relative to the native ApoA-I from human plasma, showing that engineered ApoA-I remain biologically active ( Fig. 3 ). Lower levels of cholesterol efflux to the engineered unlabeled and fluorescently labeled proteins in comparison to the activity displayed by human plasma ApoA-I are statistically significant (p < 0.05), showing that introduced modifications of ApoA-I have reduced its activity. Activities of fluorescently labeled and unlabeled N8H-Avi-ApoA-I(E136C) are comparable ( Fig. 3 ).
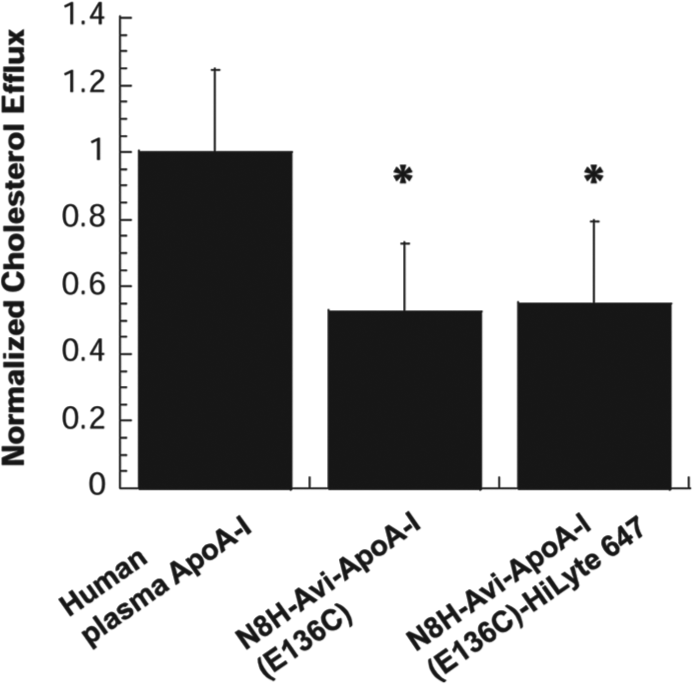
Normalized cholesterol efflux from differentiated THP1 cells induced by addition of various constructs of apolipoprotein A-I (ApoA-I) protein after a 6-h of incubation at 37 °C. Data were normalized by subtracting the background radioactivity (efflux in samples that do not contain ApoA-I) and normalizing the background-subtracted values to the efflux in samples that contain human plasma ApoA-I, which was set to 1.0. Each data point is an average of three independent experiments performed in triplicates. Error bars depict the standard deviation from the mean value. Two-tailed t-test statistical analysis was used to calculate the statistical significance of differences in cholesterol efflux levels measured for unlabeled and HiLyte 647–labeled ApoA-I in comparison to the activity of human plasma ApoA-I. *p < 0.05.
Homogeneous Time-Resolved FRET Assay Measuring the Lipidation State of ApoA-I
Recombinant HDL were prepared using either N8H-Avi-ApoA-I(E136C)–HiLyte 647 or unlabeled N8H-Avi-ApoA-I(E136C) at PL:FC:ApoA-I molar ratios ranging from 80:5:1 to 5:0:1, as described in Materials and Methods. The resultant rHDL particles were characterized by native, non-denaturing gradient gel electrophoresis ( Fig. 4A ). As expected, both the 80:5:1 and 40:3:1 PL:FC:ApoA-I ratios produced larger rHDL, in agreement with particles having two ApoA-I molecules per particle. Consistent with higher lipid content, the 80:5:1 rHDL comprising the heterogeneous population of particles were larger (10–20 nm) than the 40:3:1 rHDL (8–17 nm). The rHDL made at 20:2:1, 10:1:1, and 5:0:1 PL:FC:ApoA-I ratios were consistent with lipid-poor particles composed of a single ApoA-I and could not be distinguished from lipid-free ApoA-I by the gel system. The concentration of N8H-Avi-ApoA-I(E136C)–HiLyte 647 in rHDL particles was measured by the Bradford assay, and the accuracy of determination was evaluated by loading equal amounts of protein onto SDS-PAGE and comparing the fluorescence intensity of the N8H-Avi-ApoA-I(E136C)–HiLyte 647 band in rHDL samples ( Fig. 4A ).
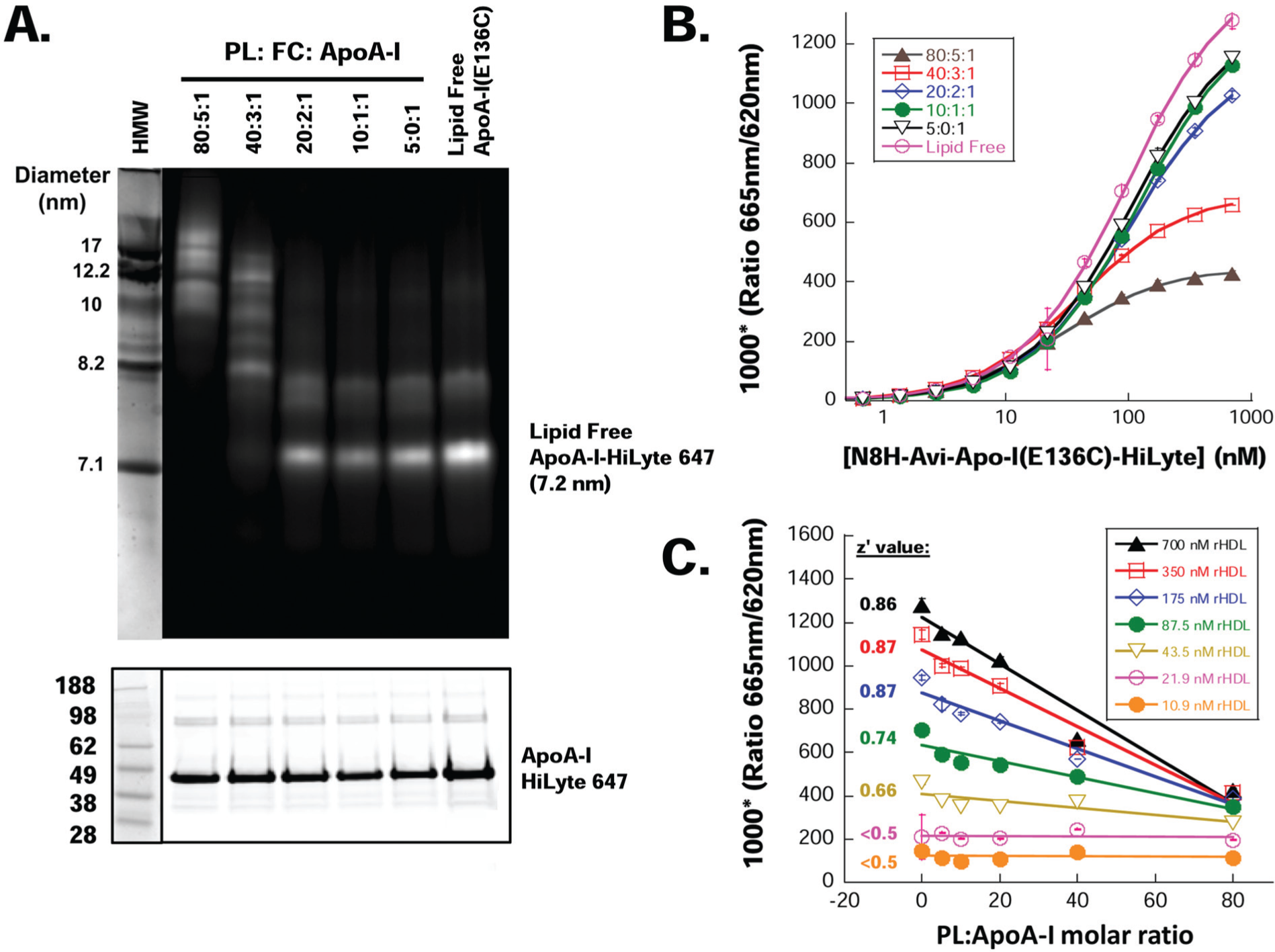
Homogeneous time-resolved fluorescence resonance energy transfer (TR-FRET) assay detects the lipidation state of apolipoprotein A-I (ApoA-I). (
TR-FRET signal was measured in a solution containing increasing concentrations of recombinant HDL particles that were assembled using N8H-Avi-ApoA-I(E136C)–HiLyte 647 and lipids at molar ratios of PL:FC:ApoA-I ranging from 80:5:1 to 5:0:1 ( Fig. 4B ). The TR-FRET signal decreased upon incorporation of ApoA-I into rHDL, and the change was proportional to the increase in the ratio of lipid to ApoA-I ( Fig. 4C ). This observation is in agreement with the expansion of the surface area of lipid molecules that are encircled within the ApoA-I belt as the ratio of lipids to ApoA-I protein increases. This increase in rHDL diameter leads to increased spatial separation of the N-terminal and central regions of the protein in rHDL and hence an increase in the distance between donor and acceptor pair, demonstrating that the homogeneous TR-FRET lipidation assay is sensitive to the amount of lipid associated with ApoA-I. The assay could discriminate various rHDL particles at ApoA-I concentrations as low as 44 nM (1.4 µg/mL). Z′ values of the assay were calculated using lipid-free N8H-Avi-ApoA-I(E136C)–HiLyte 647 and rHDL with a 80:5:1 lipid ratio, as negative and positive control, respectively. Z′ values were ≥0.66 for N8H-Avi-ApoA-I(E136C)–HiLyte 647 concentrations greater than or equal to 44 nM (1.4 µg/mL), with a dynamic window ranging from 1.7 at 44 nM of fluorescent ApoA-I to 3.0 at 700 nM of protein.
Label-Free ApoA-I Capture Assay
To complement the TR-FRET–based ApoA-I lipid-sensing assay, we also developed a lipid-sensing assay using the Corning Epic platform. The Epic system is a high-throughput, label-free platform that allows for the observation of direct biomolecular interactions. The Epic instrument incorporates an evanescent waveguide sensor within each well of its proprietary 384-well microplates. Direct binding or capture of ligands to target proteins immobilized onto the microplate well surface can be detected by exposing the waveguide to a broadband light source. The resulting resonant wavelength shift (measured in picometers [pm]) is proportional to the mass bound to the surface (
For the label-free assay, we also used recombinant N8H-Avi-ApoA-I(E136C) protein in the presence or absence of lipids. To capture biotinylated N8H-Avi-ApoA-I(E136C), we coated the Epic microplate with either streptavidin (50 µg/mL) or BSA (40 µg/mL), which resulted in an immobilized surface density of ~1600 pm (data not shown). BSA-coated Epic plates were used as a control surface to distinguish between actual and nonspecific capture of N8H-Avi-ApoA-I(E136C).
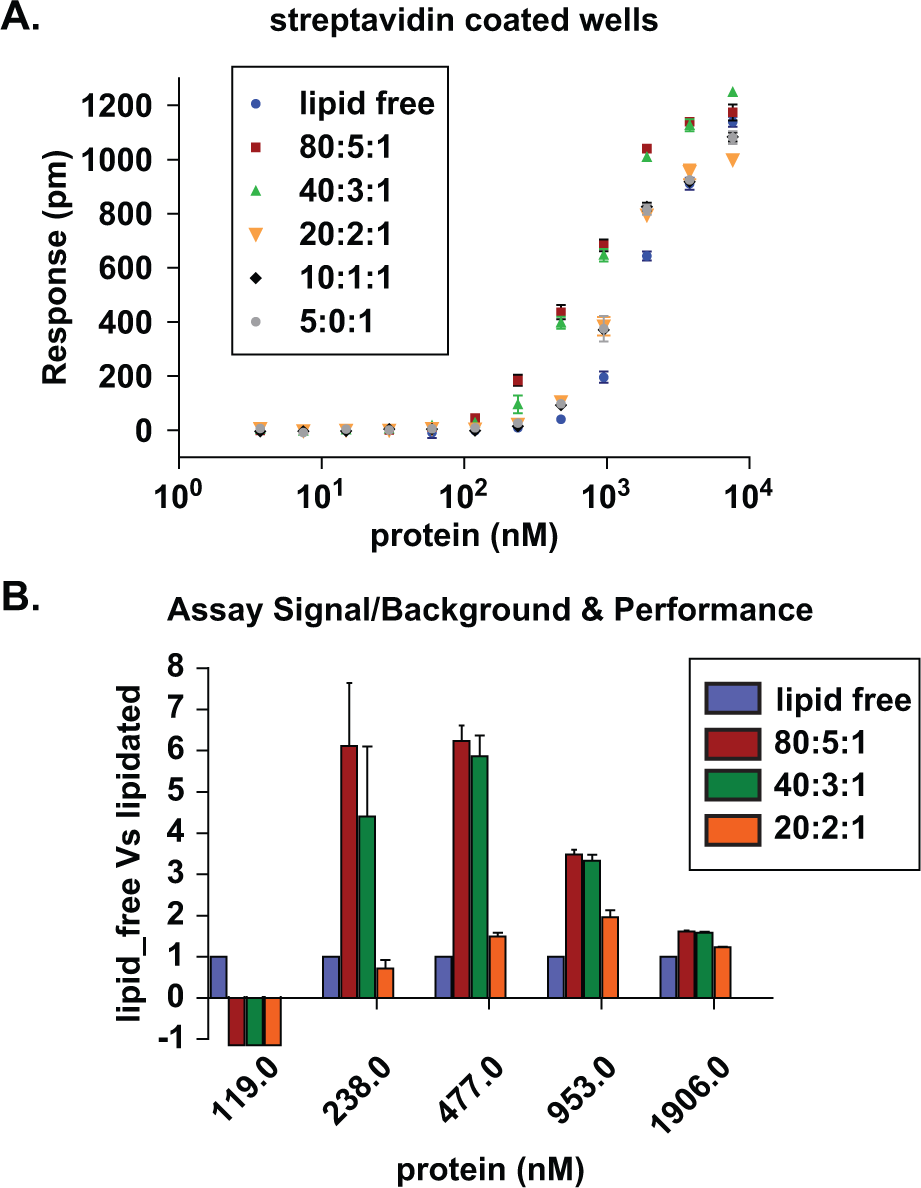
Performance of the label-free ApoA-I capture assay. (
Discussion
Cardiovascular disease is the number one cause of death in the United States and worldwide.25,26 Coronary artery disease (CAD), a major CVD, is responsible for one in every six deaths in the United States and in 2004 was responsible for 1.2 million hospital stays and >$44 billion in expenses. 27 HDL particles play an important role in protection against CAD 6 owing to their involvement in transport of free cholesterol and phospholipids from peripheral tissues to the liver and other organs for downstream processes such as excretion and steroidogenesis. 28 Recent studies suggest that modulation of cholesterol transport that is mediated by ABCA1 may be beneficial in the treatment of CAD. 7 Therefore, ABCA1 appears to be a promising target for the development of a new generation of drugs. Hence, there is a need for sensitive assays to measure ABCA1 activity that have good dynamic range and high-throughput capacities. Toward this end, we have developed two novel assays that are capable of reporting changes in the total mass of lipids associated with ApoA-I and could be employed to screen for small molecules that enhance the function of ABCA1 and increase the lipidation of ApoA-I. Both assays can be performed in an HTS format in contrast to the traditional low-throughput assays for ABCA1 function. Both assays described here are sensitive to the amount of lipids associated with ApoA-I in HDL and can detect concentrations of ApoA-I as low as 44 nM or 1.4 µg/mL (in TR-FRET format) or 240 nM (8 µg/mL) in the Epic version ( Figs. 4C and 5B ). The assays use different readout formats; the TR-FRET assay format is homogeneous and employs a fluorescent variant of recombinant human ApoA-I ( Fig. 1 ). The fluorescently labeled ApoA-I retains its biological activity as indicated by its ability to participate in cholesterol efflux from differentiated THP1 cells ( Fig. 3 ) and to form recombinant HDL particles that vary in size with the amount of lipids that are bound to ApoA-I ( Fig. 4A ). In the TR-FRET version of the assay, the amount of lipid (total lipid mass) loaded into recombinant HDL can be directly read from the magnitude of the FRET signal ( Fig. 4B , C ). The TR-FRET pair used in this assay is very well suited for small-molecule screening purposes as it does not suffer from the interference of natural fluorescence displayed by many screening compounds and unrelated proteins potentially present in the assay. For that reason, the TR-FRET lipid-sensing assay should have minimal background ( Fig. 2B ) and fewer false-positive/false-negative results. 17 The homogeneous format, high throughput, superior sensitivity, and ease of use make the TR-FRET–based ApoA-I lipidation assay very attractive as a screening platform. This assay employs a standard technology validated and widely applied in drug discovery efforts.17,29,30 Contrary to the fluorescent ApoA-I designed by Cavigiolio et al., 20 our probe required less engineering and had negligible background fluorescence. In the study by Cavigiolio et al., a Trp-AEDANS FRET pair was used, which has a rather small Förster distance (Ro) of 22 Å. The fluorescent ApoA-I exhibited a desired property—namely, a distinct fluorescence emission spectrum depending on the lipid association state. Unfortunately, the construction of Trp-AEDANS fluorescent ApoA-I required extensive protein mutagenesis: All endogenous tryptophan residues needed to be mutated to phenylalanine, Val19 was substituted with tryptophan (the FRET donor), and glutamic acid at 136 was mutated to cysteine, which was further modified with AEDAN (the FRET acceptor). This fluorescent probe suffered from interfering background fluorescence emission and required employment of a set of carefully designed control proteins. 20
The second assay developed in our study is label free and employs a high-throughput biosensor technology (
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article beyond normal compensation for employees. The research represents work done during employment and was supported by internal funding.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.