
Abstract
Cyclic adenosine monophosphate (cAMP) is a second messenger of many G-protein-coupled receptors (GPCRs) and a useful readout molecule to estimate the biological activity of various GPCR-specific agents. Here we report the development and use of a Förster resonance energy transfer (FRET) biosensor for cAMP (Epac2-camps) combined with a baculovirus-based BacMam transduction system. The constructed BacMam-Epac2-camps viral transduction system is a simple and robust tool for ligand screening at the second-messenger level in a variety of mammalian cell lines. The level of biosensor protein expression can easily be adjusted in a dose-dependent manner depending on the multiplicity of viral infection. For setting up the assay, we used a B16F10 murine melanoma cell line with endogenous expression of melanocortin-1 receptor (MC1R). The receptor activation was characterized by a set of MC1R full and partial agonists. Bivalent ions Ca2+ as well as Mg2+ modulated ligand potencies, whereas the effect was ligand and ion specific. Results obtained for MC1R indicate that the BacMam-Epac2-camps system may also be applicable for studying the activation of other GPCRs and may be implemented in routine analysis as well as in high-throughput screening.
Introduction
G-protein-coupled receptors (GPCRs), a large family of cell surface receptors involved in transmembrane signal transduction, are a broad target group for pharmaceuticals. They are activated by a variety of stimuli and mediate signals to different classes of effectors, including several types of ion channels and second-messenger-generating enzymes. The melanocortin-1 receptor (MC1R) is a GPCR that is involved in regulation of skin and hair color in mammals and in development of melanoma, in which it is overexpressed. 1 Melanocortins, the endogenous ligands of MC1R, are pituitary peptide hormones—adrenocorticotropic hormone and different forms of melanocyte-stimulating hormone (MSH). Methods to investigate the binding properties of melanocortin ligands have been developed in the past 20 years (see review by Hruby et al. 2 ). Problems encountered in whole-cell radioligand-binding studies, such as low resolution due to high nonspecific binding as well as poor reproducibility because of interfering processes such as receptor protein degradation or internalization, were solved using cell membranes instead. It enabled making the receptor sample and assay environment “cleaner” for the binding studies, so acceptable data of MCR ligand-binding affinities became available. After revealing allosteric interactions between binding sites of MCR, 3 models of tandemly arranged ligand-binding sites 4 and ago-allosteric modulation 5 were proposed. Implementation of a fluorescence anisotropy/intensity assay with a dye-labeled NDP-α-MSH for MC4R allowed monitoring of ligand binding in real time 6 and revealed complexities in the binding processes, which have a big impact on the apparent affinities. How these processes influence the biological activity of MCR ligands remains open.
As many GPCRs, including the MC1R, modulate cyclic adenosine monophosphate (cAMP) level upon receptor activation, it is a good reporter molecule to assay the biological activity of ligands downstream of MC1R binding. From a variety of cellular cAMP assays, 7 we opted for Förster resonance energy transfer (FRET)–based biosensors, which allow for fast and real-time observation of processes. Among the constructs proposed, the Epac-camps (biosensor based on the exchange protein directly activated by cAMP, Epac) 8 seemed to be suitable and promising due to its monomolecular nature and good dynamic range. The measurement relies on the relative fluorescence of two fluorescent proteins, enhanced cyan (eCFP) and yellow fluorescent protein (eYFP), genetically fused to the cAMP binding domain of Epac. The sensor protein is freely distributed in the cell cytosol, and binding of cAMP leads to a conformational change associated with an increase in the distance between the fluorophores, which results in a decrease in eYFP (acceptor) fluorescence and an increase in eCFP (donor) fluorescence. The calculated eYFP/eCFP emission ratio correlates to the change in the level of intracellular cAMP. Epac-camps has been used to characterize the activation of different GPCRs but also to monitor the action of cyclic nucleotide phosphodiesterases. There are also variants of full-length Epac protein-based cAMP FRET sensors used to monitor GPCR activation. 7 The advantage of the Epac2-camps FRET biosensor is that it does not contain any catalytic or targeting domains, which might interfere with intracellular regulatory processes. Epac2-camps is able to reflect the changes in intracellular cAMP from sub-micromolar to the high micromolar concentrations as its EC50 value is reported to be 0.9 µM, 8 which is in a suitable range for ligand screening.
The aim of this work was to find optimal conditions for the use of Epac2-camps for characterization of MCR signaling in mammalian cells, which is important for characterization of the ligands’ activity. By monitoring changes in cAMP level, we seek better understanding of the mechanism of signal transduction and clarification of some of the complexities we have observed previously in the ligand-binding processes.
Among the transfection methods tested for sufficient and reproducible expression of the Epac2-camps in B16F10 murine melanoma cell line, endogenously expressing MC1 receptors, we found BacMam technology 9 as the most suitable option. The use of BacMam-Epac2-camps system is not limited to B16F10 cells. We have successfully applied the same expression system also for monitoring cAMP elevations in HEK293 cells. BacMam technology has been reported to be compatible with a broad range of cell lines, 10 suggesting that the same approach would work well in many cell lines commonly used in GPCR studies.
Materials and Methods
Cell Lines and Cell Culture
Spodoptera frugiperda cells (Sf9) (Invitrogen Life Technologies, Paisley, UK) were cultured in suspension with EX-CELL 420 growth medium (Sigma-Aldrich GmbH, Munich, Germany) in a 27 °C incubator in a nonhumidified environment. Human embryonic kidney (HEK293; ATCC, Boras, Sweden) and murine melanoma cells (B16F10) (Invitrogen Life Technologies) were grown as an adherent monolayer and maintained at 37 °C and 5% CO2 in a humidified incubator in complete growth medium. Dulbecco’s modified Eagle’s medium (DMEM) was used for HEK293, and Roswell Park Memorial Institute medium (RPMI1640 with GlutaMAX, 25 mM HEPES) was used for B16F10 cells. Growth media (PAA Laboratories GmbH, Pasching, Austria) were supplemented with 10% fetal bovine serum (Gibco, Invitrogen Life Technologies), 100 U/mL penicillin, and 0.1 mg/mL streptomycin (PAA Laboratories GmbH). Dulbecco’s phosphate-buffered saline (DPBS) and trypsin were also from PAA.
Plasmid Construction and Generation of BacMam Virus
The expression vector for pcDNA3.1(+)-EYFP-epac2B(murine)-ECFP (referred to as Epac2-camps throughout the text) was kindly provided by Prof. M. J. Lohse group from The Institute of Pharmacology and Toxicology, University of Würzburg, Germany. The Epac2-camps construct, under the control of the cytomegalovirus (CMV) promoter, was cloned into the pFastBac1 vector (Invitrogen Life Technologies) using the restriction enzymes (Fermentas, Vilnius, Lithuania) Bst1107I (BstZ17I) and Bsp68I (NruI) for pcDNA3.1(+) and Eco105I (SnaBI) and KspAI (HpaI) for pFastBac1, respectively. 9 The polyhedrin promoter was removed from the pFastBac1 vector to ensure low promoter interference during virus amplification. The obtained pFastBac-Epac2-camps construct was transformed into DH10Bac competent cells (Invitrogen Life Technologies) for the production of recombinant bacmid DNA. PCR-verified bacmid DNA was then transfected into Sf9 insect cells using four equivalents of ExGen 500 (Fermentas) to prepare BacMam virus stocks according to the Invitrogen Life Technologies Bac-to-Bac expression system manual. P1 viral stocks were amplified and the titers were determined by plaque assay using Sf9 cells. P2 viral stocks were aliquoted and stored at −80 °C until the day of the experiment.
BacMam-Epac2-camps Assay
Near-confluent cell cultures were trypsinized, counted, and seeded on black 96-well clear-bottom cell culture plates (Corning B.V. Life Sciences, Amsterdam, the Netherlands) about an hour prior to transduction (~2 × 104 cells/well). Attached cells were transduced with 50 µL/well of viral stock dilution in serum-free growth medium at a multiplicity of infection (MOI) of 200 to 400 for 3 h. Thereafter, the medium was replaced with 100 µL/well complete growth medium containing 10 mM sodium butyrate (Sigma-Aldrich GmbH), and the cells were incubated for another 21 h. Transduction was carried out in a humidified incubator set to 37 °C and 5% CO2. The following day, the medium was replaced by DPBS an hour prior to the assay. The functional assays were performed on a PHERAstar plate reader (BMG LABTECH GmbH, Ortenberg, Germany), with excitation at 427 nm and simultaneous dual emission at 480 and 530 nm. The cells were assayed in 100 µL DPBS upon addition of 10× ligand solution. Stock solutions of MC1R ligands (Bachem, Bubendorf, Switzerland) and forskolin (Tocris Bioscience, Bristol, UK) were dissolved in DMSO (10 mM) and stored at −20 °C until the day of the assay. Ligand dilutions were made in Milli-Q grade water containing 0.1% pluronic acid (Sigma-Aldrich GmbH) to avoid ligand adsorption to assay plates and laboratory plastic. Ethylenediaminetetraacetic acid (EDTA) and ethylene glycol tetraacetic acid (EGTA) were from Sigma-Aldrich GmbH. CaCl2 and MgCl2 were from AppliChem GmbH (Darmstadt, Germany).
Data Analysis
The change in FRET ratio was calculated according to the following formula:

where CFP0 and YFP0 refer to the fluorescence emissions at 480 nm and 530 nm before and CFP and YFP after the ligand treatment, respectively. Background-corrected ∆Ratio was determined by subtracting the average ∆Ratio of vehicle-treated wells from the ∆Ratio of each well treated with ligand. The maximal FRET change (∆Ratio) ranged from 10% to 20% depending on the cell line and MOI and was normalized to 100% response for data analysis. Data are presented as mean pEC50 ± SEM. Experiments were performed at least in four independent determinations in duplicates or triplicates. Data analysis was performed using GraphPad PRISM 5.04 (GraphPad Software, San Diego, CA) using default settings for nonlinear regression analysis and curve fitting the data to the sigmoidal dose-response equation. The Z′ factor from three independent experiments with 12 parallel assay points was calculated as described previously by Zhang et al. 11 Positive control signals were registered at maximal effective concentrations of three different MC1R agonists and forskolin, and negative control signals were registered from vehicle-treated cells.
Results and Discussion
The B16F10 murine melanoma cell line is a good choice for the functional studies of MC1R because of its high endogenous expression of the receptor. The use of the BacMam system for Epac2-camps expression enabled us to monitor elevation of the second-messenger cAMP in live cells. Usually, sensor constructs such as Epac-camps are transfected into mammalian cells using lipophilic reagents (e.g., polyethylene imine). In our setup, Lipofectamine 2000 (Invitrogen Life Technologies) as well as ExGen 500 (Fermentas) resulted in low protein expression and also inconsistent expression levels between independent assays, posing problems for our assay development. Increases in the amount of DNA and lipophilic reagent caused increased cytotoxicity but not higher protein expression. Another reason why transfection reagents poorly suit high-throughput screening (HTS) assays is their relatively high cost and need for large amounts of purified plasmid DNA. We could overcome these limitations by developing a cost-efficient and reproducible assay for routine analysis by implementing BacMam technology. 9 The method employs a baculovirus vector with the CMV promoter to drive the expression of proteins in mammalian cells. This technology provides advantages such as high transduction rates; protein expression levels that can be adjusted by viral dose; low cytotoxicity to host cells; safety in production and handling (Biosafety Level 1); compatibility with a broad range of cells, including primary and stem cells; and, importantly, the ease and convenience of use (see review by Kost and Condreay 10 ).
After obtaining the virus (for bacmid construction and virus production, see Materials and Methods), the conditions for cell transduction were optimized for the B16F10 cell line, relying on previous studies related to the BacMam system.9,12 Optimal sensor expression levels were achieved at an MOI of 200 to 400 and by incubation of the mammalian cells with baculovirus for 2 to 4 h. To enhance protein expression after the transduction step, we used a histone deacetylase inhibitor, sodium butyrate, at 10 mM. Sufficient protein expression for functional assays was achieved 24 h posttransduction and remained such for a further 24 h. This shows that the assay is flexible and has a convenient time frame between the transduction of cells and the measurement.
The effects of forskolin, a direct activator of adenylate cyclase, were measured in two different mammalian cell lines. Forskolin induced a concentration-dependent elevation in cAMP levels that was detectable with the Epac2-camp sensor. For B16F10 and HEK293 cells, the EC50 values were around 1 µM and almost identical ( Fig. 1 ), indicating that the sensor responds to changes in cAMP levels in both cell lines. The change in FRET (∆Ratio) upon forskolin treatment remained between 15% and 20% depending on the MOI. Because of the high signal-to-noise ratio and the simple and robust ratiometric nature of this assay, we did not correct for photobleaching and fluorescence bleedthrough. Therefore, the current method only reflects qualitative changes in cAMP concentrations.
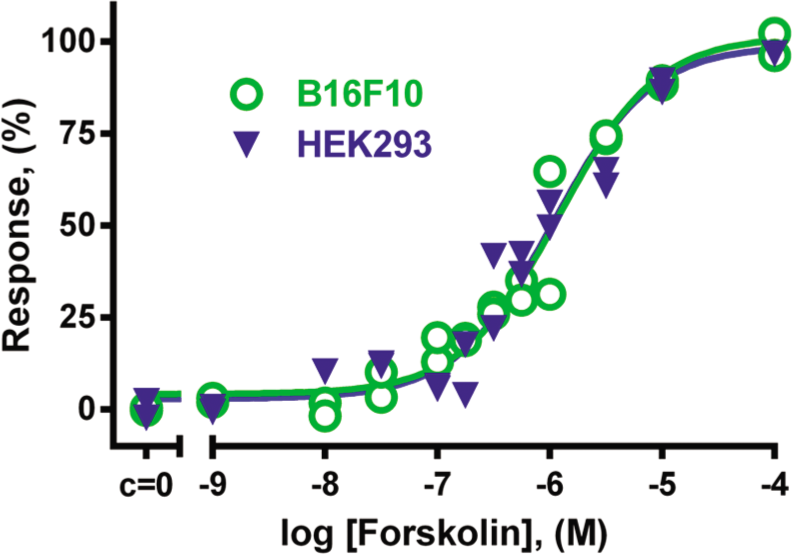
Influence of different concentrations of forskolin on cAMP levels in B16F10 cells and HEK293 cells. Cells were transduced with BacMam-Epac2-camps virus for 3 h and further incubated for 21 h in complete growth medium supplemented with 10 mM sodium butyrate. Cells were treated with adenylate cyclase activator forskolin for 10 min at 37 °C. The maximal Förster resonance energy transfer (FRET) change (∆Ratio) was normalized to 100% response. Activity of forskolin in B16F10 was characterized with pEC50 = 5.90 ± 0.06 (n = 4); in HEK293 cells, pEC50 = 6.04 ± 0.16 (n = 4). Graph showing data from a representative experiment.
After verifying that our assay system is able to characterize changes in cellular cAMP levels depending on the concentration of the biologically active compound, receptor-mediated agonist effects were investigated next. In B16F10 cells, all studied MC1R agonists caused a concentration-dependent increase in cAMP concentration, which was registered and characterized using the Epac2-camp sensor ( Fig. 2 ). α-MSH, β-MSH, and NDP-α-MSH behaved as full agonists for MC1R with similar sensor activation profiles, whereas their activation level remained 75% of the level achieved by forskolin. Partial agonists (SHU-9119, MS-05, HS-024) achieved a level of 75% of full agonist activation. JKC-363, known as an MC4R antagonist, showed no receptor activation at sub-micromolar concentrations. MC1R activation by full agonists was blocked in the presence of 10 nM JKC-363 (data not shown), confirming the antagonistic properties of this ligand. The selective low molecular weight MC4R agonist I-THIQ had no effect on MC1R activation in our system. Our results are in agreement with previously published efficacies for these ligands. The pEC50 values characterizing the activation of MC1R of tested compounds are presented in Table 1 and Figure 2 .
Effect of Bivalent Cations on MC1R Activation by Its Agonists
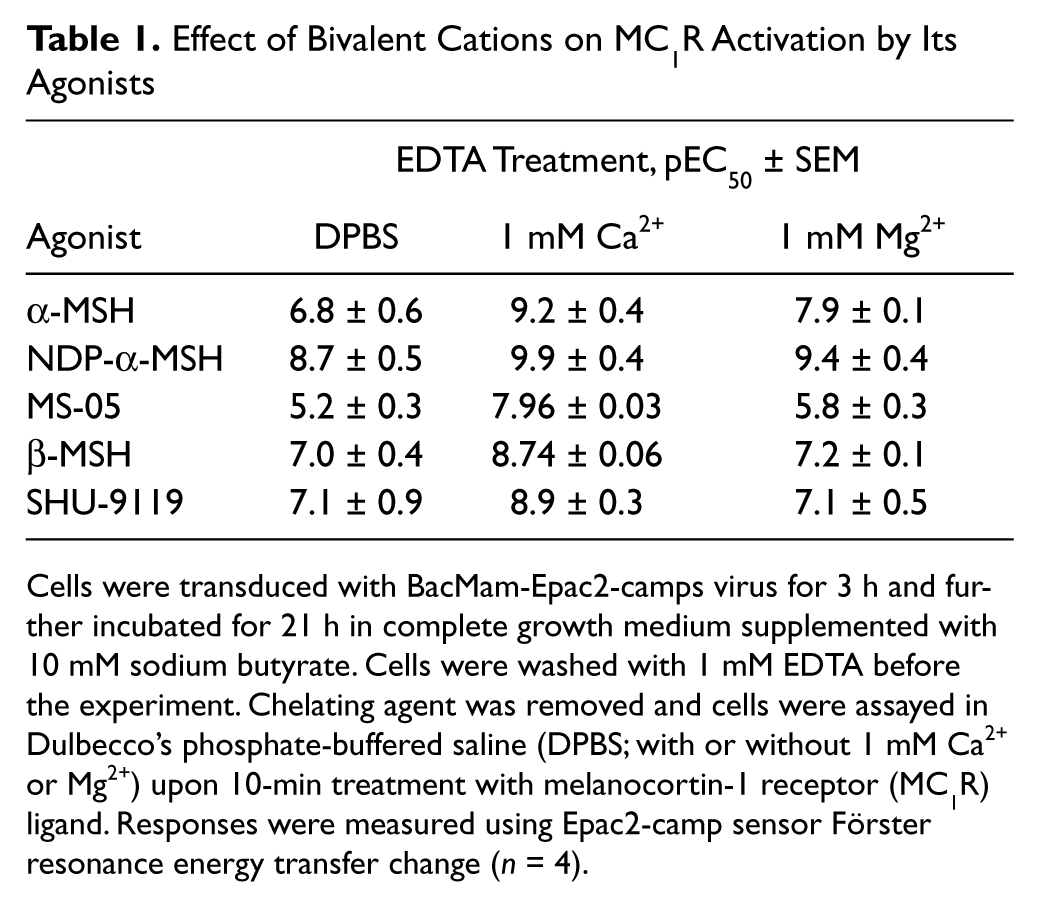
Cells were transduced with BacMam-Epac2-camps virus for 3 h and further incubated for 21 h in complete growth medium supplemented with 10 mM sodium butyrate. Cells were washed with 1 mM EDTA before the experiment. Chelating agent was removed and cells were assayed in Dulbecco’s phosphate-buffered saline (DPBS; with or without 1 mM Ca2+ or Mg2+) upon 10-min treatment with melanocortin-1 receptor (MC1R) ligand. Responses were measured using Epac2-camp sensor Förster resonance energy transfer change (n = 4).
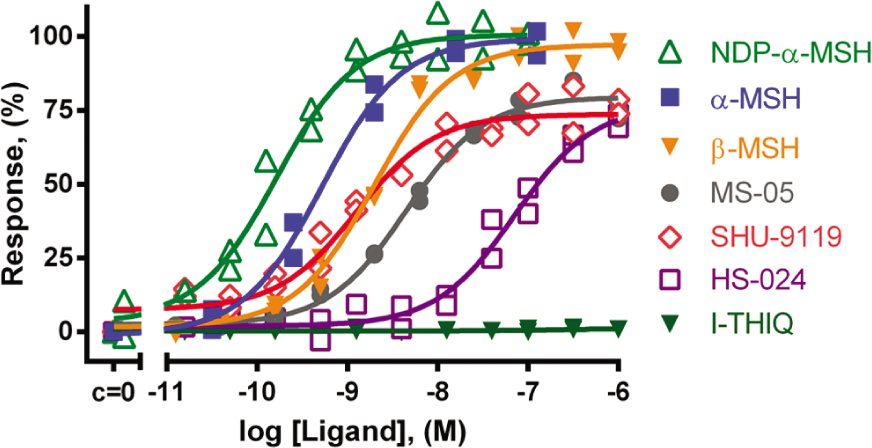
Influence of different concentrations of MC1R ligands on cAMP levels in B16F10 cells. Cells were transduced with BacMam-Epac2-camps virus for 3 h and further incubated for 21 h in complete growth medium supplemented with 10 mM sodium butyrate. Cells were treated with ligands for 10 min at 37 °C. Measurements were performed in Dulbecco’s phosphate-buffered saline (DPBS) + 1 mM Ca2+ for full receptor activation. The maximal Förster resonance energy transfer (FRET) change (∆Ratio) was normalized to 100% response. pEC50 ± SEM (n = 4): NDP-α-MSH, 9.9 ± 0.4; α-MSH, 9.2 ± 0.4; β-MSH, 8.74 ± 0.06; SHU-9119, 8.9 ± 0.3; MS-05, 7.96 ± 0.03; HS-024, 7.27 ± 0.13. I-THIQ showed no activation (n = 2). Graph showing data from a representative experiment.
The ligand affinities reported in the literature have been measured either directly by binding of a radioactively labeled ligand to the receptor or indirectly by displacing a MC1R-bound labeled ligand (e.g., [125I]NDP-α-MSH) with an unlabeled ligand.3,13 For endogenous peptide α-MSH, the affinities were in the sub-nanomolar range (pKi = 9.3–9.9), whereas synthetic agonist NDP-α-MSH had a slightly higher affinity (pKi = 9.9–10.1), and MS-05 had a lower affinity (pKi = 9.1).3,13 The pEC50 values obtained from functional assays were 8 to 8.6 for α-MSH and 7.9 for NDP-α-MSH.14,15 Functional responses in these studies have been determined using [3H]adenine in a [3H]cAMP accumulation assay to quantify cAMP changes in B16F10 cells 15 and in COS-7 cells. 14 The latter method provides a direct readout of [3H]cAMP accumulation. It is sensitive and has a large dynamic range over which cAMP responses can be measured. This method, alas, is time-consuming, provides endpoint readouts, and requires the use of radioactively labeled precursors. With our BacMam-Epac2-camps system, we use a monomolecular FRET pair as the indicator and fluorescence intensity ratio as the readout, and baculovirus gene delivery makes this method simpler and more convenient to use. Despite a loss in dynamic range and sensitivity, considerable improvements are achieved in terms of real-time detection and the possibility of an HTS format. The readout in the radiolabeled cAMP accumulation assay is a measure of turnover rather than of absolute cAMP levels. Our cellular biosensor, however, measures real-time physiological changes in cAMP levels, so slight differences in pEC50 values measured with different methods are explicable. The relative hierarchy of previously published affinities and potency values of the MC1R agonists remains unchanged and is comparable to the data obtained using Epac2-camps in our BacMam setup. It is thus evident that our approach is suited for monitoring GPCR activation downstream of the binding event, providing reliable, pharmacologically relevant data for the tested compounds.
Bivalent metal cations are known to play an important role in GPCR-mediated signal transduction, but the exact mechanisms still remain elusive and vary depending on the ion and on the receptor in question. The groups of T. W. Schwartz and J. E. Wikberg have stated that Zn2+ and Ca2+ improve binding of ligands to the MC1R.3,14 To determine the effect of bivalent cations on activation of second messengers with our assay system in B16F10 cells, we first removed the endogenous ions using EGTA or EDTA and then replaced them with fixed concentrations of Ca2+ or Mg2+.
Upon removal of bivalent cations from the reaction buffer with 1 mM EDTA, the potency of the MC1R agonists was significantly lowered ( Fig. 3 , Table 1 ). Addition of 1 mM Ca2+ was found to be necessary for high-potency agonist effect, whereas 1 mM Mg2+ also increased the potency of agonists but to a lesser extent. After treatment with EGTA, which binds Ca2+ with much higher affinity than Mg2+, the obtained results were comparable to data observed after EDTA treatment (data not shown). These results together demonstrated that the pEC50 values strongly depend on the ionic composition of the assay buffer. The positive influence of Ca2+ and Mg2+ on MC1R activation may be the reason behind the differences in ligand affinities reported previously.3,14,15 The characteristic hierarchy of values for ligand binding, as well as for downstream functional response, is still maintained. In this light, we decided to carry out all of the following experiments on MCR in DPBS in the presence of 1 mM Ca2+, if not stated otherwise.
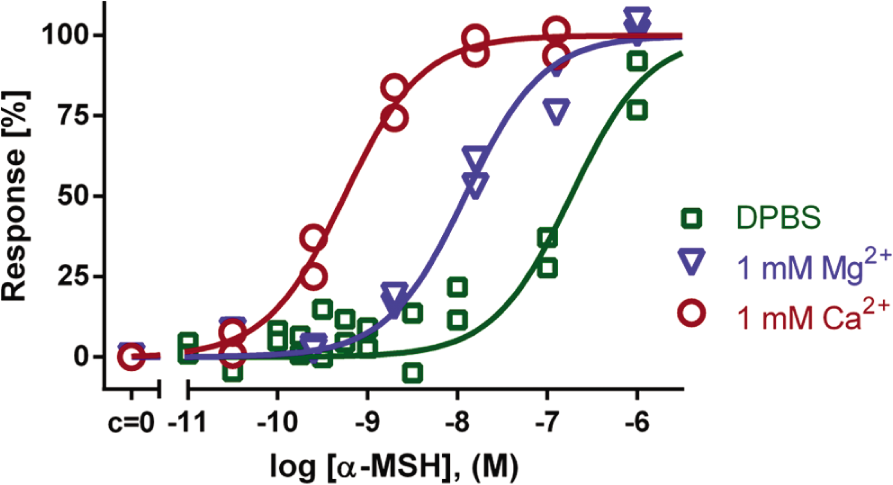
The effect of Ca2+ and Mg2+ on MC1R activation by α-MSH. B16F10 murine melanoma cells were transduced with BacMam-Epac2-camps virus for 3 h and further incubated for 21 h in complete growth medium supplemented with 10 mM sodium butyrate. Cells were washed with Dulbecco’s phosphate-buffered saline (DPBS) containing 1 mM EDTA prior to experiment. Chelating agents were removed and cells were assayed in DPBS (with or without 1 mM Ca2+ or Mg2+) upon 10-min treatment with a full agonist, α-MSH. The maximal Förster resonance energy transfer (FRET) change (∆Ratio) was normalized to 100% response. Without Ca2+ and Mg2+: pEC50 = 6.8 ± 0.6; in presence of 1 mM Mg2+: pEC50 = 7.9 ± 0.1; in presence of 1 mM Ca2+: pEC50 = 9.2 ± 0.4; (n = 4). Graph showing data from a representative experiment.
After optimization of our BacMam-Epac2-camps system, we have obtained a reliable tool with a reasonable signal-to-noise ratio and detection window. The pharmacological validity of the biosensor was confirmed by MC1R activation with a set of known agonists and partial agonists. In parallel, we have also been performing experiments for detecting activation of other GPCRs with our cAMP-sensor system. We have tested the activation of the recombinant dopamine D1 receptor expressed in a stable line of HEK293 cells with various dopaminergic agonists. Also, we measured the activation of an endogenous β2-adrenergic receptor in HEK293 cells. We obtained reasonable potency and efficacy values for all ligands tested (data not shown). This suggests that the proposed assay format should be amenable to HTS. We calculated the Z′ factor values 11 for our assay system, which revealed values >0.6 for all MC1R-specific agonists and forskolin studied in all of our independent experiments. We conclude that our assay represents a new cost-efficient and robust cell-based HTS assay system that reports receptor activation on a second-messenger level and can be established in any cell culture laboratory. Of course, assay conditions for any particular receptor of interest might require additional optimization, but there are many GPCR-expressing stable cell lines, which are reported to be compatible with the BacMam system. 10 This system, together with fluorescence anisotropy/intensity measurements 6 and/or radioligand binding studies for characterization of ligand-binding affinities, generates a set of methods that allow us to determine compounds’ affinities, potencies, and efficacies and adds up to a reliable platform for HTS of new drugs.
Footnotes
Acknowledgements
We thank Professor Martin J. Lohse’s group at the University of Würzburg for providing us the Epac-camp sensor plasmids.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was funded by the Estonian Science Foundation (7569, 8011, 8314) by the Estonian Ministry of Education and Science (SF0180032s12) and by the European Union through the European Regional Development Fund (TK114, 30020).