
Abstract
Early success of kinase inhibitors has validated their use as drugs. However, discovery efforts have also suffered from high attrition rates due to lack of cellular activity. We reasoned that screening for such candidates in live cells would identify novel cell-permeable modulators for development. For this purpose, we have used our recently optimized epidermal growth factor receptor (EGFR) biosensor assay to screen for modulators of EGFR activity. Here, we report on its validation under high-throughput screening (HTS) conditions displaying a signal-to-noise ratio of 21 and a Z′ value of 0.56—attributes of a robust cell-based assay. We performed a pilot screen against a library of 6912 compounds demonstrating good reproducibility and identifying 82 inhibitors and 66 activators with initial hit rates of 1.2% and 0.95%, respectively. Follow-up dose-response studies revealed that 12 of the 13 known EGFR inhibitors in the library were confirmed as hits. ZM-306416, a vascular endothelial growth factor receptor (VEGFR) antagonist, was identified as a potent inhibitor of EGFR function. Flurandrenolide, beclomethasone, and ebastine were confirmed as activators of EGFR function. Taken together, our results validate this novel approach and demonstrate its utility in the discovery of novel kinase modulators with potential use in the clinic.
Introduction
The critical role of protein phosphorylation in the development and progression of many cancers has driven considerable efforts to discover therapeutic agents targeting aberrant signaling events. Receptor tyrosine kinases (RTKs) such as epidermal growth factor receptor (EGFR) play a well-established role in several cancers and have become a crucial class of targets for the development of small-molecule anticancer agents. 1 Besides high-profile successes such as Iressa (gefitinib) and Tarceva (erlotinib), progress in identifying new drugs inhibiting RTKs has been slow in recent years. A major obstacle hampering the rapid discovery of new effective drugs inhibiting RTKs is the lack of cellular activity of potent and selective candidates originally identified in screens relying on assays using recombinant kinase domains. Such RTK inhibitors very often fail the transition from being potent toward purified recombinant protein to being active in cells, believed to be due mainly to lack of cellular permeability. As a consequence, time-consuming exploratory chemistry efforts are needed to enhance the cell permeability of drug candidates. Therefore, the ability to screen directly for potent RTK inhibitors in cells is highly sought after.
Furthermore, significant setbacks have been encountered with the current generation of approved inhibitors, resulting from rapid acquisition of resistance mutations in the kinase domain. 2 This observation highlights the need for identifying RTK inhibitors with an alternative mechanism of action, distinct from targeting the kinase activity of RTK. Interestingly, a strong link between endocytosis and signaling is emerging, with growing evidence revealing the key role of endocytosis in the compartmentalization of cell signaling components. Although receptor endocytosis has long been known as a mechanism to attenuate ligand effect and to transport and recycle receptors, receptor trafficking is now increasingly seen as playing a direct role in triggering transduction signals.3–6 Receptor signaling has been shown to continue in endosomal compartments following receptor activation; furthermore, certain signaling events have been demonstrated to require endocytosis. 5 Receptor trafficking can control the timing, amplitude, and specificity of signaling. 5 For this reason, the field would highly benefit from efficient methods to rapidly identify inhibitors of RTK activation and trafficking in cells.
Live-cell-based assays have crucial advantages compared with in vitro assays relying on the use of purified recombinant proteins. Live cells recapitulate the endogenous environment surrounding RTKs, including their cell signaling networks with proteins expressed at physiological levels. In addition, because cell populations are heterogeneous in nature, assays measuring the overall response of the cell population in a well are prone to error. For this reason, high-content assays are preferred because they allow us to perform cell-by-cell analysis. 7 Therefore, cell-based assays are necessary for the identification of cell-potent inhibitors of RTK activation, potentially targeting events distinct from tyrosine kinase phosphorylation.
We recently described the development of a novel cell-based biosensor assay allowing the identification of EGFR modulators in high-throughput formats. 8 The assay relies on the expression, in A549 EGFR biosensor cells (A549-EGFRB cells), of a SRC homology 2 domain (SH2) of GRB2 that specifically binds to activated EGFR, fused to green fluorescent protein (GFP). Upon receptor activation following ligand stimulation, EGFR clustering, internalization, and trafficking are visualized, and granule formation imaged on the GFP channel is quantified as a surrogate for endogenous RTK activity in live cells ( Fig. 1 ). In addition, stained nuclei are imaged and quantified as a measure of cell number and cytotoxicity.
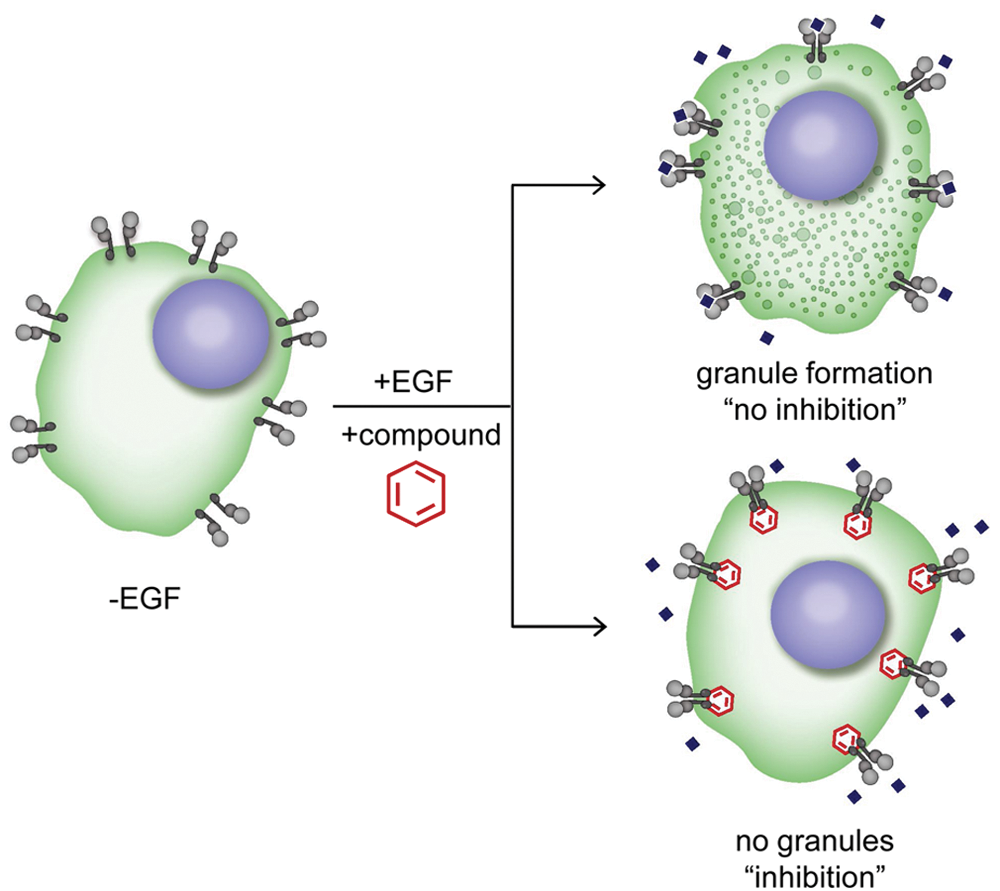
Principles of the epidermal growth factor receptor biosensor (EGFRB) assay. Schematics of the EGFRB assay with the A549 EGFR biosensor cell line (A549-EGFRB). In the absence of epidermal growth factor (EGF) stimulation, diffused green fluorescent protein (GFP) is observed in the cytoplasm of cells. In contrast, EGF addition triggers EGFR activation and subsequent clustering and internalization as observed by the formation of granules (vesicles) in the GFP channel, corresponding to “no inhibition.” Granule formation upon EGF stimulation is prevented by EGFR small-molecule inhibitors (“inhibition”), allowing the identification of novel EGFR inhibitors by high-throughput screening.
In this study, we sought to validate our domain-based biosensor assay for the identification of novel small-molecule EGFR modulators by high-throughput screening (HTS). We conducted a control run aimed at evaluating the robustness of the optimized EGFRB assay in the conditions used for automated screening, followed by a pilot screen of approximately 7000 compounds tested in duplicate to assess the reproducibility and robustness of the assay, as well as its ability to identify inhibitors of EGFR activity in live cells. We confirmed the dose-dependent activity of obtained positives in the EGFRB assay and further assessed their activities in cell-based viability assays as well as against an in vitro kinase panel. The results of this comprehensive study are described below.
Materials and Methods
Materials
Reagents used for the EGFRB assay and for EGFR protein immunostaining and EGFR knockdown experiments were obtained as previously described. 8 A549 cells were purchased from ATCC (cat. CCL-185; ATCC, Manassas, VA). H2030, H3255, and HCC4011 non–small cell lung cancer cells (NSCLCs) were obtained from Dr. Romel Somwar (The Varmus Lab, Memorial Sloan-Kettering Cancer Center [MSKCC], New York, NY). DRAQ5 DNA dye was purchased from Biostatus (Leicestershire, UK). The “killer mix” used as a low control in viability assays consists of a proprietary mixture of cytotoxic compounds as previously described.9–11 For the luminescence adenosine diphosphate (ADP) production kinase assay, HEPES, β-glycerol phosphate, MgCl2, tris(2-carboxyethyl)phosphine (TCEP), sodium orthovanadate, Poly(Glu,Tyr) peptide, and Tween-80 were purchased from Sigma-Aldrich (St. Louis, MO). The ADP-Glo Kinase Assay Kit and adenosine triphosphate (ATP) were purchased from Promega (Madison, WI). SRC, ABL, and VEGFR1 kinase were purchased from Life Technologies (Grand Island, NY). EGFR kinase was purchased from Carna Biosciences (Kobe, Japan). Staurosporine was purchased from LC Laboratories (Woburn, MA).
Assay Control Run
The performance of the EGFRB assay in the HTS conditions was assessed as previously described 8 in a control run consisting of three 384-well microplates that contained 1% DMSO (v/v) for the high control and three 384-well microplates that contained 10 µM gefitinib in 1% DMSO (v/v) for the low control. Then, 5 µL of 10% DMSO (v/v) was added to the high control plates, and 5 µL of 100 µM gefitinib in 10% DMSO (v/v) was added to the low control plates with a custom-designed 384 head on a PP-384-M Personal Pipettor (Apricot Designs, Monrovia, CA). A549-EGFRB cells were added to the plates in the 45-µL cell culture media using an automated Multidrop 384 dispenser (Thermo Scientific, Waltham, MA) at the previously optimized cell density of 5000 cells per well and incubated in Cytomat automated temperature- and humidity-controlled incubator (Thermo Scientific) at 37 °C and 5% CO2 for 16 h. The cell culture media were then aspirated using an automated plate washer ELx405 (BioTek Instruments, Winooski, VT) and replaced with media containing 500 nM epidermal growth factor (EGF). Plates were further incubated in a Cytomat for 70 min, and cells were fixed following media aspiration using a BioTek washer and dispensing of 50 µL 4% paraformaldehyde (v/v) in phosphate-buffered saline (PBS) using a Multidrop. After a 20-min incubation at room temperature and 1 wash with PBS using the Multidrop and Biotek washer, cell nuclei were stained with 50 µL of 2.5 µM DRAQ5 in PBS added using the Multidrop. After a 15-min incubation at room temperature, cells were washed twice with 50 µL PBS using the Multidrop and BioTek washer.
Pilot Screen
A pilot screen against the chemical library of 6912 compounds described above was performed in duplicate according to the assay workflow described for the assay control run and at a screening concentration of 10 µM compound in 1% DMSO (v/v). Controls present in each assay plate consisted of 1% DMSO (v/v) (high control) and 10 µM gefitinib in 1% DMSO (v/v) (low control) final concentration. Plate fixing was automated on a linear track robotic platform (CRS F3 Robot System; Thermo Scientific) with an integrated BioTek washer and Multidrop for plate washing and liquid dispensing. For automated INCA2000 imaging, plate handling was conducted using the Orbitor RS Microplate Mover (Thermo Scientific). Screening data files resulting from the automated image analysis described above with a granule and nuclei count for each well were subsequently loaded onto the HTS Core Screening Data Management System, a custom-built suite of modules for compound registration, plating, and data management powered by ChemAxon Cheminformatic tools (ChemAxon, Budapest, Hungary). The percentage inhibition in the granule count and nuclei count was calculated for each compound based on control values present in each assay plate as follows.
The percentage inhibition in the granule count was calculated based on both high and low control averages as follows:

The percentage inhibition in the nuclei count was calculated as follows based only on high controls as low control nuclei count values are not lower than high control values:
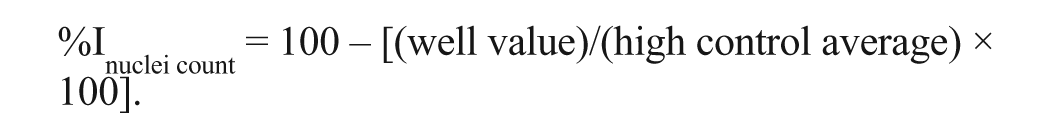
Assessment of the Antiproliferative Effects of Hits against a Panel of Established Cell Lines
The antiproliferative effect of confirmed hits was assessed against a panel of established cell lines that included those harboring wild-type EGFR (A549-EGFRB, A549, and H2030 cell lines) and those harboring the activating L858R EGFR mutation (H3255 and HCC4011 cell lines). A549-EGFRB cells were cultured as previously described. 8 H2030, H3255, and HCC4011 cells were cultured as previously described. 12 A549 cells were cultured in F12K media (cat. 21127-022; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; cat. A15-201; PAA Laboratories GmbH, Pasching, Austria). The antiproliferative effect of each compound was assessed in dose-response studies in a 384-well format using 12 doubling dilutions in duplicate with a 10-µM and 1-µM compound concentration as the upper limit and using the Alamar Blue viability assay as previously described. 13 Control wells consisted of 1% DMSO (v/v) (high control) and 1 µM “killer mix” in 1% DMSO (v/v) (low control). Final compound incubation time with cells was 120 h. In dose-response curves plotted using SigmaPlot 9.0 (Systat Software, San Jose, CA), the mean data from duplicates are presented and the error bars correspond to the standard error of the regression.
Assessment of the Inhibitory Activity of Hits against a Panel of Kinases
The activity of confirmed positives was assessed in a panel of kinases consisting of EGFR, VEGFR1, SRC, and ABL kinase using a luminescence ADP production kinase assay as previously described.14–16 The potency of each compound was measured in dose-response studies in a 384-well format using 12 doubling dilutions in duplicate with a 10-µM and 1-µM compound concentration as the upper limit. All reagents transfers were performed using the PP-384-M Personal Pipettor (Apricot Designs, Monrovia, CA). Tested compounds or controls were added to the wells at a volume of 1 µL to white 384-well microplates (Corning #3570; Corning, New York, NY). Controls consisted of 1% DMSO (v/v) (high control) and 30 µM staurosporine in 1% DMSO (v/v) (low controls). The assay buffer was 25 mM HEPES/NaOH (pH 7.5) and contained 10 mM MgCl2, 2 mM TCEP, 20 mM β-glycerol phosphate, and 100 µM Na3VO4; for each kinase on the panel, 4 µL kinase dilution in assay buffer was added to the wells to reach a final concentration of 50 nM enzyme. Of note, the specific activity of the kinases of the panel is unknown, and therefore the concentration of active enzyme in the preparation is unknown. After enzyme addition, kinase and compound were preincubated for 10 min at room temperature. Then, 5 µL of a mix containing ATP and Poly(Glu, Tyr) substrates in solution in assay buffer was added to the wells to reach a final concentration of 200 µM. After a 45-min reaction at room temperature, 10 µL ADP-Glo Reagent was added to each well. After a 40-min incubation, 20 µL Kinase Detection Reagent was added followed by a 60-min incubation. The luminescence signal output was measured on the LEADseeker multimodality imaging system (GE Healthcare, Piscataway, NJ). Dose-response curves were plotted using SigmaPlot and represent the mean data from duplicates, and the error bars correspond to the standard error of the regression. The low limit for calculating compound IC50 in the assay conditions was 10 nM.
Results
We have previously established a proof of concept for a novel domain-based biosensor assay that allows us to screen for EGFR modulators in live cells. 8 Our goal in this study is to validate the optimized EGFRB assay for chemical screening and to assess whether it would allow for the identification of EGFR kinase modulators, to include activators and inhibitors, as well as those with an alternative mechanism of action.
Assay Control Well Assessment of the Optimized EGFRB Assay in HTS Format
For this purpose, we first evaluated the robustness of the assay performed in the conditions of screening in a control run consisting of 1152 high control wells containing 1% DMSO (v/v) and 1152 low control wells containing 10 µM gefitinib in 1% DMSO (v/v). As expected, granule formation was triggered by stimulation with 500 nM EGF in the high control wells ( Fig. 2A1 & 4 ), whereas it was inhibited in the low control wells ( Fig. 2A2 & 5 ), mimicking the absence of EGF stimulation ( Fig. 2A3 & 6 ). Quantification of granule formation revealed a large signal window between high and low controls with an average granule count of 10 737 for high control wells compared with 507 for low control wells ( Fig. 2B , C ). This large signal window was accompanied with an acceptable variability for both the high and low controls, with a coefficient of variation (CV) of 13% and 14%, respectively. Combined with a signal-to-noise ratio of 21:1, this low variability translated into a calculated Z′ value of 0.56, indicative of good assay performance and robustness ( Fig. 2C ). As expected, nuclei count values were not significantly different between high and low controls, with an average imaged nuclei count of 1219 for high controls compared with 1200 for low controls and with CVs of 11% and 9%, respectively ( Fig. 2B , C ). This result demonstrates that the observed difference in granule count between the two conditions is not due to a difference in cell number but rather the direct consequence of EGFR activity inhibition by gefitinib. Overall, the results from this control run demonstrate that the robustness of the EGFRB assay is compatible with HTS.
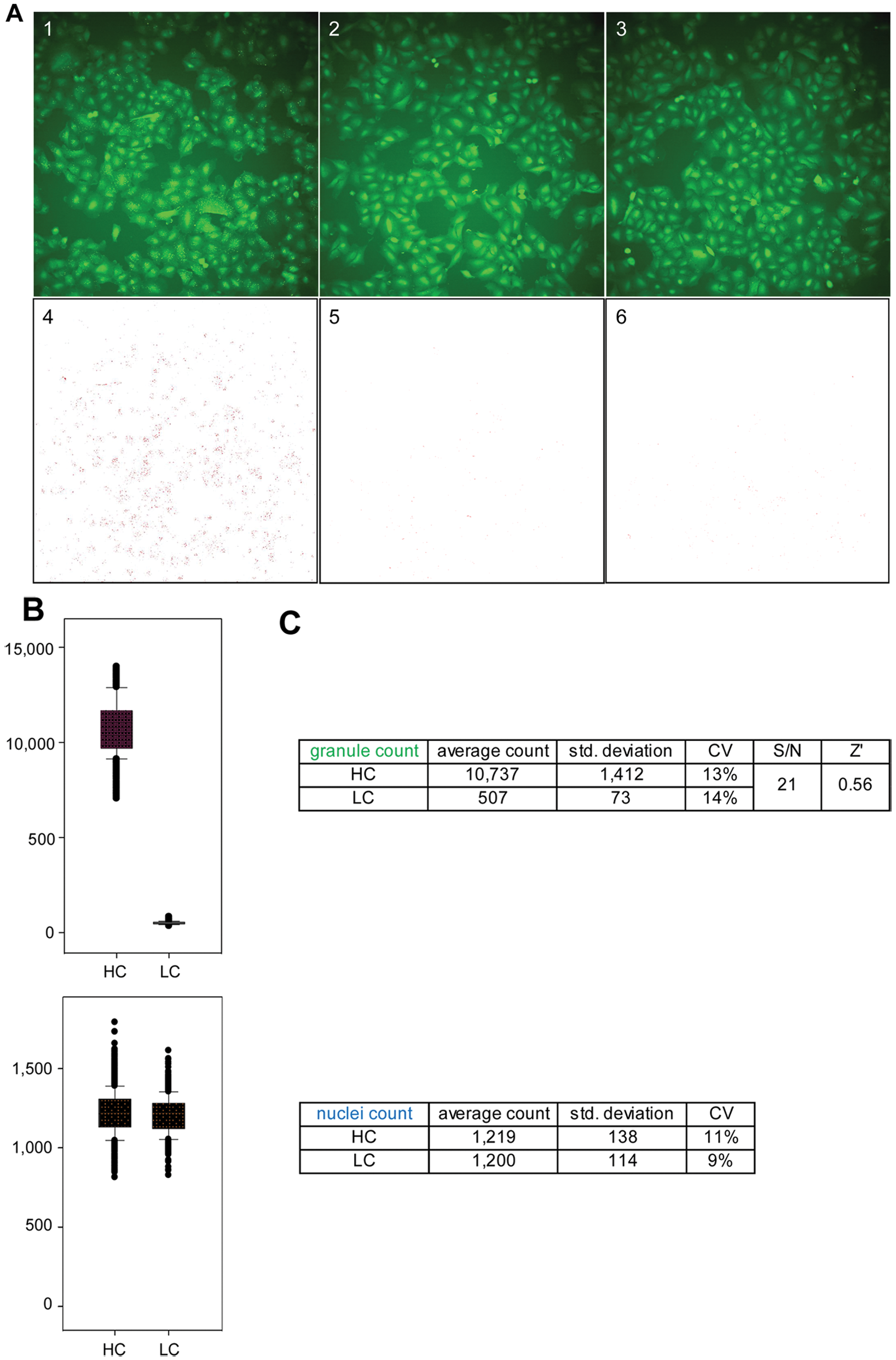
Control well images and control run assessment of the epidermal growth factor receptor biosensor (EGFRB) assay. (
Pilot Screen against a Library of 6912 Compounds
Following this positive result, we performed a pilot screen against a library of 6912 Food and Drug Administration (FDA)–approved and known bioactive compounds in duplicate and at a compound screening concentration of 10 µM in 1% DMSO (v/v). We evaluated the reproducibility of the screen by plotting the granule count values induced by each compound and for each set of data as a scatter plot ( Fig. 3A ). As expected, most compounds had no effect and clustered around a granule count value of 10 000 to 12 000, consistent with the average granule count for high controls of 10 737 observed in the assay control run; in contrast, few compounds induced granule count values below 5000. When plotting as a scatter plot the nuclei count values induced by each compound and for each set of data, most compounds had little effect on cell count, with the cloud of compounds centered on a nuclei count of about 1200 ( Fig. 3B ), consistent with the average nuclei count value of 1219 and 1200 observed for both high and low controls, respectively, in the assay control run. This result is expected because most cytotoxic compounds present in the library are not expected to be potent within the 17-h time frame of the assay, inferior to the doubling time of A549 cells. The linear shape of the cloud of compounds and the presence of a few outliers for both the granule count and nuclei count scatter plots demonstrate the good reproducibility of our assay in the conditions of screening.
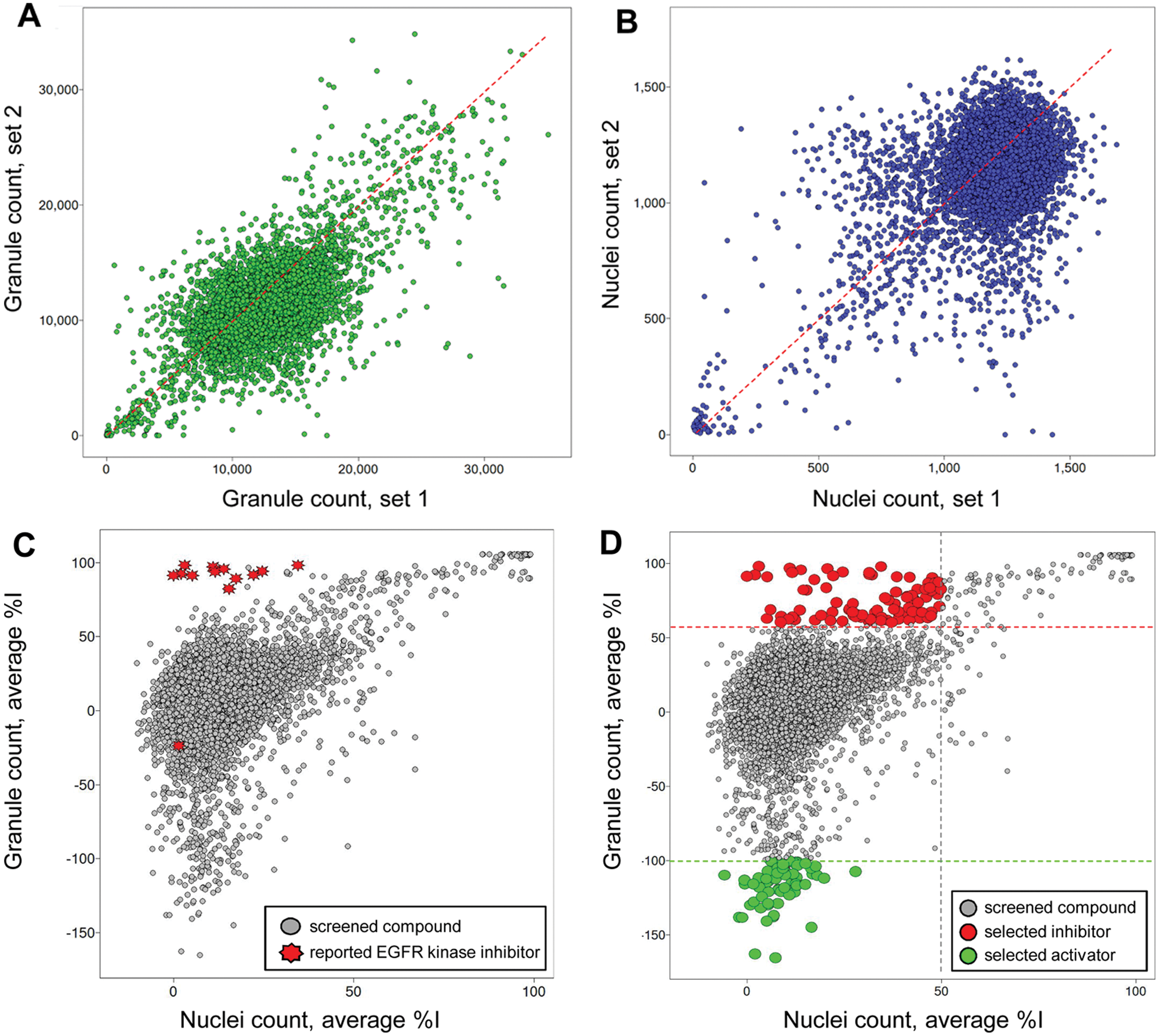
Scatter plot analysis of the pilot screen performed in duplicate. A library of 6912 compounds was screened in duplicate to evaluate assay reproducibility and performance. (
To assess whether the EGFRB assay was able to identify EGFR inhibitors in live cells, we highlighted all described EGFR kinase inhibitors present in the library in the scatter plot of the average percentage inhibition in granule count for each compound against the average percentage inhibition it induced in nuclei count (
Fig. 3C
). As an important result, 12 of 13 reported EGFR kinase inhibitors are clustered in this scatter plot as inducing a high percentage inhibition in granule count and low percentage in nuclei count, as expected (
Fig. 3C
and
On the basis of the performance of each compound in the granule count and nuclei count readout, we identified two populations of compounds: those compounds mimicking the performance of EGFR kinase inhibitors that inhibited granule count in the absence of toxicity, as well as compounds that apparently induced an increase in granule formation in the absence of an increase in cell count (
Fig. 3D
). We selected 82 positives for inhibition of granule formation in our pilot screen as those compounds inducing greater than 60% inhibition of granule formation and less than 50% inhibition in nuclei count, resulting in an initial hit rate of 1.2% (
Dose-Response Studies of the Confirmed EGFR Inhibitors and Activators
For confirmation of the identified positives in the EGFRB assay, we assessed the activity of the 42 resupplied positives in the EGFRB assay in dose-response studies over 12 doubling dilutions from a high concentration of 10 µM compound. Thirteen of the 27 identified inhibitors of granule formation were confirmed as inhibitors in the EGFRB assay (48% confirmation rate) with a calculated IC50 lower than 10 µM for inhibition of granule formation, along with a calculated IC50 greater than 10 µM for nuclei count, indicating that the observed decrease in granule count is not due to a decrease in the number of cells (
Table 1
and
Performance of 16 Hits and Erbstatin Analog in the EGFR Biosensor Assay
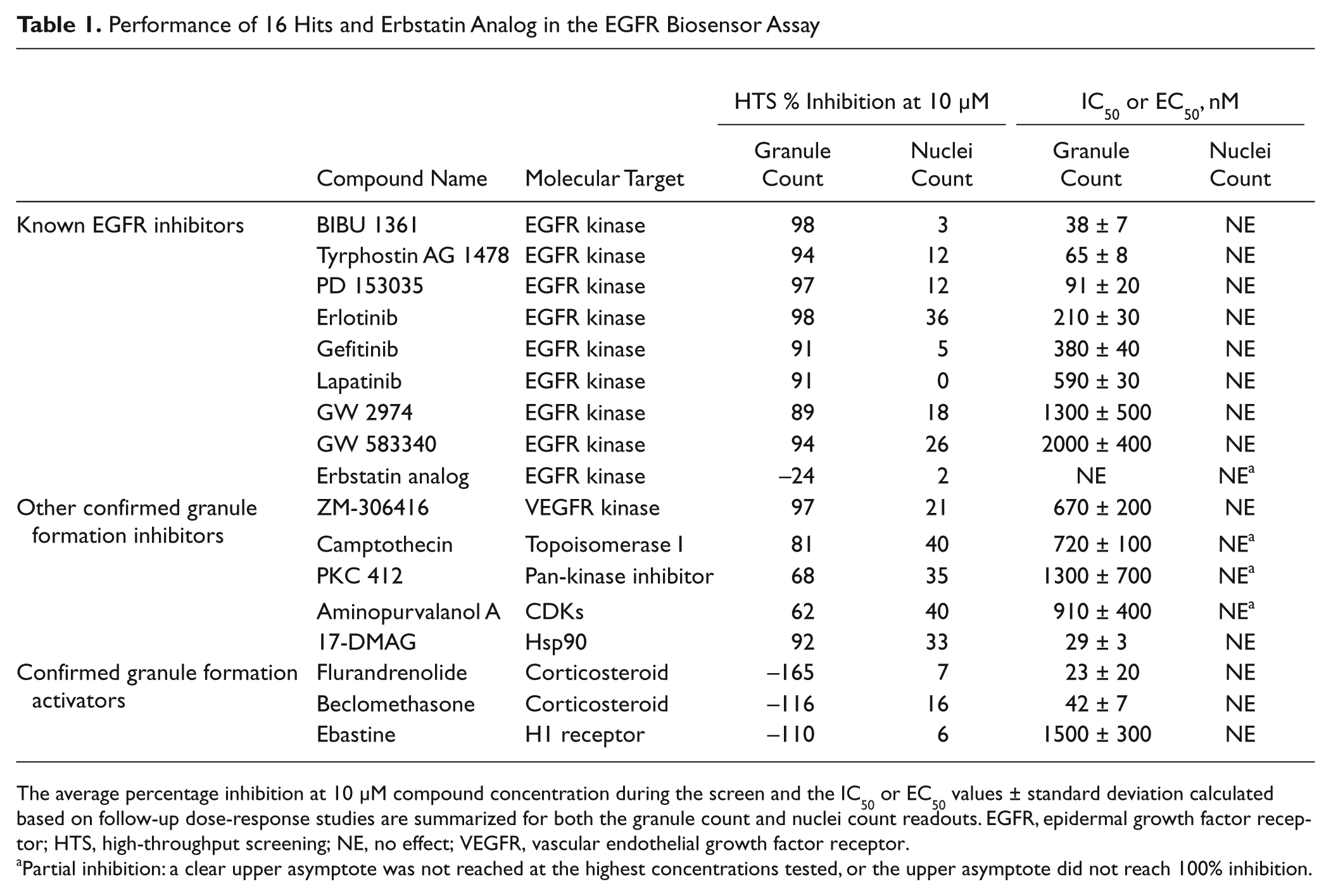
The average percentage inhibition at 10 µM compound concentration during the screen and the IC50 or EC50 values ± standard deviation calculated based on follow-up dose-response studies are summarized for both the granule count and nuclei count readouts. EGFR, epidermal growth factor receptor; HTS, high-throughput screening; NE, no effect; VEGFR, vascular endothelial growth factor receptor.
Partial inhibition: a clear upper asymptote was not reached at the highest concentrations tested, or the upper asymptote did not reach 100% inhibition.
Among the 15 resupplied activators of granule formation, 3 were confirmed as activators in the EGFRB assay (20% confirmation rate) with a calculated EC50 lower than 10 µM for activation of granule formation, along with a calculated EC50 greater than 10 µM for nuclei count. As a control, granules were not observed when we imaged A549 parental cells in the GFP channel after treatment with the identified activators of granule formation, ruling out the possibility that the observed increase in granules results from an artifact. The three confirmed activators were the steroids flurandrenolide (EC50 = 0.023 ± 0.02 µM) (
Cytotoxicity Assessment of the Confirmed EGFR Inhibitors and Activators
To further characterize the activity of the confirmed positives in the EGFRB assay, we assessed the dose response in the Alamar Blue viability assay of confirmed granule formation inhibitors and activators toward wild-type EGFR cells (A549-EGFRB, A549, and H2030 cells), as well as the EGFR-addicted H3255 and HCC4011 human NSCLC cells harboring the activating L858R EGFR mutation. Not surprisingly, all known EGFR inhibitors confirmed in our assay had potent cytotoxic activity toward the H3255 and HCC4011 cell lines such as gefitinib, with an IC50 of <0.01 µM and 0.028 ± 0.003 µM toward H3255 and HCC4011 cells, respectively, while having no or low effect toward the wild-type EGFR cell lines A549 and H2030 (IC50 > 10 µM) ( Fig. 4 and Table 2 ). An important result was that the reported EGFR kinase inhibitor erbstatin analog did not have any significant antiproliferative effect toward any of the cell lines tested, inducing only partial inhibition in the Alamar Blue viability assay up to 10 µM ( Fig. 4 and Table 2 ). This result is in agreement with our previous findings, according to which erbstatin analog exhibited no activity in the EGFRB assay. Confirming the newly identified inhibitory activity of ZM-306416 toward EGFR, this compound induced selective antiproliferative effect toward the EGFR-addicted NSCLC cell lines H3255 and HCC4011 (IC50 = 0.09 ± 0.007 µM and 0.072 ± 0.001 µM, respectively), while sparing the wild-type EGFR cell lines A549 and H2030 (IC50 > 10 µM) ( Fig. 4 ).
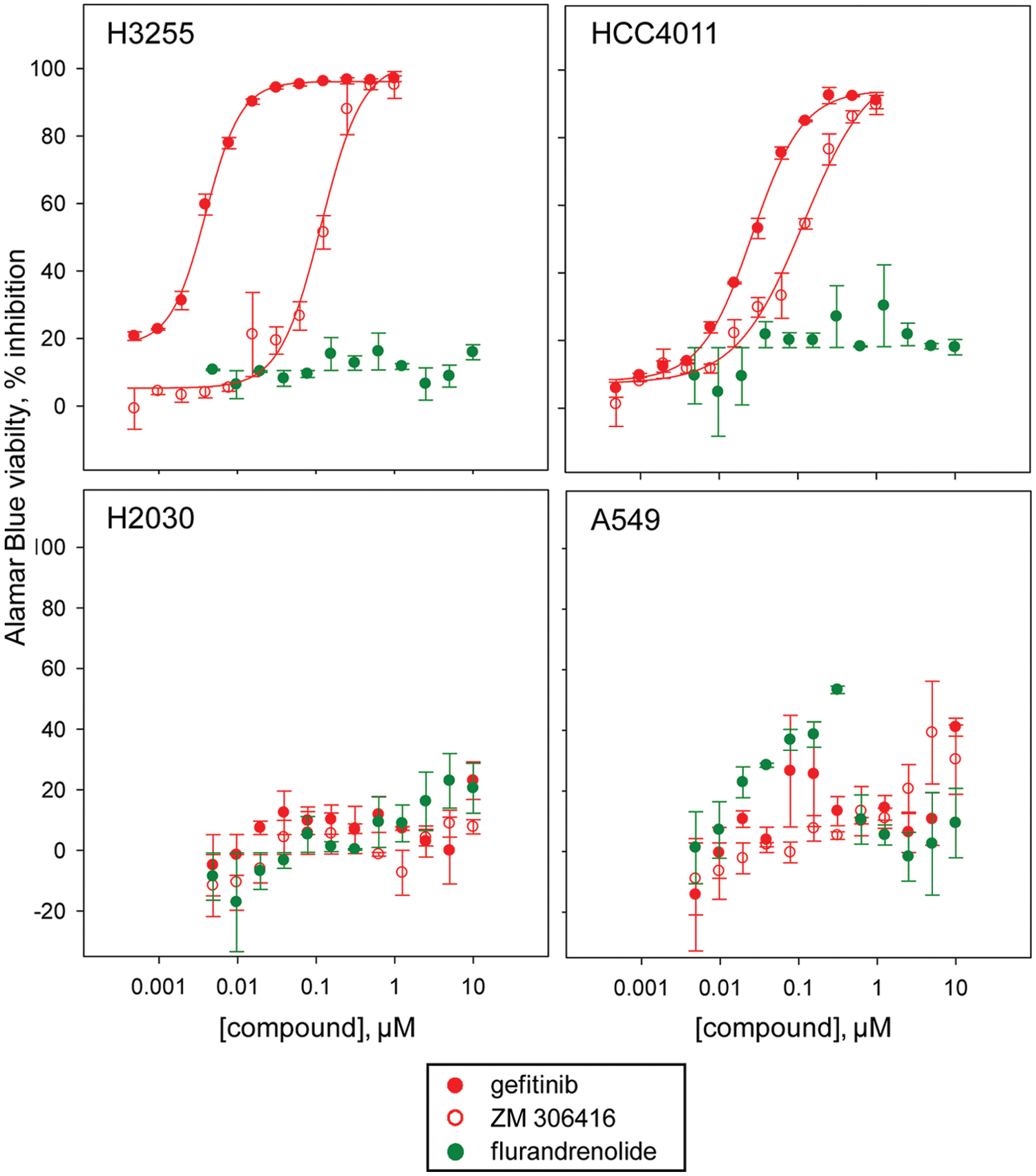
Assessment of the selective antiproliferative effect of confirmed positives toward epidermal growth factor receptor (EGFR) mutant cell lines. Dose-response studies of the compounds gefitinib, ZM-306416, and flurandrenolide in the Alamar Blue viability assay against the H3255 and the HCC4011 cell lines harboring the activating EGFR mutation L858R as compared with the wild-type EGFR cell lines H2030 and A549. The EGFR kinase inhibitor gefitinib is selectively potent toward the EGFR mutant H3255 and HCC4011 cells, with a calculated IC50 of <0.01 µM and 0.028 ± 0.003 µM, respectively. Similarly, ZM-306416 is selectively potent toward the EGFR mutant H3255 and HCC4011 cells, with a calculated IC50 of 0.09 ± 0.007 µM and 0.072 ± 0.01 µM, respectively. Both gefitinib and ZM-306416 are inactive toward the wild-type EGFR cell lines H2030 and A549. The confirmed activator of granule formation, flurandrenolide, is inactive toward both the EGFR mutant and wild-type cell lines tested.
Of note, none of the confirmed granule activators induced any significant effect toward the viability of our cell panel (
Table 2
). This is an expected result since these compounds did not significantly affect the nuclei count during the screen (
Performance of 16 Hits and Erbstatin Analog in the Alamar Blue Viability Assay
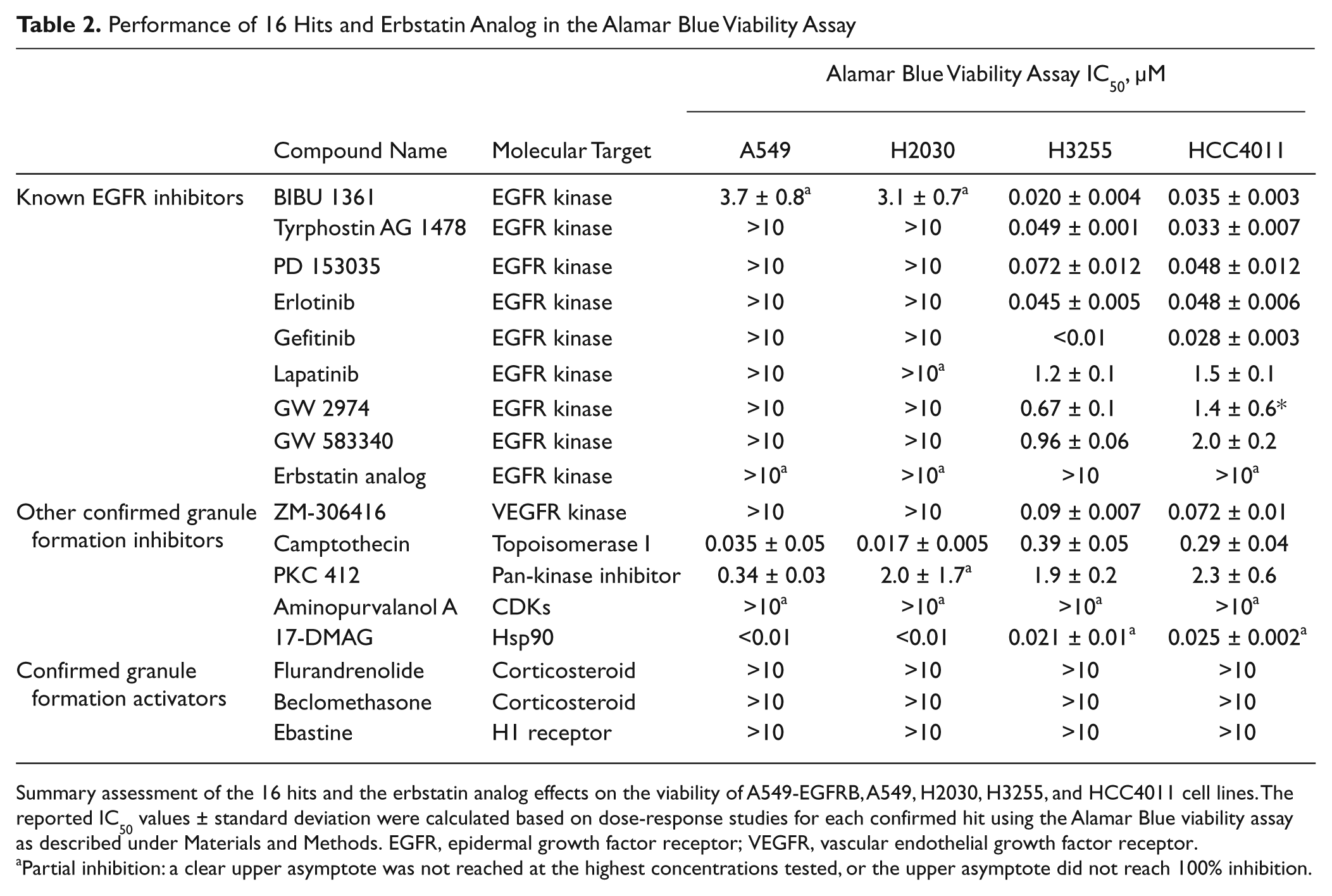
Summary assessment of the 16 hits and the erbstatin analog effects on the viability of A549-EGFRB, A549, H2030, H3255, and HCC4011 cell lines. The reported IC50 values ± standard deviation were calculated based on dose-response studies for each confirmed hit using the Alamar Blue viability assay as described under Materials and Methods. EGFR, epidermal growth factor receptor; VEGFR, vascular endothelial growth factor receptor.
Partial inhibition: a clear upper asymptote was not reached at the highest concentrations tested, or the upper asymptote did not reach 100% inhibition.
Kinase Activity Assessment of the Confirmed Hits against a Panel of Four Kinases
We further characterized confirmed positives by assessing their potency in a luminescence ADP production kinase toward a panel of kinases that included EGFR, VEGFR1, ABL, and SRC kinases. As expected, the eight resupplied known EGFR kinase inhibitors inhibited EGFR kinase activity with IC50 values consistently in the nanomolar range ( Table 3 ). Interestingly, the described EGFR kinase erbstatin analog was inactive against all kinases tested, including its target, EGFR. This result confirms our observation that this compound was inactive in the EGFRB assay and had no observed cytotoxicity effects on the H3255 and HCC4011 cell lines harboring the activating L858R EGFR mutation. ZM-306416 was found to be very potent toward the EGFR kinase, with an IC50 value lower than 10 nM, reaching our assay detection limit and confirming our result using the EGFRB assay (see above). ZM-306416 exhibited inhibitory activity across all three kinases of the panel, yielding IC50 values of 0.33 ± 0.08 µM for SRC, 0.33 ± 0.04 µM for VEGFR1, and 1.3 ± 0.2 µM for ABL kinases ( Fig. 5 and Table 3 ). Although the other confirmed inhibitors of granule formation distinct from described EGFR kinase inhibitors have a calculated IC50 toward EGFR kinase greater than 10 µM, it is important to note that they do induce partial inhibition of EGFR kinase activity, which could potentially explain their potency in the EGFRB assay. Partial inhibition of nuclei count induced by these compounds could also contribute to the observed decrease in granule count ( Table 1 ).
Performance of 15 Hits and Erbstatin Analog against a Panel of Kinases
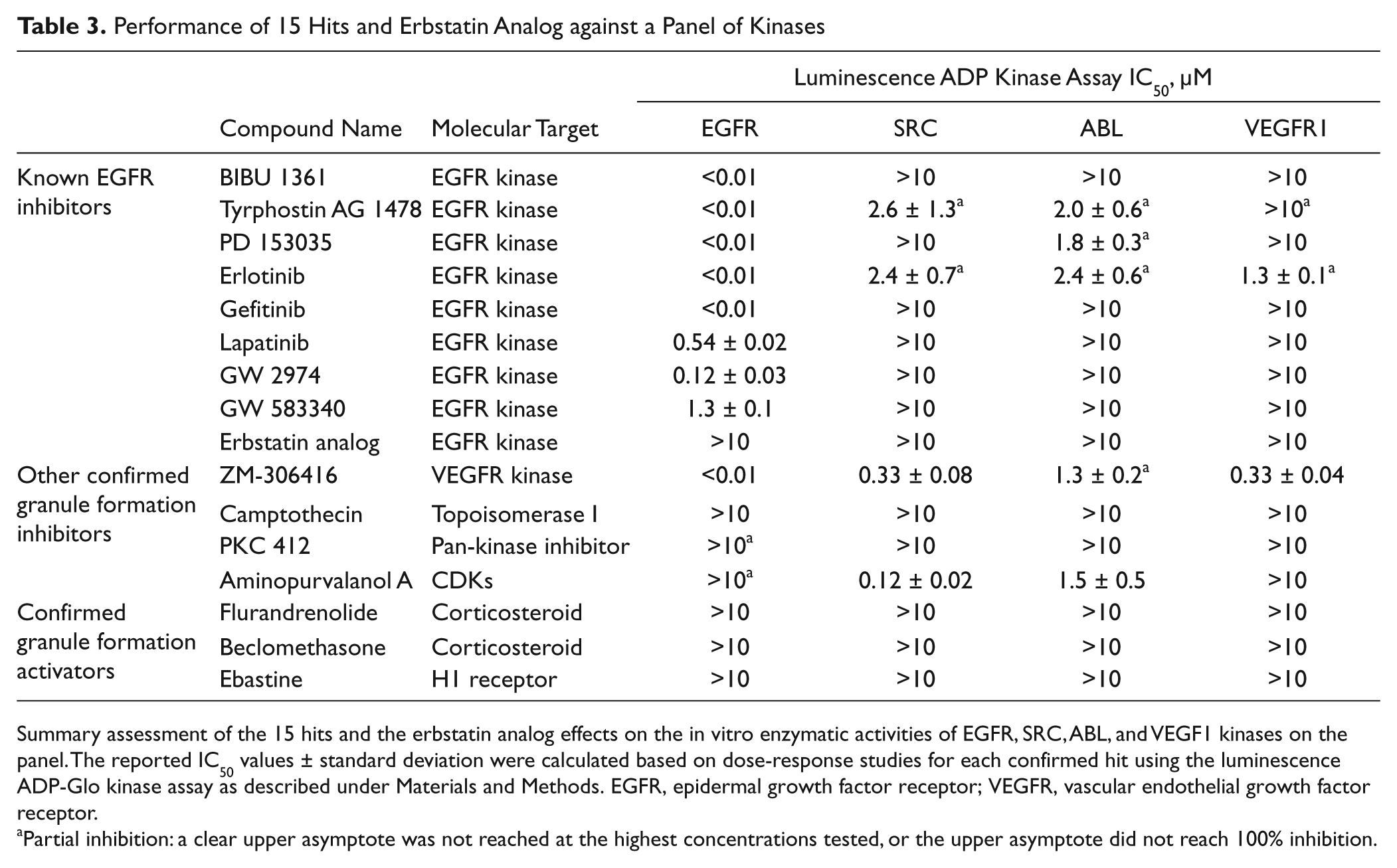
Summary assessment of the 15 hits and the erbstatin analog effects on the in vitro enzymatic activities of EGFR, SRC, ABL, and VEGF1 kinases on the panel. The reported IC50 values ± standard deviation were calculated based on dose-response studies for each confirmed hit using the luminescence ADP-Glo kinase assay as described under Materials and Methods. EGFR, epidermal growth factor receptor; VEGFR, vascular endothelial growth factor receptor.
Partial inhibition: a clear upper asymptote was not reached at the highest concentrations tested, or the upper asymptote did not reach 100% inhibition.
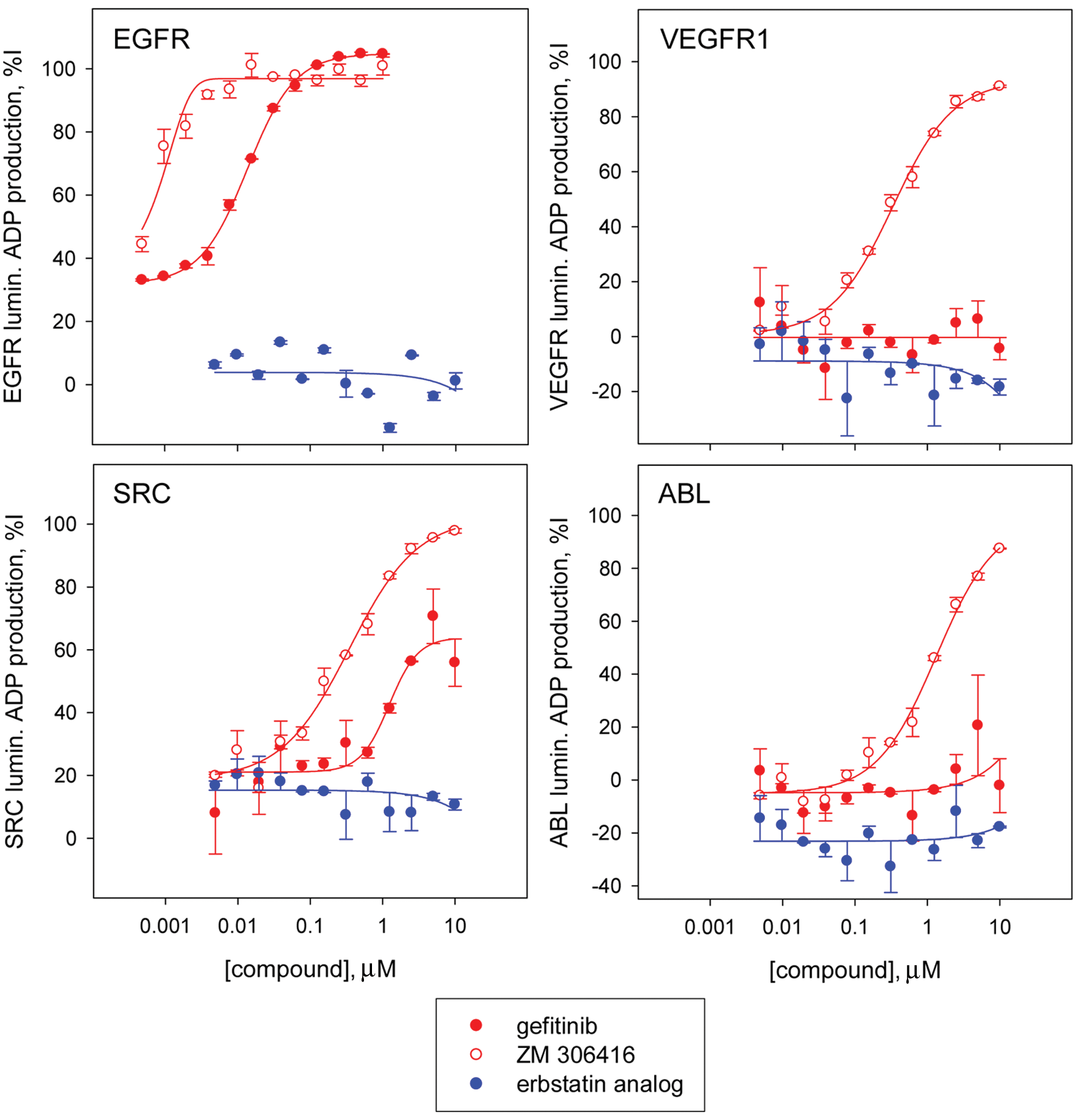
Assessment of potency of selected hits in a panel of kinases using the luminescence adenosine diphosphate (ADP) production kinase assay. Dose-response studies of gefitinib, ZM-306416, and the erbstatin analog against a kinase panel consisting of the kinases EGFR, VEGFR1, SRC, and ABL. The epidermal growth factor receptor (EGFR) kinase inhibitor gefitinib is selectively potent toward EGFR, with a calculated IC50 of <0.01 µM, and low or no activity toward VEGFR1 (IC50 > 10 µM), SRC (IC50 > 10 µM), and ABL (IC50 > 10 µM). In contrast, ZM-306416 is potent toward all kinases of the panel: EGFR (IC50 < 0.01 µM), VEGFR1 (IC50 = 0.33 ± 0.04 µM), SRC (IC50 = 0.33 ± 0.08 µM), and ABL (IC50 = 1.3 ± 0.2 µM). Erbstatin analog is inactive (IC50 > 10 µM) toward all kinases in the panel, including EGFR kinase.
Of note, the three confirmed activators of granule formation—flurandrenolide, beclomethasone, and ebastine—have no inhibitory effects on the enzymatic activity of the kinases on the panel ( Table 3 ). This is an expected result since the increase in granule count induced by these compounds is indicative of a stimulation of EGFR activation rather than its inhibition, and to our knowledge, those compounds have no reported inhibitory activity toward kinases.
Discussion
Targeting RTKs has proven to constitute a successful strategy for the development of novel antitumor agents potent in the clinic. As an example, there are currently three small-molecule drugs approved by the FDA that target EGFR: gefitinib (Iressa), erlotinib (Tarceva), and lapatinib (Tykerb, Tyverb). All three drugs are 4-anilinoquinazoline-based chemicals and share the same inhibitory mechanism of action (
Current drugs eventually become inactive in those patients developing mutations, and as such, there is a need to rapidly identify new drug candidates overcoming resistance. However, current approaches to identify new drug candidates targeting RTKs are rather slow with a high attrition rate of leads, hampering the discovery of novel candidates. The failure of many lead candidates during development is due to the fact that they are identified in HTS screens relying on assays measuring the kinase activity of recombinant kinases. Very often, potent molecules in vitro fail the transition to being potent when tested in cellular assays since such assays are highly artificial compared with physiological protein expression levels, together with the complexity of the cellular environment and the presence of interconnected signaling pathways. The current low success rate of drug candidates targeting RTKs can therefore be attributed to the lack of cell-based assays that would allow direct identification of RTK inhibitors. In addition, since currently available assays amenable to HTS all measure the kinase activity of the receptor, all drug candidates discovered through this process share the same limitations of kinase inhibitors with regard to the appearance of resistance in patients.
For this reason, we sought to explore the use of domain-based biosensors of RTK activation, and after developing a domain-based EGFR biosensor as a proof of concept,
8
we aimed at validating this new technology for HTS (
Fig. 1
). In this article, we show that our miniaturized assay in a 384-well format is robust, with a Z′ of 0.56 in a control run performed in the conditions of screening (
Fig. 2B
,
C
). In addition, scatter plot representation of the performance of each compound tested in duplicate in our screen of 6912 bioactive compounds is indicative of the good reproducibility of our assay (
Fig. 3A
,
B
). For positive compounds present in the library as multiple instances provided by different suppliers, the observation that we picked as positives, several of each instance of those compounds, further demonstrates the good reproducibility of the EGFRB assay in the conditions of screening, as this was the case, for example, with tyrphostin AG 1478, PD 153035, camptothecin, cycloheximide, lycorine, and emetine for the identified inhibitors of granule formation (
Our results validated our approach, in that we have developed an assay that allowed the identification of 12 of 13 reported EGFR kinase inhibitors present in the screened library, including the three FDA-approved EGFR kinase inhibitors: gefitinib, erlotinib, and lapatinib (
Table 1
and
In addition, we have discovered that the VEGFR (Flt and KDR) kinase inhibitor ZM-306416,
21
a previously not described inhibitor of EGFR, was an inhibitor of granule formation in the EGFRB assay (
Besides the known EGFR inhibitors and the discovery of ZM-306416 as an EGFR inhibitor, several confirmed hits with distinct biological activities were also identified in the pilot screen. Among them were camptothecin, a topoisomerase I inhibitor and potent cytotoxic agent
24
; PKC412, a pan-active kinase inhibitor reported to exhibit weak activity against EGFR (IC50 = 3.0 µM)
20
; aminopurvalanol A, an inhibitor of various CDKs
25
; and 17-DMAG, an HSP90 inhibitor and potent cyotoxic agent.
22
Camptothecin, PKC412, and aminopurvalanol A were not found to be potent toward EGFR kinase or induced only partial inhibition of EGFR kinase activity up to 10 µM (
Table 3
); those three compounds also induced partial inhibition of EGFRB cells’ nuclei count (
Table 1
), indicating that the observed reduction in granule count in the EGFRB assay may result from a combination of partial inhibition of kinase activity and cell count. 17-DMAG, however, induced potent inhibition of granule formation (IC50 = 0.029 ± 0.003 µM) in the absence of any effect on nuclei count (
Table 1
and
In addition, our results demonstrate the versatility of our approach, in that we have unexpectedly identified confirmed activators of granule formation such as flurandrenolide (
In conclusion, we anticipate that our approach allows us to accelerate the discovery of potent drugs targeting RTKs. The ability to screen for modulators of mutated endogenous RTK in cells derived from patients opens the door to the rapid identification of drug candidates overcoming the specific mechanism of resistance developed in each patient. Furthermore, assays relying on a domain-based biosensor can easily be adapted to conducting RNAi screening, allowing the identification of genes involved in signaling pathways in relation to the RTK of interest. For these reasons, we expect that the validated domain-based biosensor technology that we describe in this article will accelerate drug discovery as well the understanding of complex signaling pathways related to RTKs.
Footnotes
Acknowledgements
The authors thank Constantin Radu, Alun Bermingham, and members of the HTS Core Facility for their help during the course of this study, as well as their colleagues Dmitry Malkov, Keming Song, and John Fetter at Sigma-Aldrich. The authors also thank Tony Riley and Terry Helms for their help with the artwork in this manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The HTS Core Facility is partially supported by Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, the William Randolph Hearst Fund in Experimental Therapeutics, the Lillian S. Wells Foundation, and NIH/NCI Cancer Center Support Grant 5 P30 CA008748-44.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.