
Abstract
Alzheimer’s disease (AD) is a devastating neurodegenerative disease affecting millions of people. The amyloid hypothesis suggests that the pathogenesis of AD is related to the accumulation of amyloid beta (Aβ) in the brain. Herein, the authors quantify Aβ-mediated changes in neuronal morphology in primary cultures using the Cellomics neuronal profiling version 3.5 (NPv3.5) BioApplication. We observed that Aβ caused a 33% decrease in neurite length in primary human cortical cultures after 24 h of treatment compared with control-treated cultures. We also determined that quantifying changes of neuronal morphology was a more sensitive indicator of nonlethal cell injury than traditional cytotoxicity assays. Aβ-mediated neuronal deficits observed in human cortical cultures were also observed in primary rat hippocampal cultures, where we demonstrated that the integrin-blocking antibody, 17E6, completely abrogated Aβ-mediated cytotoxicity. Finally, we showed that Aβ challenge to 21 days in vitro rat hippocampal cultures reduced synapsin staining to 14% of control-treated cultures. These results are consistent with the finding that loss of presynaptic integrity is one of the initial deficits observed in AD. The implementation of phenotypic screens to identify compounds that block Aβ-mediated cytotoxicity in primary neuronal cultures may lead to the development of novel strategies to prevent AD.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by irreversible loss of memory, cognitive decline, and ultimately death. It is estimated that more than 26 million people worldwide live with AD, where it is the leading cause of dementia in the aging population. Although AD is mostly associated with the elderly population, an aggressive earlier-onset form of the disease also occurs. Several defining hallmarks of AD include the presence of senile plaques consisting of insoluble Aβ, neurofibrillary tangles composed of hyperphosphorylated tau, and dystrophic neuritis. 1 These structural and morphological changes are associated with loss of synaptic density and cognitive function.
From a compilation of biochemical, genetic, and animal model data, it is thought that the pathogenesis of AD is related to the accumulation of Aβ1-40 and Aβ1-42, more formally known as the amyloid hypothesis. 2 Initially, it was thought that the toxicity of amyloid beta (Aβ) was mediated by the extracellular fibrillar forms of Aβ. However, several lines of evidence have emerged from in vitro and in vivo models to suggest that low n-oligomers of human Aβ or protofibrils are also synaptotoxic. 3 It is likely that both deposited and soluble forms of Aβ play a role in the pathogenesis of AD, where soluble Aβ species are more relevant to synaptic toxicity and insoluble species may be more relevant to neurite dystrophy.
The molecular mechanisms by which Aβ mediates neurodegeneration in AD are not completely clear at this time. It has been hypothesized that Aβ causes mitochondrial and lysosomal dysfunction, could form porelike structures with channel activity, or may create aberrant signaling that leads to altered synaptic plasticity. 4 In another proposed mechanism, aberrant integrin signaling may contribute to Aβ−mediated neurodegeneration. In support of this hypothesis, integrin-blocking antibodies have been shown to inhibit Aβ deposition onto human neurons, suggesting that integrins may be one of the cellular receptors for Aβ. 5 Furthermore, Aβ−mediated toxicity in vitro was shown to be dependent on integrin-signaling pathways where Aβ−induced integrin signaling led to the activation of Pyk2 kinase. 5 It is thought that sustained rather than transient Pyk2 signaling may lead to neurotoxicity. Hyperactivation of cdk5 and other kinases has also been linked to neurodegeneration in AD. Moreover, Aβ-induced increases of intracellular calcium result in the activation of the calcium-dependent protease calpain, which leads to increased cdk5 activity and increased phosphorylation of tau. Recently, small-molecule inhibitors of calpain activation were identified in a cell-based high-throughput screen that blocked Aβ-mediated cytotoxicity. 6 Whatever the mechanism by which Aβ mediates synaptotoxicity in transgenic animal models and in AD patients, loss of presynaptic integrity characterized by a loss of presynaptic synaptophysin and synapsin levels and pronounced neurite dystrophy are early manifestations of the disease.1,2
Current clinical therapeutic strategies are aimed at enhancing neurotransmission with acetylcholinesterase inhibitors and NMDA receptor antagonists, yet these therapies are palliative in nature and not disease modifying. Antioxidant therapies have been a popular topic of discussion, yet to date, there have not been clear data supporting the effectiveness of this strategy in the clinic. In addition, there have been and are currently a number of compounds whose mechanism involves inhibition of Aβ aggregation; it remains to be seen if this strategy will be effective in the clinical setting. 7 There are also clinical trials ongoing that involve β and γ-secretase inhibitors, which decrease the production of Aβ. An important test of the amyloid hypothesis will come when the clinical trial data for solanezumab and bapineuzumab, monoclonal antibodies that bind and help clear Aβ from the brain, are unveiled.
In vitro cellular models that can recapitulate key features of AD allow for the development of phenotypic screens aimed at identifying new classes of therapeutics for AD. High-content screening has been performed in primary neuronal cultures, embryonic stem cells, and induced pluripotent stem cells. This technology acquires large data sets of fluorescence images on cell shape and morphology, intensities, and organelle localization, thus providing a phenotypic signature of the state of the cell. 8 Herein, we have developed a high-content analysis (HCA) assay in which we have used the Cellomics NPv3.5 BioApplication to quantify Aβ-mediated deficits of neuronal morphology and synaptic integrity in human and rat primary neuronal cultures. The results of our studies could lead to the implementation of phenotypic screens using HCA to identify new compounds and strategies for AD therapy and prevention.
Materials and Methods
Reagents
Aβ1-40 catalog No. 641-10 and Aβ1-42 catalog No. 62-0-80A were obtained from California Peptide Research (Napa, CA) and the American Peptide Company (Sunnyvale, CA), respectively. The β3 tubulin antibody, No. G712A, used for staining of neurites was obtained from Promega (Fitchburg, WI). The monoclonal antibody for staining of Aβ was developed, expressed, and purified at Elan Pharmaceuticals (San Francisco, CA). The 17E6 integrin-blocking antibody was obtained from EMD-Bio No. 407286. Glutamine, penicillin, B27, Hanks balanced salt solution (HBSS), and streptomycin were all purchased from Gibco-Invitrogen (Carlsbad, CA). Alamar blue No. DAL1100 and Hoechst dye were also purchased from Invitrogen and were used according to the manufacturer’s suggestions. Fetal bovine serum, No. F0926, was obtained from Sigma (St. Louis, MO). FITC, TRITC, and Cy3 goat anti-rabbit and mouse secondary antibodies were purchased from Jackson Immuno Laboratories (West Grove, PA). Human fetal brain tissue was obtained from Advanced Bioscience Resources (Alameda, CA). The protocol for obtaining fetal brain tissue complied with federal guidelines for fetal research and with the Uniformed Anatomical Gift Act. All other reagents were acquired from Sigma.
Cell Culture
Primary rat hippocampal cultures were prepared from fetal rats at embryonic day 17 to 18. Hippocampi were dissected in Ca2+/Mg2+–free HBSS buffered with 10 mM HEPES, dissociated in the same solution, and plated on poly-D-lysine–coated culture plates. Cells were cultured in neurobasal medium, which consisted of MEM supplemented with B27, 0.5 mM glutamine, penicillin (50 U/mL), and streptomycin (50 µg/mL). Cultures were incubated at 37 °C in a humidified atmosphere with 5% CO2 and treated with cytosine arabinoside (10 µM) from day 3 to day 5 to inhibit nonneuronal proliferation. For development of human primary cortical cultures, 1 to 3 g of tissue was washed twice in Ca2+/Mg2+–free HBSS and then dissociated by repeated pipetting in 10 mL of cold HBSS with 1 mL of DNase, and the solution was passed through a 100-µm nylon cell strainer. The cells obtained were then centrifuged for 5 min at 200 × g, resuspended in a trypsin/EDTA solution (0.05% trypsin and 0.53 mM EDTA in HBSS), and incubated for 20 min at 37 °C. After centrifugation, cells were resuspended in neuronal medium (MEM supplemented with B27, 1% glucose, 1 mM sodium pyruvate, and 1 mM glutamine) and plated onto poly-D-lysine–coated 96-well plates at a density of 25 000 or 50 000 cells per well.
Automated Image Analysis
The Cellomics ArrayScan VTI was used for automated image acquisition and analyses. Images were acquired using a Zeiss 20× objective (0.4 NA) and ORCA-ER CCD camera. After fixing neurons with 4% paraformaldehyde and washing, cells were permeabilized using 0.01% Triton X-100 in phosphate-buffered saline (PBS). An antibody to β3 tubulin and FITC-labeled secondary antibody was used to stain the cell body and neurites. Fixed cells were treated with Hoechst dye for 30 min at room temperature to label nuclei. Automated image analysis was performed using the Cellomics NPv3.5 BioApplication, where the algorithm independently creates nuclear, cell body, and neurite masks and then assimilates these areas to create an image. The nuclear mask was created using the area defined by the Hoechst nuclear stain, where a nucleus was identified as valid if it registered relevant features of size, intensity, and shape. A cell body mask was created from β3 tubulin staining, where a cell body was accepted as a valid object if it registered relevant area, staining intensity, and appropriate shape. Based on positional information, nuclei were then associated with the correct cell body. Neurites were then identified also based on the β3 tubulin stain, where thresholds for neurite features of width, shape, and intensity were manually set. Neurites were individually traced and then associated with the appropriate cell body. Cutoffs for nuclear, cell body, and neurite features were arbitrarily determined based on inspection of fixed neurons by at least two investigators. To effectively trace neurites in our system, the neurite threshold modifier was manually set to −0.79. Parameters of neuronal morphology such as neurite average length, neurite max length with branches, branch point average count, and branch point average distance from the cell body were automatically calculated by the BioApplication.
For synapsin staining, after cells were fixed and permeabilized, the synapsin antibody was incubated with cells for 60 min and then washed 3 times with PBS. A goat anti-rabbit secondary–TRITC-labeled antibody was then incubated with fixed cells for 60 min and then washed three times with PBS. To quantify synapsin staining using a third channel, the spot detect radius and spot detect method were manually set to 1. The neurite mask was created with the β3 tubulin stain, and this area was expanded to define the region to measure punctate synapsin staining, where spots between 1 and 18 um2 in total area were quantified. Punctate synapsin spots smaller or larger than these arbitrarily set cutoffs were eliminated from analysis.
Defining Parameters of Neuronal Morphology
The parameter “Neurite Average Length” = neurite total length
Aβ Preparations and Treatments
Aβ1-40/2 was prepared as described by the method of Wogulis et al. 9 Briefly, for the seed preparation, Aβ peptide was weighed out in a 4 mL glass vial and made to a 1 mM solution in water. This solution was incubated for three days at 37 °C. The seed Aβ was then dispensed into 4 mL glass vials and snap frozen. Soluble Aβ was prepared in 100% DMSO at a 20 mM concentration and then aliquotted into 4 mL glass vials and snap frozen. Two component Aβ treatments involved placing aggregated or seed Aβ onto cells for 60 min followed by washing and then adding DMSO solubilized Aβ for 24 to 72 h. In experiments using primary rodent hippocampal cultures, DMSO solubilized Aβ1-40 and Aβ1-42 were directly applied from DMSO stock into media and then applied to cultures in the absence of seed Aβ.
Alamar Blue Assay
In the Alamar blue assay, viable cells convert the nonfluorescent oxidized compound resazurin to the highly fluorescent-reduced resorufin, which is an indicator of cellular metabolic activity. Assays were performed by replacing the culture media with a 10% Alamar blue solution in DMEM. Reduction of Alamar blue was determined spectrofluorometrically after 2 h using a Millipore Cytofluor plate reader (excitation 440 nm, emission 485 nm).
Statistical Analysis Methods
Compound effects on each neuronal morphology parameter were statistically analyzed using a one-factor analysis of variance model. Dose-responsive changes were examined using a two-sided sequential trend test. Homogeneity of variance was evaluated using Levene’s test at the 0.01 significance level. Compound effects on BDNF were statistically analyzed using a two-sided two-sample t-test at the 0.05 significance level, where homogeneity of variance was evaluated using Levene’s test at the 0.01 significance level.
Results and Discussion
Alterations of neuronal morphology and neuritic dystrophy are early hallmarks of AD, where Aβ has been hypothesized to be the causative agent. Our initial studies were aimed at quantifying the effect of Aβ1-40 on parameters of neuronal morphology in human primary cortical cultures. The Aβ peptide can be difficult to manipulate because of its physical chemical characteristics, and thus results can be variable between investigators. In our initial studies, we have used the two-component method of Aβ preparation in which we have observed consistent results. In the two-component Aβ model, 1 µM of fibrillar Aβ1-40 is added to primary human cortical cultures for 1 h, the media are then changed, and 2.5, 5, 10, or 20 µM soluble Aβ is added for 24 h. In Figure 1 , we have quantified Aβ-mediated changes in neuronal morphology after 24 h of treatment, where we observed that Aβ1-40 induced a dose-dependent decrease in a number of parameters of neuronal morphology including neurite average length, neurite max length with branches, the branch point average count, and branch point average distance from cell body. The 20 µM Aβ1-40 challenge caused a reduction of neurite average length (67% of control), a decrease in neurite max length with branches (54% of control), markedly fewer branch points (42% of control), and a decrease in branch point average distance from the cell body (46% of control). The data would suggest that branch point average count was the most sensitive parameter, which showed a significant decrease compared with control at a 5 µM Aβ1–40 concentration ( Fig. 1 ).
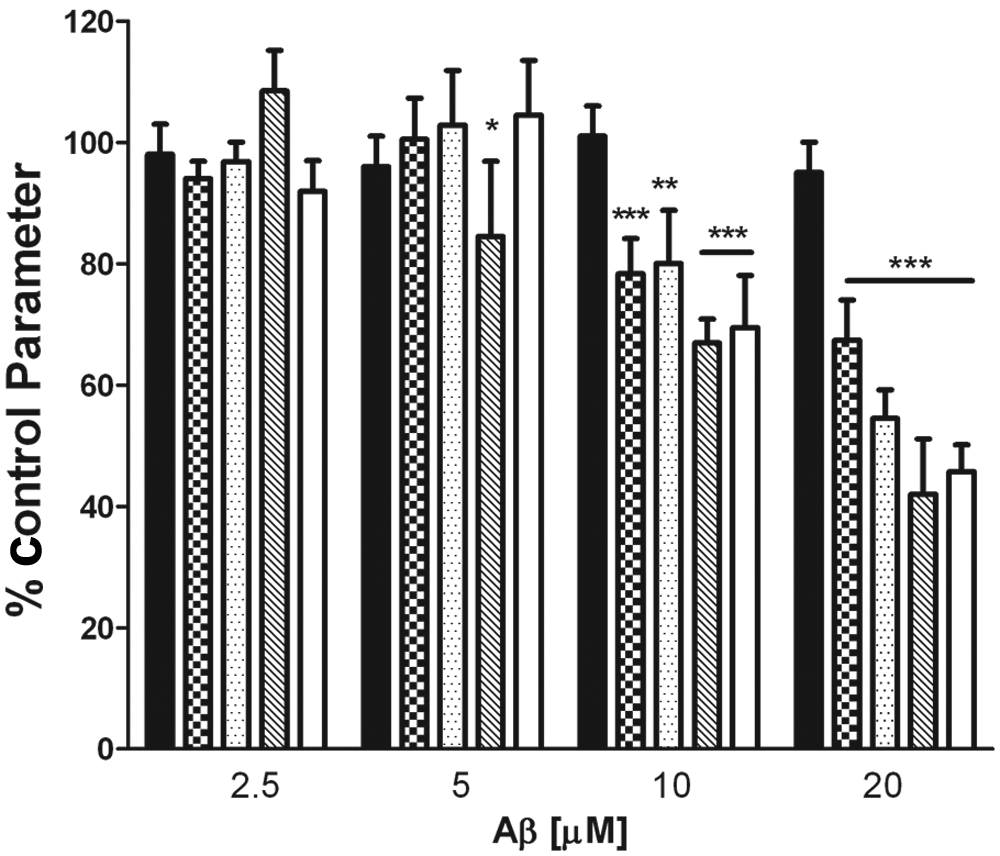
Characterization of Aβ1-40–induced changes of neuronal morphology in human cortical cultures. Human primary cortical neurons were prepared and cultured for 7 days in vitro. Aβ1-40, at the concentrations indicated in the figure, was incubated with primary neurons for 24 h, and then cultures were fixed and stained as outlined in the Materials and Methods section. Neurite average length (checkered), neurite max length with branches (dotted), branch point average count (hatched), and branch point average distance from the cell body (white) were determined using the Cellomics NPv3.5 BioApplication and compared with control (0.1% DMSO–treated cultures). The Alamar blue readout (dark panel) was determined on a replicate plate and compared with DMSO-treated cells. Data represent the mean ± SD, n = 3. These data have been repeated on three different occasions with similar results. ***p < 0.001; **p < 0.01; *p < .05 versus DMSO-treated control parameter.
We hypothesized that measuring parameters of neuronal morphology would be a more sensitive measure of nonlethal neuronal cell injury compared with traditional cytotoxicity assays, such as the Alamar blue assay. The Alamar blue assay measures reduction of a tetrazolium dye, where decreases in Alamar blue fluorescence correlates with cell death.
10
In contrast to the marked and significant changes of neuronal morphology induced by Aβ1-40 at 24 h of incubation, we observed no significant Aβ-mediated decreases in the Alamar blue assay readout at 24 h (
Figs. 1
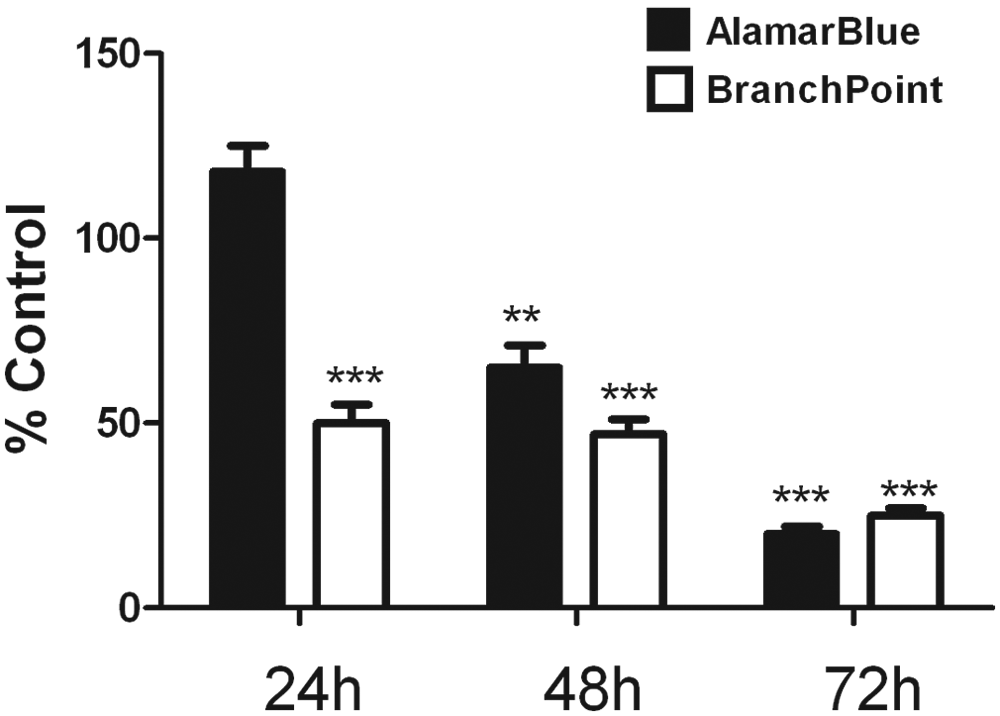
Time course of Aβ1-40 induced changes of neuronal morphology and Alamar blue signal. Human primary cortical neurons were prepared and then cultured for 7 days in vitro. A total of 20 µM Aβ1-40 was incubated with primary neurons for 24, 48, or 72 h, and then cultures were fixed and stained as outlined in the Materials and Methods section. The readout parameter, branch point average distance from cell body (white panel), was determined and compared with control (0.1% DMSO–treated cultures). The Alamar blue readout (dark panel) was determined on a replicate plate and compared with control DMSO-treated cells. Data represent the mean ± SD, n = 3. These data were repeated on three different occasions with similar results. ***p < 0.001, **p < 0.01 versus DMSO-treated control parameter.
There are a number of points to consider when working with primary human cultures. First availability can be limited and cultures are periodically contaminated with virus, making a Biosafety Level II hood critical to use for this work. Considerable variability can also be observed when working with these cultures. In addition, there are ethical issues that can limit the extent to which human fetal tissues can or should be used. Therefore, we also generated rat primary cortical and hippocampal cultures from embryonic day 18 embryos. As a control to show that we can modulate and quantify changes in neuronal morphology in rat hippocampal cultures, we treated primary hippocampal neurons with BDNF, which has been shown to stimulate dendritic growth in primary rodent hippocampal cultures.
11
Indeed, we observed a significant enhancement of branch point average count upon treatment with BDNF (
(
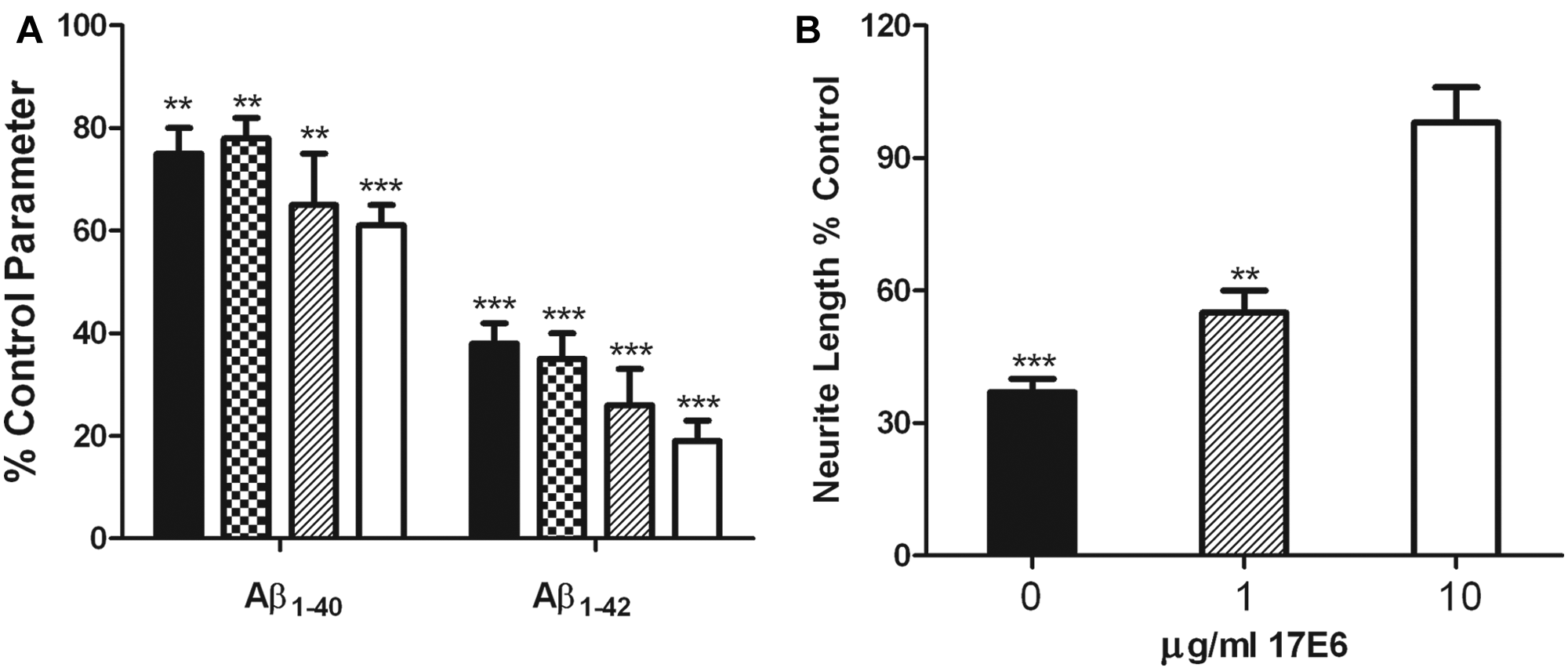
Recently, it has been demonstrated that active immunization with Aβ (AN1792) protected against AD-related deficits in neurite morphology when measured in postmortem tissue. 14 We also wanted to determine whether the Aβ1-42- mediated deficits of neuronal morphology in our in vitro model could be reversed pharmacologically. Aβ has been shown to bind to neurons through integrins, where this interaction can be blocked by α2, αV, and β1 integrin-blocking antibodies. 5 In Figure 3B , we show that the integrin-blocking antibody 17E6 dose-dependently blocked Aβ1-42-induced deficits in neuronal morphology in primary rat hippocampal cultures. Similar results were obtained in rat cortical cultures and with Aβ1-40 (data not shown). Although integrin-blocking antibodies have had tremendous success in the clinic for the treatment of multiple sclerosis, it remains to be determined whether they will show efficacy for other neurodegenerative diseases. In in vitro models, α2β1 and αVβ1 integrin-signaling pathways may be key components in the neurodegeneration caused by Aβ, where Pyk2 and JNK may be two kinases that mediate Aβ cytotoxicity. 6 It should also be noted that we consistently observe a Z′ value >0.5 for all parameters of neuronal morphology in primary rodent hippocampal and cortical cultures using 10 or 20 µM soluble Aβ1-42 (data not shown). Taken together, these results form the basis of a functional cellular screen of sufficient sensitivity and reproducibility to identify antibodies or small molecules that block Aβ-mediated neuronal deficits.
Synapsins are a family of presynaptic proteins that have been implicated in the regulation of neurotransmitter release at the synapse. Structurally, a synapse can be defined by punctate synapsin staining in vitro and in vivo, where losses of synapses are associated with AD. In rat hippocampal cultures, we did not observe synapsin staining at 7 or 14 days in vitro (DIV; data not shown), consistent with reports in which culture of 21 DIV is needed to observe synapsin staining. Here we have performed immunofluorescence staining for the presence of synapsin in rat hippocampal neurons at 21 DIV, where we observed clear and distinct punctate synapsin staining that was quantified by measuring the spot total intensity/neurite parameter (
Fig. 4A
-
C
). In defining the region in which we measured synapsin staining, we have used the region defined by the β3 tubulin stain along the neurite to form a mask and extended that area by two pixels (
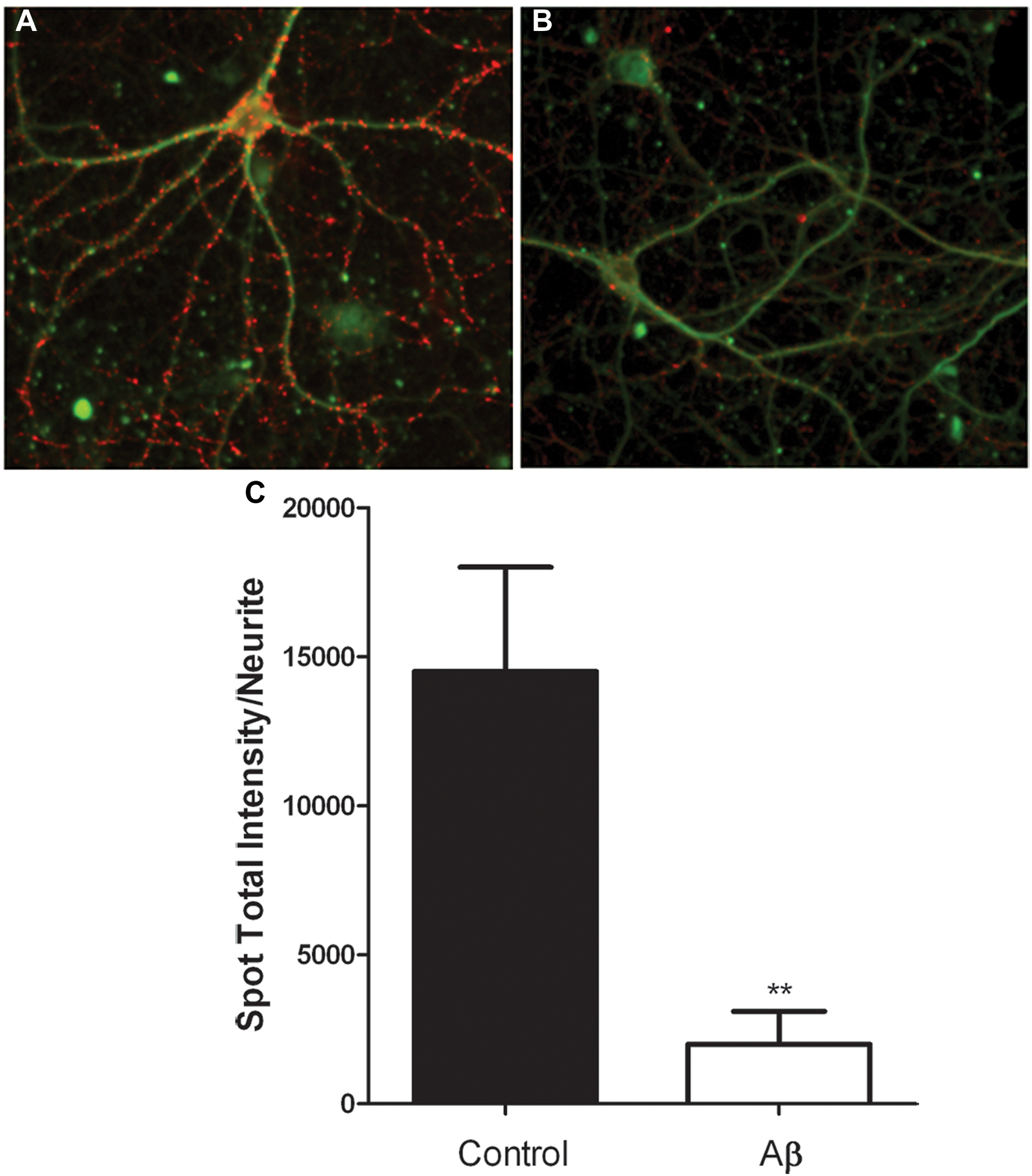
Quantification of synapsin immunostaining in mature rat hippocampal cultures. Rat hippocampal cultures were prepared and then cultured for 21 days in vitro. Cultures were fixed, permeabilized, and immunostained as described in the Materials and Methods section. (
The NPv3.5 BioApplication represents an advance in that in addition to measuring neurite length and branch points, a neurite mask can be defined based on the area defined by the tubulin stain and spot numbers and intensities within that mask can be quantified. This feature allows for quantification of pre- and postsynaptic nerve terminals in primary neuronal culture systems (
Fig. 4C
). Additional markers such as synaptophysin, tyrosine hydroxylase, and postsynaptic density protein 95 can also be measured. Recently, in a cellular model of developmental toxicity, the chemical effects of a number of model toxins on synaptogenesis in mixed rat cortical cultures were measured using the NPv3.5 BioApplication, where the researchers observed that this method was comparable with results obtained using manual methods.
15
Furthermore, the ArrayScan platform performed comparably to previously published manual methods in characterizing the effects of stem cell–conditioned media on neurite outgrowth.
16
In two other reports, automated fluorescence imaging platforms coupled to MetaMorph imaging analysis software were used to quantify Aβ1-42-mediated deficits in neurite outgrowth and demonstrated that α7 nicotinic receptor ligands or small peptides could block these effects.17,18 One of the advantages of the Cellomics ArrayScan platform and NP v3.5 BioApplication is its ease of use for screening. Images are captured and immediately analyzed with the NPv3.5 BioApplication, where the ArayScan platform allows for enhanced throughput and stability compared with other platforms. We have determined that the ArrayScan platform performs similarly to previously published methods in terms of characterizing parameters of neuronal morphology (
Our in vitro model of Aβ-induced changes of neuronal morphology in primary rat hippocampal cultures has correctly recapitulated a number of features of AD. First, presynaptic deficits are an early indicator of AD, where in our in vitro cell model, we have observed marked deficits in presynaptic nerve terminal levels after 24 h of treatment with 10 µM Aβ. We are also the first to show that Aβ-mediated deficits in parameters of neuronal morphology can be abrogated by integrin-blocking antibodies and can be quantified in a screenable assay format. Finally, we have shown that measuring parameters of neuronal morphology is a more sensitive indicator of nonlethal neuronal cell injury than using traditional cytotoxicity methods. By incorporating a neuronal morphology assay into a functional screening paradigm of Aβ-mediated cell injury, we may discover pharmacologically relevant inhibitors that would not be identified by other methodologies, thus leading to new classes of therapeutics to treat AD.
Footnotes
Acknowledgements
We would like to acknowledge Frederique Bard for thoughtful discussions and Kang Hu for his technical expertise. We would also like to thank Neil Durso, Sarah Tencza, Amy Peters, and Rebecca Henderson of Cellomics/Thermofisher.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no external financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.