
Abstract
The rise in organisms resistant to existing drugs has added urgency to the search for new antimicrobial agents. Aspartate β-semialdehyde dehydrogenase (ASADH) catalyzes a critical step in an essential microbial pathway that is absent in mammals. Our laboratory is using fragment library screening to identify efficient and selective ASADH inhibitors. These preliminary agents are then tested to identify compounds with desired antimicrobial properties for further refinement. Toward this end, we have established a microplate-based, dual-assay approach using a single reagent to evaluate antibiotic activity and mammalian cell toxicity during early stage development. The bacterial assay uses nonpathogenic bacteria to allow efficacy testing without a dedicated microbial laboratory. Toxicity assays are performed with a panel of mammalian cells derived from representative susceptible tissues. These assays can be adapted to target other microbial systems, such as fungi and biofilms, and additional mammalian cell lines can be added as needed. Application of this screening approach to antibiotic standards demonstrates the ability of these assays to identify bacterial selectivity and potential toxicity issues. Tests with selected agents from the ASADH inhibitor fragment library show some compounds with antibiotic activity, but as expected, most of these early agents display higher than desired mammalian cell toxicity.
Keywords
Introduction
Although cardiovascular diseases and cancer continue to receive well-deserved attention from the research community, the threats that we face from the microbial world are perhaps not as widely appreciated. It is estimated that about a third of the world’s population carries the tuberculosis-causing microorganism Mycobacterium tuberculosis, and bacterial infections are responsible for a quarter of the deaths worldwide each year. Although significant advances are being made against many diseases, the microbial threat to human health is continuing to grow due to the dramatic increase in the number of multidrug-resistant organisms. Resistance to antibiotics such as erythromycin and tetracycline was first discovered in Enterococcus faecalis during the early 1970s. 1 Plasmid encoding of resistance genes has allowed the facile transfer of multidrug resistance plasmids between different bacterial species. Encounters of methicillin-resistant strains of Staphylococcus aureus (MRSA) have risen from less than 20% in 1999 to greater than 50% in 2003, and community-associated MRSA strains are now the predominant isolates found in U.S. hospitals. 2 Multidrug-resistant isolates of M. tuberculosis have become the most prevalent strains, and a growing percentage of new tuberculosis patients are infected with extremely drug-resistant strains (i.e., infections that do not respond to treatment by more than three of the six available second-line drugs). 3 Multidrug-resistant Acinetobacter species have also emerged as a major health threat, 4 responsible for an increasing number of fatal bloodstream and urinary tract infections as well as the most frequent infections found in war-related injuries.
As a consequence of these increases in microbial drug resistance, the decline in effective antibiotics available to treat these growing threats has given greater urgency to the search for new antibiotics. Modifications of formerly effective compounds to create new, effective versions of these drugs have inevitably led to a rapid response from the microbial world and to more widespread drug resistance among infectious species. The ease by which even distantly related bacteria can transfer genetic elements such as plasmids and transposons through conjugation suggests a reserve capacity in the microbial world to rapidly acquire new antibiotic resistance. To combat this growing threat of microbial resistance, unique microbial targets must be identified for validation and new drug development. Aspartate β-semialdehyde dehydrogenase (ASADH) is an essential enzyme 5 that functions at a critical step in a vital microbial pathway. 6 ASADH enzymes from various microbial sources share the same binding and active site catalytic groups. 7 The highest level of ASADH similarity is found within subgroups of organisms, such as gram-negative bacteria, gram-positive bacteria, or fungi, with structural variation between these different classes of microorganisms being more substantial. An initial group of compounds evaluated through fragment library screening shows a range of inhibitory activities against ASADH enzyme from gram-negative (Vibrio cholerae) and gram-positive (Streptococcus pneumoniae) bacteria, as well as from a yeast species (Candida albicans). A few agents in this fragment library exhibit high ligand efficiencies, reasonable potency, and selectivity against specific members of this enzyme family. 7 For the current study, selected agents from this ASADH inhibitor library will be examined for their efficacy against bacterial cell growth and toxicity against representative mammalian cell lines.
The key to the successful adoption of a new antimicrobial drug is to demonstrate the agent has specific toxicity at low concentrations against the targeted microbes, while displaying little or no toxicity to humans or test animals unless exposed at substantially higher concentrations. In later stages of preclinical testing, efficacy is established through standardized protocols performed in dedicated microbiology laboratories with specialized equipment and facilities designed for the safe handling and testing of pathogenic organisms. The minimum agent concentration that effectively prevents microbe growth, known as the minimum inhibitory concentration (MIC), is generally the primary measure of in vitro efficacy. In vitro toxicity analyses during this stage typically focus on specific mechanisms of toxicity, such as human ether-à-go-go related gene (hERG) ion channel assays or hepatic enzyme bioactivation assays, as described in the Food and Drug Administration (FDA) guidance documents S7B Non-Clinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals (www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm129122.pdf) and M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals (www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm073246.pdf). In vivo efficacy and toxicity evaluations are then performed in appropriate animal models, yielding measures of the effective dose for 50% of the population tested (ED50), as well as the lethal dose (LD50) or dose causing nonlethal toxic side effects (TD50) in 50% of the test animal population. One common way of quantifying the relative efficacy versus toxicity for in vivo testing is to evaluate the therapeutic index, which is given by the ratio of LD50/ED50 or TD50/ED50. Other ratios have also been defined, such as the more conservative certain safety factor, which is given by the ratio of the dose that is lethal or toxic to 1% of the population (LD1 or TD1) to the dose that is effective in 99% of the population (ED99).
Although these advanced types of compound evaluations are required during later preclinical testing stages, they are not necessarily appropriate, convenient, or affordable for the early stages of drug discovery. This is particularly true for the early stage screening of novel classes of compounds, where a large number of analogues must be evaluated and their specific mechanisms of toxicity are unknown. In addition, if resources are limited, such as in the case of small startup companies and academic research, these tests would typically be performed in laboratories that are not devoted to antimicrobial research. In these circumstances, in vitro assays for efficacy against related nonpathogenic microbes and toxicity tests against a variety of representative mammalian cell types can provide a readily obtainable and cost-effective means of establishing structure-activity relationships (SAR), designing improved analogues, and selecting promising agents for more rigorous testing. Toward this end, the dual-assay methods described herein represent a system designed to evaluate promising compounds from our ASADH inhibitor fragment screening. For the purposes of method development and preliminary agent assessment, a series of antibiotic standards and preliminary library compounds with a range of ASADH inhibitory activity have been examined by this screening system.
Materials and Methods
Reagents and Stock Solutions
Commercial antibiotic standards (listed in Table 1 ) were purchased from Sigma-Aldrich (St. Louis, MO). Enzyme inhibitors (listed with abbreviations in Table 2 ) were obtained from a variety of commercial sources, except for NGS, which was synthesized by reacting glycine with succinic anhydride in acetic acid. Stock solutions of the test agents were prepared in DMSO or sterile water, depending on their solubility, and stored at −80 °C until use. Media components not otherwise specified were purchased from Sigma-Aldrich. alamarBlue cell viability reagent was acquired from Invitrogen (Carlsbad, CA).
Summary of Measured Therapeutic Ratio (TR) and Safety Ratio (SR) for Antibiotic Standards
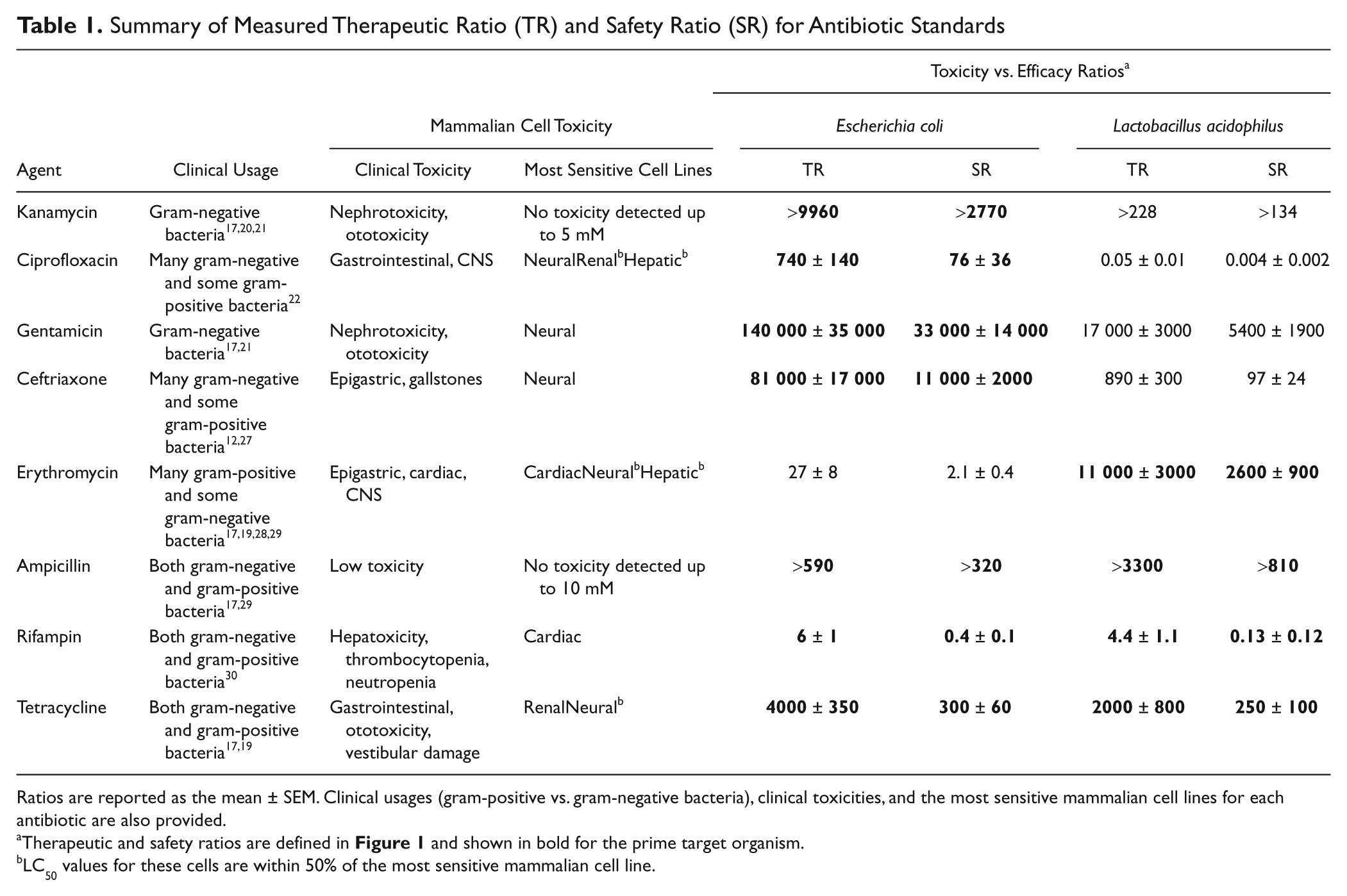
Ratios are reported as the mean ± SEM. Clinical usages (gram-positive vs. gram-negative bacteria), clinical toxicities, and the most sensitive mammalian cell lines for each antibiotic are also provided.
Therapeutic and safety ratios are defined in Figure 1 and shown in bold for the prime target organism.
LC50 values for these cells are within 50% of the most sensitive mammalian cell line.
Summary of Measured Therapeutic Ratio (TR) and Safety Ratio (SR) for Aspartate β-Semialdehyde Dehydrogenase (ASADH) Inhibitors
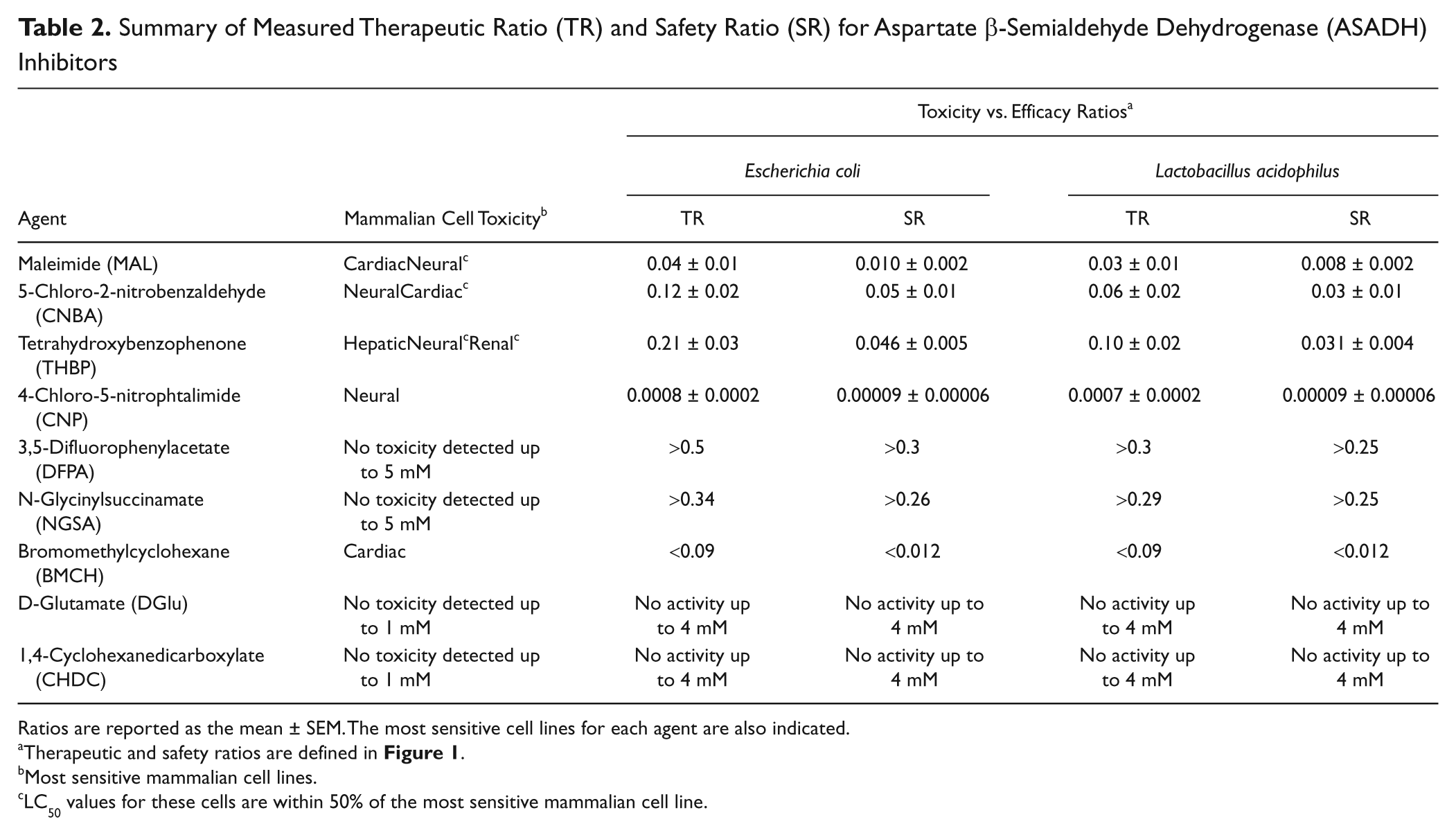
Ratios are reported as the mean ± SEM. The most sensitive cell lines for each agent are also indicated.
Therapeutic and safety ratios are defined in Figure 1 .
Most sensitive mammalian cell lines.
LC50 values for these cells are within 50% of the most sensitive mammalian cell line.
Cell Lines
Nonpathogenic, gram-negative Escherichia coli bacteria (JM109) from Promega (Madison, WI) were cultured and tested in Luria broth (Fisher Scientific, Pittsburgh, PA). Gram-positive Lactobacillus acidophilus bacteria (NCBI 1899 NCDO 2), purchased from American Type Culture Collection (ATCC, Manassas, VA), were cultured and tested in MRS broth (Fisher Scientific). Each of the four mammalian cell lines used in this study were generously provided by colleagues at the University of Toledo, including the HepG2 human hepatoma cell line, the 293 human embryonic kidney cell line, the SH-SY5Y human neuroblastoma cell line, and the H9c2 embryonic rat myoblast cell line. Mammalian cell lines were cultured and tested at 37 °C in a 5% CO2/100% humidity environment in RPMI-1640 media containing 2.0 mM L-glutamine supplemented with 20 mM HEPES, 0.2% sodium bicarbonate, 10% fetal bovine serum, and 50 µg/mL of gentamicin.
Antibiotic Activity Assay
Prior to each antibiotic activity assay, bacteria were grown overnight in 5 mL of broth at 37 °C with 230 rpm shaking. Cultures were then transferred to a flask containing 25 mL broth and grown at 37 °C with 230 rpm shaking for 2 to 3 h until the OD600 was between 0.2 and 0.5. Test agent stock solutions were diluted in broth to the desired maximum test concentration, with DMSO added as needed to yield a final level of 2% DMSO. Nine serial twofold (1:1) dilutions were prepared by addition of vehicle-only broth with a matching 2% DMSO level. Each of the 10 test agent broth concentrations were added in replicate (50 µL/well) to four wells of a Corning (3603; Corning, Inc., Lowell, MA) sterile black tissue culture–treated polystyrene 96-well plate, with the highest concentration broth added to a second set of four wells for bacteria-free agent background readings and vehicle-only broth added to four wells to measure uninhibited bacterial growth. This experimental setup requires a total of 48 wells per agent, allowing the testing of two agents per plate. Bacterial cultures were diluted according to preliminary colony counting measurements versus OD 8 so that ~5 × 10 5 bacteria in 50 µL broth was added to each well, except for the bacteria-free agent background wells, which received 50 µL/well of bacteria-free broth. Plates were incubated overnight, with each well containing 1% DMSO during this exposure period. After 24-h exposure, a 100-µL solution containing 80% broth/20% alamarBlue reagent was added to each well, and the fluorescence emission was read every 5 min for 1 h on a Molecular Devices (Sunnyvale, CA) SpectraMax M5 plate reader at 570 nm excitation and 585 nm emission with a 570-nm cutoff filter. Fluorescence readings near the maximum of the linear range were background corrected and normalized to vehicle-only controls to determine the fraction of control growth for each agent concentration. Three additional parameters were then estimated for each test agent on each plate. The MIC was determined per standard procedures with alamarBlue methods9,10 as the lowest agent concentration that produced bacterial growth less than or equal to 10% of vehicle-only control growth. Estimates were also made of the agent concentration yielding 50% and 90% inhibition of growth versus controls, labeled GI50 and GI90 values, respectively ( Fig. 1 ). These latter two values were determined by linear interpolation of fraction of control growth versus concentration for the test agent concentrations just above and below 50% of control growth (50% inhibition) for the GI50 and just above and below 10% of control growth (90% inhibition) for the GI90.
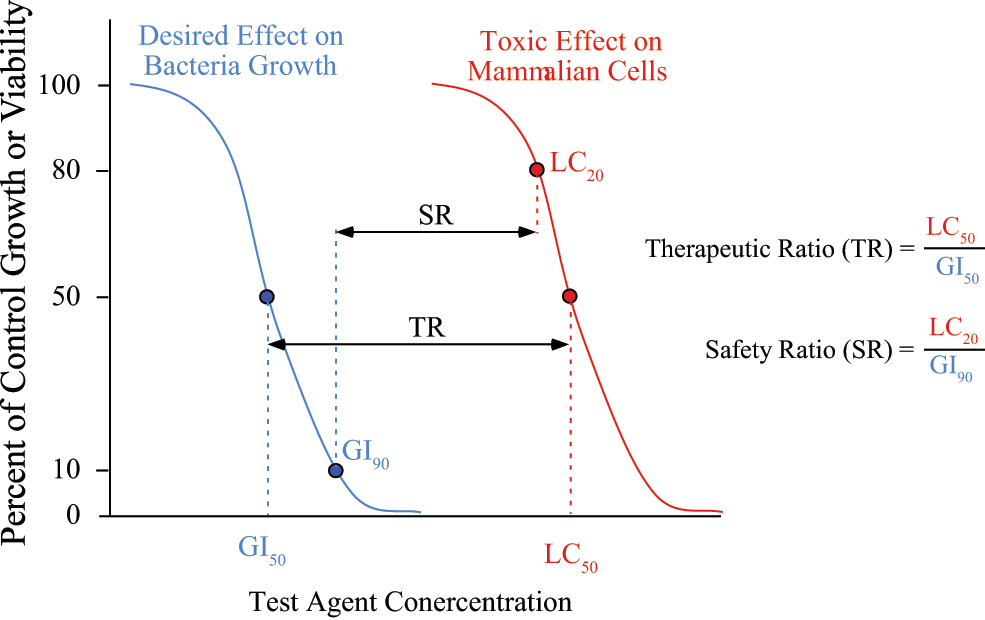
Hypothetical growth curves showing the determination of the GI50 and GI90 values for bacterial efficacy, the LC20 and LC50 values for mammalian cell toxicity, and the calculation of the therapeutic ratio (TR) and the safety ratio (SR).
Mammalian Cell Toxicity Assay
Mammalian cells were maintained in routine culture with weekly passaging, and cells were used up to the 20th passage. Immediately before loading, the cells were trypsinized, centrifuged, resuspended in cell media, counted on a hemacytometer, and diluted to an appropriate cell density to give the desired number of cells per 100 µL media. Two cell lines were then loaded into three rows (36 wells) on each Costar (3595; Corning, Inc.) sterile tissue culture–treated polystyrene 96-well plate via 100 µL/well using densities of 4000 cells per well (cpw) for HepG2 (hepatic), 293 (renal), and SH-SY5Y (neural) cells and 700 cpw for H9c2 (cardiac) cells. These cell densities were determined in preliminary tests to give linear alamarBlue readings over a 90-min period after 4 days of growth. The two remaining rows of each plate were loaded with 100 µL/well of cell-free media to provide background readings. Plates were cultured overnight, after which test agent solutions were prepared in media at the highest test concentration and at four serial 10-fold (1:9) dilutions, each containing 0.5% DMSO. The five agent concentrations were then loaded in separate columns via 100 µL/well, with solutions for two different agents included on each plate. Vehicle-only (0.5% DMSO) media were added to the two remaining columns on each plate to provide control viability readings, with all wells then containing a final vehicle concentration of 0.25% DMSO. The plates were incubated for a 72-h exposure period, after which 20 µL of alamarBlue reagent was added to each well, the plates were incubated another 60 min, and absorbance was read at 580 nm and 600 nm. Normalized values representing the amount of alamarBlue converted to its oxidized form were calculated for each well using background-corrected absorbances as adapted from William et al. 11 and described by the alamarBlue protocol information packet available from Invitrogen Technical Support. The fraction of control viability for each agent concentration with each cell line was then determined by the ratio of normalized values for the treated wells to the control wells. Note that the normalized values calculated as described were found to provide more consistent results for mammalian cell viability than the fluorescent detection method used with the bacterial growth inhibition assay. Estimates were then made of the agent concentration yielding 50% and 20% reduction in cell viability, labeled LC50 and LC20, respectively ( Fig. 1 ). These values were determined by linear interpolation of fraction of control growth versus concentration for the test agent concentrations just above and below 50% of control viability (50% lethality) for the LC50 and just above and below 80% of control viability (20% lethality) for the LC20.
Data Analysis
Fraction of control growth values for antibacterial assays and fraction of control viability for mammalian cell toxicity assays are reported as arithmetic mean ± standard error of the mean (SEM). The MIC, GI50, GI90, LC50, and LC20 values follow a lognormal distribution, so these values are reported as geometric mean ± SEM. Two ratio values have been defined to indicate relative antibiotic activity versus mammalian cell toxicity. A therapeutic ratio (TR), given by LC50/GI50, is defined as an in vitro analogy to the therapeutic index (LD50/ED50) ( Fig. 1 ). A more conservative safety ratio (SR), given by LC20/GI90, is defined to be analogous to the certain safety factor (LD1/ED99). The LC50 and LC20 values used to evaluate TR and SR were taken from the most sensitive mammalian cell line for each agent.
Results
Methods Development
Initial efficacy testing was performed using representative nonpathogenic gram-negative and gram-positive bacterial strains. Early indications of toxicity were determined with immortalized mammalian cell lines of hepatic, renal, neural, and cardiac tissue origin. The antibacterial and mammalian toxicity assays both use the same commercial alamarBlue reagent as an indicator of cell viability, further simplifying the experimental setup. To test sensitivity and selectivity, several different clinical antibiotics and several inhibitors of our target enzyme were examined using this method. A full summary of all measured activity and toxicity parameters is provided in
Representative concentration versus activity results for the antibiotic standard ceftriaxone and ASADH inhibitor tetrahydroxybenzophenone (THBP) are provided in Figure 2 . Ceftriaxone is known to be effective against many gram-negative bacteria but also shows efficacy against selective gram-positive bacteria. 12 When examined against our representative bacterial cell lines, this antibiotic demonstrated high potency against E. coli but was significantly less effective against L. acidophilus ( Fig. 2A ), consistent with its known bacterial profile. As expected for a clinical antibiotic, toxicity against mammalian cells was observed only at very high concentrations. In contrast, THBP, a very effective inhibitor (Ki = 40 µM) against the gram-negative bacterial form of ASADH, 7 showed much weaker potency against both bacterial cell lines while possessing comparable toxicity against all of the mammalian cells tested ( Fig. 2B ).
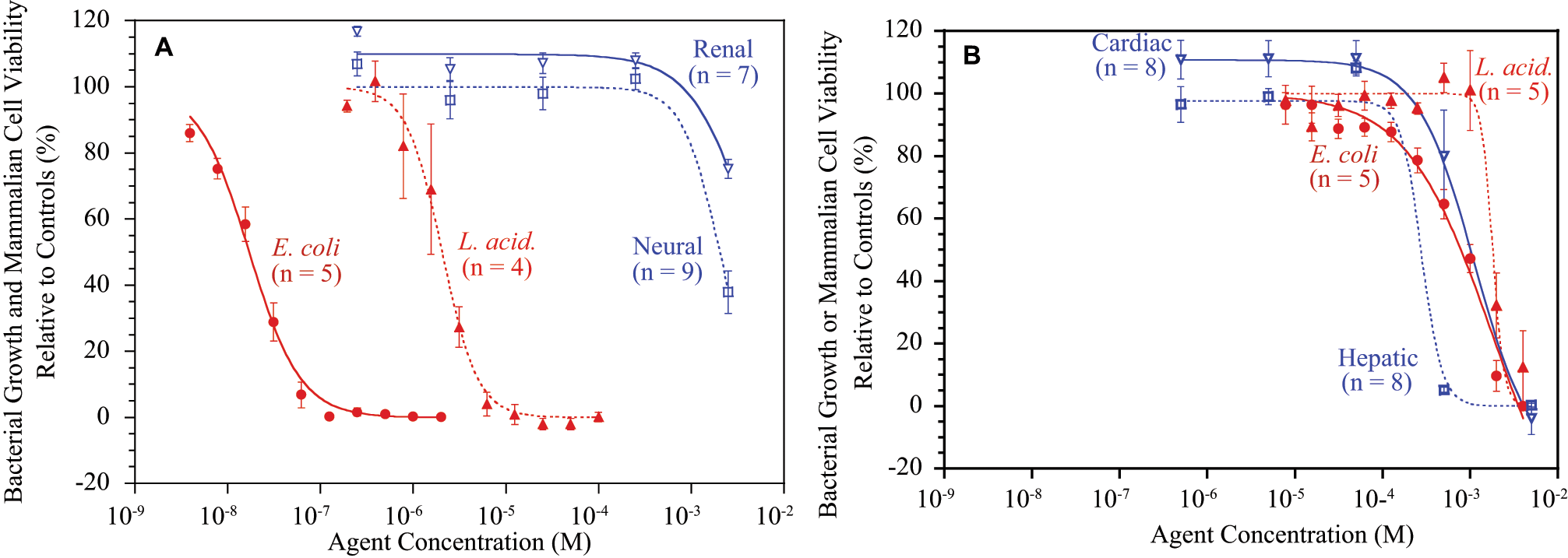
Representative examples of bacterial growth inhibition and mammalian cell cytotoxicity measurements for (
Method Validation with Clinical Antibiotics
A wide range of antibiotics are available with different degrees of efficacy against a variety of microbial infections. To examine the sensitivity and selectivity of our antibiotic activity assay, we tested clinical antibiotics against our representative gram-negative (E. coli) and gram-positive (L. acidophilus) bacteria. Antibiotics that are known to be effective against gram-negative bacterial infections13,14 were found to inhibit E. coli growth with GI50 values (defined in Fig. 1 ) ranging from 0.5 µM (kanamycin) to as low as 20 nM (ceftriaxone and gentamicin) ( Fig. 3A ). These same antibiotics also showed discrimination for gram-negative versus gram-positive bacteria in our assay ranging from 20-fold for gentamicin to as high as four orders of magnitude for ciprofloxacin ( Fig. 3A ). For an antibiotic that is known to be selective for gram-positive bacterial infections,14,15 erythromycin showed inhibition of L. acidophilus growth with a GI50 value of <0.1 µM and gave nearly a three-order-of-magnitude discrimination compared with the levels required to inhibit E. coli growth ( Fig. 3A ). Several known broad-spectrum antibiotics were also examined,14,16,17 with GI50 values ranging from 20 µM for ampicillin to as low as 10 nM for tetracycline for the inhibition of E. coli growth. As expected, these drugs showed minimal discrimination between gram-negative and gram-positive bacterial growth inhibition ( Fig. 3A , Table 1 ).
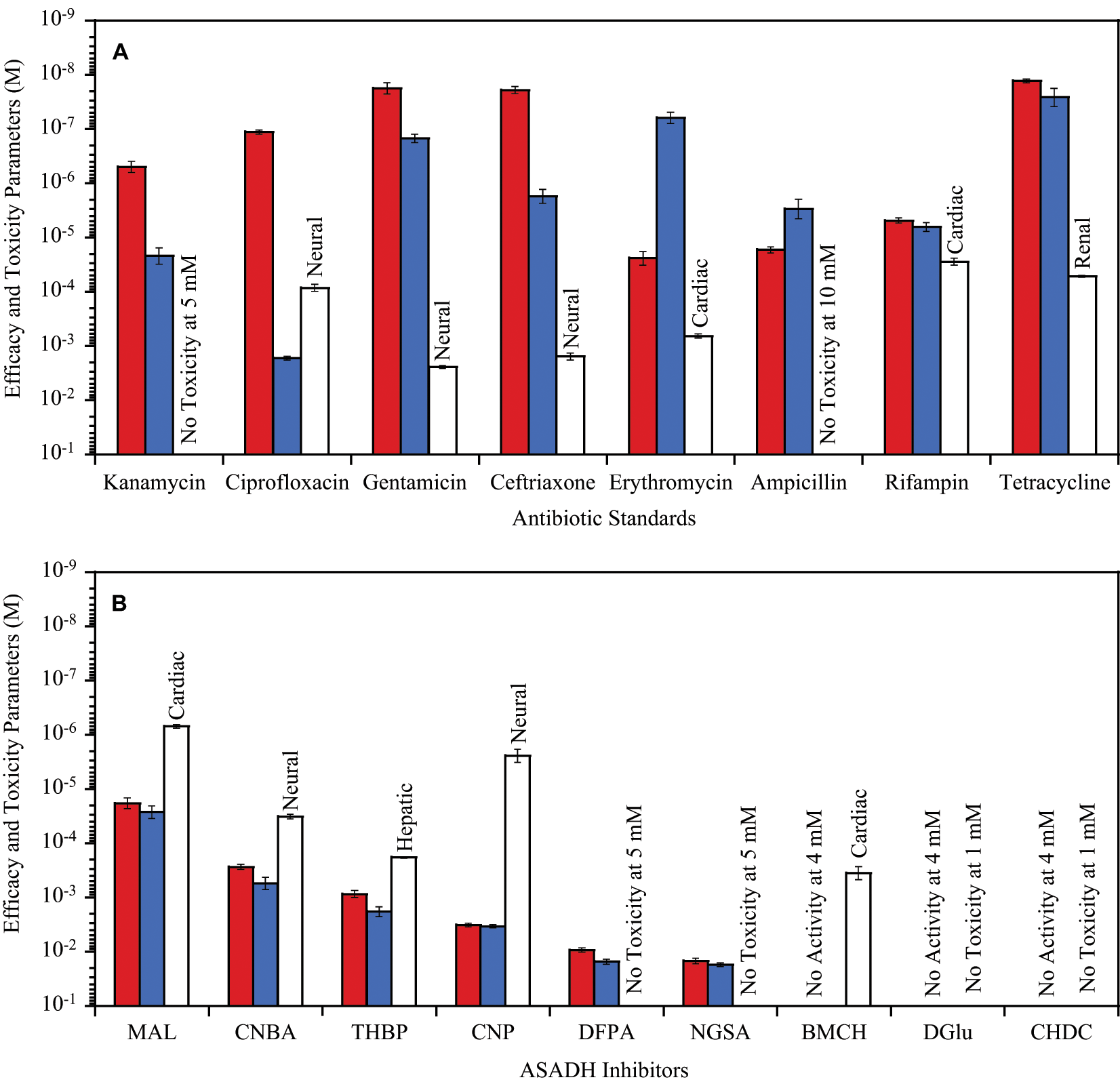
Antibacterial GI50 (Escherichia coli in dark gray, Lactobacillus acidophilus in light gray) and mammalian cell toxicity LC50 (most sensitive mammalian cell type for each agent as white bar) for (
Examination of this panel of antibiotics against representative mammalian cell lines provides information on cell toxicity under the same conditions as the bacterial efficacy studies. Antibiotics such as ampicillin and kanamycin that are known to exhibit low human toxicity13,18 showed negligible growth inhibition against these mammalian cells, even when examined at concentrations greater than 1 mM ( Fig. 3A ). Antibiotics with recognized human toxicity 19 reduced the viability of all the human cell lines with LC50 values (defined in Fig. 1 ) ranging from about 1 mM for erythromycin to as low as 50 µM for tetracycline ( Fig. 3A ). A few clinical antibiotics also began to show some selective toxicity against specific mammalian cell types when examined at higher concentrations. For example, rifampin caused a nearly 10-fold higher inhibition of cardiac cells compared with the other tissue types, and ceftriaxone gave greater inhibition of neural cells ( Fig. 3A , Table 1 ).
Assessment of Target Enzyme Inhibitors
Several early stage compounds identified from fragment library screening have been shown to be moderately effective inhibitors of our target enzyme, ASADH, and have also demonstrated some selectivity against different forms of this enzyme isolated from gram-negative and gram-positive bacteria and from a fungal species.
7
Representative members of these enzyme inhibitors, ranging from moderately potent and selective to very weak inhibitors, were tested to measure their toxicity against mammalian cells and also to determine if this assay method is sensitive enough to detect bacterial growth inhibition with compounds that have not been refined and optimized as antimicrobial agents. The structures of these compounds are shown in
MAL and CNBA showed the highest efficacy against these bacterial cells, with GI50 values ranging from 0.5 mM to 20 nM ( Fig. 3B ). CNBA is not an inhibitor of the gram-positive form of ASADH and is only a weak inhibitor of the gram-negative enzyme, with a Ki that is the same as its GI50 value. MAL does not inhibit either bacterial enzyme form but is a moderately effective inhibitor (Ki = 140 µM) against the fungal ASADH. 7 The next best bacterial growth inhibitors are each moderately effective bacterial enzyme inhibitors with Ki values of 150 to 180 µM for CNP and a Ki value of 40 µM for THBP against the gram-negative ASADH. 7 However, each of these compounds has relatively high mammalian cell toxicity, with an LC50 of 0.4 µM against neural cells for CNP and 0.2 mM against hepatic cells for THBP. Two very weak bacterial enzyme inhibitors, DFPA and NGSA, also demonstrated fairly weak antibacterial activity against both E. coli and L. acidophilus but did so with no detectable mammalian cell toxicity when tested at concentrations up to 5 mM ( Fig. 3B ). Two other ASADH inhibitors, CHDC, a very weak bacterial enzyme inhibitor, and DGlu, a moderately effective inhibitor (Ki = 87 µM) of gram-positive ASADH, each had only minimal toxicity against mammalian cells but were equally ineffective as bacterial growth inhibitors ( Fig. 3B , Table 2 ).
Discussion
Efficacy and Toxicology Screening Approach
The tremendous effort and costs associated with drug development have made it essential to assemble a variety of different data on compound viability, as early as possible in the developmental pipeline. With thousands of compounds being examined in the early stages of a project, it is neither feasible nor cost-effective to carry out the detailed efficacy and toxicology studies that are required for advanced lead compounds. However, some early indications of these properties can be useful to guide the direction of compound development, to identify particularly promising core structures, or to flag classes of compounds with especially unfavorable properties. The goal of the work reported in this article is to describe straightforward screening methods that can serve as a useful model for other laboratories in setting up early stage antimicrobial drug discovery screening approaches.
The screening of clinical antibiotics against two nonpathogenic bacterial species in our assay has readily identified drugs with gram-negative, gram-positive, or broader spectrum efficacy that closely correlates with their well-established antibacterial efficacies. Examination of these antibiotics against immortalized human cell lines also revealed drugs with different toxicity levels. Antibiotics with known low human toxicity such as kanamycin and ampicillin showed no toxicity against the four cell lines used in this study.20,21 Ciprofloxacin, erythromycin, and rifampin, which have known toxicities against CNS and cardiac tissues, indicated toxicity in both neural and cardiac cells in our system. 22 However, not all of the antibiotics matched exactly with their established toxicities. These differences are due, in part, to the difficulty of replicating whole-animal in vivo toxicity studies with cell-based assays and partly due to the limitations in the number of cell types used in our screening approach, which, for example, did not include gastrointestinal, epithelial, or cochlear cells.
The therapeutic and safety ratios provide useful measures to compare the selective efficacy of experimental compounds against their bacterial targets. Clearly, the higher these ratios, the more selective the action of any potential antibacterial agent. The TR values are all much greater than 1 for the primary clinical usage of all of the standard antibiotics (shown in bold in Table 1 ). Only rifampin has a relatively low TR for its prime target as a broad-spectrum antibiotic. This is somewhat surprising for an approved and frequently used clinical antibiotic, but rifampin is recognized as having potential therapeutic ratio issues in some situations. 23 Conversely, although useful for final drug testing, the safety ratio may be overly conservative, especially for early antimicrobial development. If this criterion were to be strictly applied, then it could have caused successful agents such as rifampin to be dropped from further consideration before they are fully evaluated. However, this ratio can be useful after several stages of drug design manipulations when there are many successful hit compounds that need to be ranked.
Early Stage Screening of Enzyme Inhibitors
As expected, the fragment library inhibitors of ASADH were much less effective as antibacterial agents, but this screening approach was able to identify some important differences in properties between these compounds. Several compounds had significantly higher cell toxicity against mammalian cells than against bacterial cells, whereas a few other compounds showed comparable bacterial efficacy and mammalian toxicity with TR values approaching 1 ( Table 2 ). As illustrated in Figure 3B , the ASADH inhibitors can broadly be divided into four different categories based on their relative efficacy and their mammalian cell toxicity profiles. Class I inhibitors are those that demonstrate moderate efficacy but with relatively high toxicity, such as maleimide and CNP. Class II inhibitors also show moderate efficacy but have only moderate toxicity, with CNBA and THBP belonging to this group. Class III inhibitors such as BMCH exhibit very low efficacy but are still fairly toxic to mammalian cells. Class IV inhibitors are those that have relatively low efficacy but also show negligible levels of toxicity, as exemplified by NGSA and DFPA. The last category represents the best class of early stage small fragment inhibitors, with properties that support their inclusion as starting structures for the next round of improved enzyme inhibitors based on these cell-based assay results.
Some significant differences are observed between these cell-based efficacy assays and the enzyme-based assays for different test compounds. For example, MAL does not inhibit either of the bacterial ASADH enzymes tested yet shows moderate efficacy against these representative bacterial cells, suggesting the presence of additional mechanisms for MAL cell growth inhibition. In contrast, THBP is one of the more potent inhibitors of the ASADHs from gram-positive and gram-negative bacteria yet shows no potency against either bacterial or mammalian cells in our assay. This ineffectiveness is most likely due to the inability of this compound to gain access into these cells.
Compounds such as NGSA, DFPA, and CNP were included in these early stage toxicity screening studies not only to validate the method, since they each showed low levels of ASADH inhibition, but also because of key structural features. These structural features will serve as positions for fragment linking and for further elaborations guided by molecular modeling and structural studies. For example, the aromatic halide functionality of CNP can provide a useful synthetic route, through various coupling reactions, to a series of more elaborate derivatives of this core structure. However, the high toxicity profile of this compound serves as a useful early “red flag” that will require careful evaluation as this class of compounds is developed. Likewise, DFPA is another compound that shows a preferred early efficacy and toxicity profile while also possessing two relatively acidic α-hydrogen atoms. Again, this structural feature can serve as a built-in “gateway” toward its regioselective linking to other structural moieties.
This early stage toxicity screening has served a key purpose. It has helped not only to identify two interesting new structural scaffolds with promising efficacy and toxicity profiles but also to streamline our efforts toward the development of more efficacious and less toxic species-selective inhibitors of ASADH. The toxicity and efficacy data obtained on the fragment library inhibitors will be used as benchmarks against which to measure the next round of improved enzyme inhibitors.
Screening Expansion
The efficacy and toxicity methods are described in this article for a 96-well plate format that can be performed either manually or by using an automated liquid dispensing system, but these assays can be readily adapted to higher throughput formats, other microbial targets, and additional mammalian cell types. As indicated by the alamarBlue product information insert (Invitrogen), this reagent can be reliably used in either 96- or 384-well plates. Antibacterial activity assays performed in a 96-well plate format as indicated in the Materials and Methods section allow testing of two agents at 10 different concentrations with one bacterial strain on each plate. Direct conversion of this plate layout to a 384-well format allows the corresponding analysis of eight agents per plate. In a corresponding manner, 96-well toxicity assays as described include two agents at five different concentrations with two cell lines per plate, whereas direct conversion to 384-well plates allows eight agents with two cell lines or four agents with four cell lines per plate. In actual practice, consolidation of background and control wells when converting to 384-well plates will afford further increases in throughput. Likewise, decreasing the number of agent concentrations for very preliminary screening will result in dramatically higher throughput. Performing the first stage of screening with only a single concentration of each agent increases the throughput to 24 agents per plate for 96 wells or 96 agents per plate for 384 wells, with further increases possible if using fewer than 4 wells per agent.
The reliability of such a single-agent approach can be estimated from the current results using a Z factor statistic. Suppose these early single-point screens are to be performed at a 1-mM agent concentration. Positive and negative controls for the toxicity assay can be defined as erythromycin, which has well-established clinical toxicity issues that match those observed in vitro here ( Table 1 ), and ampicillin, which has no expected or observed toxicity. Neural cells, which are most sensitive to erythromycin toxicity in these tests, gave a viability relative to vehicle-only wells of −6.1% with a standard deviation of ±5.9% for erythromycin at 1 mM, whereas ampicillin gave the corresponding values of 102.3% viability with a standard deviation of ±6.9% at 1 mM. This yields a Z factor of 0.65 for this single-point toxicity assay scenario, which is above the desired 0.5 Z factor minimum. For the antibacterial assay, erythromycin is again taken as the positive control since it was one of the few antibiotic standards tested near 1 mM (tested at 0.7 mM for gram-negative E. coli in this case), whereas the very weak ASADH inhibitor CHDC is taken as the negative control. Erythromycin gave 0.4% of E. coli vehicle-only growth with a standard deviation of ±0.4% at 0.7 mM, whereas CHDC gave 98.6% with a standard deviation of ±4.8% at 1 mM, providing a value of 0.84 for the single-point antibiotic assay Z factor. These Z factor calculations indicate that both the antibiotic and mammalian toxicity assays could be suitable for single-point screening.
The antibiotic assays can also be easily adapted for use with other bacterial strains or with completely different types of microbial targets such as fungi, 24 protozoa, 25 or biofilms. 26 Additional mammalian cell types can be added to detect other types of tissue toxicities, such as leukocytes for immunotoxicity, cochlear cells for ototoxicity, or vascular epithelial cells for localized intravenous injection toxicity. Thus, the described methods are applicable across a wide range of compounds that can have various physicochemical properties and functional groups. These methods can be readily adapted to the type of setup, throughput, microbial target, and mammalian toxicity screening needed for a variety of early stage antimicrobial drug development needs.
Footnotes
Acknowledgements
We thank the contributions from our colleagues at the University of Toledo, including Dr. Kenneth Bachmann (Pharmacology), Dr. Manohar Ratnam (Biochemistry and Cancer Biology), Dr. Linda Dokas (Pharmacology), and Dr. Jiang Tian (Medicine), for providing the mammalian cell lines that were used in our toxicity assays.
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by a grant from the National Institutes of Health (AI077720).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.