
Abstract
The biological complexity associated with the regulation of histone demethylases makes it desirable to configure a cellular mechanistic assay format that simultaneously encompasses as many of the relevant cellular processes as possible. In this report, the authors describe the configuration of a JMJD3 high-content cellular mechanistic imaging assay that uses single-cell multiparameter measurements to accurately assess cellular viability and the enzyme-dependent demethylation of the H3K27(Me)3 mark by exogenously expressed JMJD3. This approach couples robust statistical analyses with the spatial resolving power of cellular imaging. This enables segregation of expressing and nonexpressing cells into discrete subpopulations and consequently pharmacological quantification of compounds of interest in the expressing population at varying JMJD3 expression levels. Moreover, the authors demonstrate the utility of this hit identification strategy through the successful prosecution of a medium-throughput focused campaign of an 87 500-compound file, which has enabled the identification of JMJD3 cellular-active chemotypes. This study represents the first report of a demethylase high-content imaging assay with the ability to capture a repertoire of pharmacological tools, which are likely both to inform our mechanistic understanding of how JMJD3 is modulated and, more important, to contribute to the identification of novel therapeutic modalities for this demethylase enzyme.
Introduction
The emerging area of epigenetics offers potentially exciting new intervention opportunities across a range of therapeutic areas, including metabolic, oncology, and inflammatory pathological disorders. However, the infancy of the biology across the target class, as well as the lack of validated pharmacological tools, makes it challenging to delineate the mechanisms and signaling pathways that modulate these enzymes in development and pathophysiology. Consequently, current efforts within the epigenetic arena are focused on identification of probe molecules for epigenetic modifying enzymes to investigate the role of these enzymes in normal physiology and the etiology of diseases.
Lysine methylation was thought to be an irreversible process, but with the discovery of the flavin-dependent monoamine oxidase lysine-specific demethylase (LSD-1) in 2004 and the 2-oxoglutarate (2OG)–dependent oxygenase histone demethylase (HDM) subfamily shortly after, this process is now considered to be dynamically regulated. JMJD3 (KDM6B) is a member of the histone demethylase family, which has been shown to demethylate the repressive H3K27(Me)3 mark.1–3 A number of studies have demonstrated that JMJD3 is rapidly upregulated in macrophages in response to lipopolysaccharide (LPS), resulting in upregulation of proinflammatory genes as a consequence of the removal of the polycomb-mediated repressive mark H3K27(Me)3.4–6 In addition, a number of other biological functions, including lineage commitment and terminal differentiation of neural stem cells and of keratinocytes during wound healing, have been ascribed to JMJD3.7–12 Moreover, JMJD3 has been implicated in the pathogenesis of a number of disorders, including antineutrophil cytoplasmic autoantibody (ANCA) vasculitis, where aberrant expression of proteinase 3 (PR3) and myeloperoxidase (MPO) is associated with reduced levels of the chromatin modification H3K27(Me)3 at PR3 and MPO loci in ANCA patients compared with healthy controls. Consistent with this observation, JMJD3 is upregulated in ANCA patients compared to healthy controls. 13 JMJD3 has also been implicated in the pathogenesis of Hodgkin lymphoma, where it has been shown to be upregulated by Epstein-Barr virus. 14 Taken together, these studies suggest that JMJD3 inhibitors may potentially have therapeutic potential in both of these disease settings. Identification of JMJD3 probe molecules will therefore enable us to understand the JMJD3 demethylase-dependent events in normal physiology and in a disease setting.
To date, a number of rational design strategies and hit identification campaigns have been prosecuted for the demethylase family. These campaigns have largely employed in vitro biochemical assay formats that use truncated catalytic domains in the presence of cofactors and 15 to 20 amino acid peptide substrates.15–22 Although these strategies have been successful in identifying scaffolds for the 2-oxygenase family members, there is a potential risk that screening these enzymes in isolation outside of their natural environment may potentially limit the chemical space we can access. In addition, identification of chemotypes with the in vitro approaches above does not necessarily guarantee that the compounds will have cellular activity. Therefore, complementary cellular approaches that enable screening of the demethylase enzymes in their native state are likely to unlock currently untapped chemical space that will also deliver probes with the requisite cellular activity to enable delineation of the biological role of this new enzyme class. Thus far, a quantitative mass spectrometry–based method has been described for the quantification of intracellular histone demethylase inhibition. 23 Although this represents an exquisitely sensitive technique, it does have capacity limitations and is unlikely to be suitable for medium- to high-throughput screening (HTS) campaigns.
Herein, we describe a high-content imaging approach that has been used to prosecute a medium-throughput screen (87 500 compounds). More important, we identify a selection of JMJD3 chemotypes with inherent cellular permeability and good physicochemical properties, which will ultimately provide JMJD3 probe molecules that will be invaluable in delineating the biological roles of this demethylase enzyme.
Materials and Methods
Antibodies and reagents
Monoclonal anti-Flag antibody-mouse (F3165; Sigma-Aldrich, Gillingham, Dorset, UK) and modified histone H3-specific antibodies were purchased from Abcam (Cambridge, UK) and Millipore (Billerica, MA): H3K27(Me)3 (Millipore, #07-449), H3K27(Me)2 (Millipore, #07-452), H3K27(Me)1 (Millipore, #07-448), H3K9(Me)3 (Abcam, ab8898), H3K4(Me)2 (Millipore, #07-030), and H3K36(Me)3 (Abcam, ab9050). Species-specific fluorescent conjugated secondary antibodies were purchased from Invitrogen (Carlsbad, CA): Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 594 donkey anti-rabbit IgG (#A21207 and #A11001). Chemicals and reagents were purchased from the following vendors: Dulbecco’s modified Eagle’s medium (DMEM)/HAMSF12 media with 10% Australian fetal bovine serum (FBS), Alexa Fluor 594 donkey anti-rabbit IgG, and Hoechst 33342 trihydrochloride, trihydrate—Fluor Pure Grade were purchased from Invitrogen. Formaldehyde was sourced from VWR (Radnor, PA) and phosphate-buffered saline (PBS) without Ca and Mg from Gibco (Carlsbad, CA). Triton X-100, Tween, and bovine serum albumin (BSA) were purchased from Sigma-Aldrich. Ascorbic acid, α-ketoglutarate, DMSO, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Hepes), 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), and BSA were purchased from Sigma-Aldrich. Trifluoroacetic acid (TFA) was from Applied Biosystems (Warrington, UK). Acetonitrile and ammonium iron (II) sulfate were purchased from Fisher Scientific (Loughborough, UK). Peptide LATKAARK(trimethyl)SAPATGGVK (H3K27(Me)3) was synthesized and high-performance liquid chromatography (HPLC) purified to >95% purity by Cambridge Research Biochemicals (Cambridge, UK).
Plasmid constructs
JMJD3 DNA encoding the catalytic domain (residues 1141–1682; GenBank) and a similar truncated gene fragment lacking residues 1637–1675 (a splice variant deletion; GenBank BC009994) was amplified by PCR and subcloned into pCDNA3.1 neo (+) using BamHI and EcoRI. A Flag tag was included in the 5′ primer, downstream of the BamHI cloning site. A demethylase inactive variant was created by Quickchange (Agilent) mutagenesis, resulting in a Histidine1390 to Alanine (H1390A) conversion. The JMJD3 full-length gene was also subcloned into a pFast-Bacmam vector 24 containing a C-terminal fusion of eYFP using BamHI and NotI. The stop codon of JMJD3 was removed by QuickChange mutagenesis.
Full-length JMJD3-YFP BacMam generation
DH10Bac chemically competent Escherichia coli (Invitrogen, #10361-012) cells were transfected with pFB-JmjD3-eYFP plasmid DNA. Blue/white screening was employed to identify recombinant DNA colonies to be taken forward for Bacmid DNA purification (as described in the Invitrogen Bac-to-Bac Baculovirus protocol). A 6-well plate was seeded with SF9 cells at 9 × 10e5 cells/well in EX cell 420 media (cat. #14420; SAFC Biosciences, Hampshire, UK). Cells were transfected with 5 µL JmjD3 Bacmid DNA using Cellfectin II transfection reagent as advised by Invitrogen (cat. #10362100). Following a 5-day incubation at 27 °C, P0 viruses were harvested by centrifugation (3000 rpm, 5 min). Viral amplification was carried out by infecting 1 mL of P0 virus into Sf9 cells seeded at 1 × 10e6 cells/mL in a total volume of 150 mL. Cells were incubated at 27 °C and monitored for signs of viral amplification/cell lysis. Following cell lysis, P1 virus was harvested on day 4 when only ~40% of cells remained viable. Further viral amplifications were carried out based on a viral multiplicity of infection (MOI) of 0.1 being added to SF9 cells seeded at 1 × 10e6 cells/mL and harvesting virus when ~40% of cells remained viable.
Transient transfection
HEK293 MSRII cells were thawed from frozen stocks and transiently transfected using FuGENE HD transfection reagent (according to the manufacturer’s instructions; Roche, Basel, Switzerland). Cells were incubated at 37 °C for 48 h prior to fixation and imaging analysis.
Assessment of histone modification antibody specificity
Single-modification peptides were obtained from Abcam and Millipore. Histone modification peptides were spotted onto MSD 384-well high bind plates at three different concentrations (150, 1500, and 1500 pg). The plate was blocked in nonfat milk, incubated with primary antibodies (diluted 1:1000 in 1% nonfat milk), washed, incubated with species-specific detection antibody, washed, incubated with MSD read buffer, and analyzed using an MSD sector imager. Percent specificity was determined by normalizing the ECL counts for the respective peptides to background prior to expressing each peptide as a percentage to all modified peptides.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde, permeabilized using 0.2% Triton, and blocked with 1% BSA. Cells were incubated with anti-Flag and modified histone H3-specific antibodies, followed by species-specific fluorescent conjugated secondary antibodies. Hoechst dye was also included (1:10 000) to stain nuclei. All antibodies were made up in 1% BSA/PBS with Tween 20 (PBST) and incubated for a minimum of 1 h at room temperature. Antibodies were added at 30 µL/well and 20 µL/well for 96- and 384-well plates, respectively. Cells were washed three times in PBST after each antibody incubation, and PBST was added per well prior to analysis on a fluorescent microscope and imaging platforms.
Image acquisition on Cellomics ArrayScan
Quantitative imaging analysis of truncated JMJD3-expressing cells was carried out using ArrayScan II (Cellomics, Pittsburgh, PA), camera (Hamamatsu, Bridgewater, NJ), and software (version 3.6.1.3, BioApplications Software, Cellomics). Images were captured using three channels: Hoechst stain (nuclei), Alexa Fluor 488 (FLAG/JMJD expression), and Alexa Fluor 594 (modified H3-specific staining). Images were analyzed using Cellomics’ Cell Health II Bio application. Briefly, cell nuclei were defined by Hoechst stain. Cells were then selected based on FLAG-JMJD3 staining intensity within this nuclear region. The associated average nuclear staining intensity using histone modification-specific antibodies was then measured in the selected cell populations. Exposure times were set for individual experiments to avoid saturation and allow for quantification in the linear range. A 10× objective was used, and a minimum of 200 selected cells or maximum six field views were analyzed per well.
JMJD3 high-content imaging assay configuration
HEK MSR II cells at a density of 1.5 × 10e5 were transduced with 5% (v/v) JMJD3-YFP BacMam in DMEM/HAMSF12 media with 10% Australian FBS. The cells were then seeded at 7500 cells/50 µL/well using a Multidrop (Thermo Scientific, Waltham, MA) into 384-well poly-D-lysine-coated, black-walled, µClear plates (Greiner Bio-one, Stonehouse, UK) containing 0.5 µL of test compound or DMSO. Following overnight incubation of the plates at 37 °C, 5% CO2, cells were fixed with 20 µL/well of 8% formaldehyde (VWR). After a 10-min incubation at room temperature, the media were aspirated off using an ELX405 plate washer (BioTek, Winooski, VT), and cells were permeabilized by addition of 20 µL/well PBS, 0.2% Triton X-100. After a further 10-min incubation, the Triton solution was removed, and 20 µL/well of PBS, 0.1% Tween, 1% BSA was added to block any nonspecific binding sites. The plates were placed on a shaker for 30 min at room temperature. The blocking solution was then removed and replaced with 10 µL/well of the primary antibody solution (rabbit anti-H3K27(Me)3, 1:500 dilution in blocking solution). The plates were replaced on the shaker at room temperature for 1 h. The primary antibody solution was aspirated off, and the cells were washed three times with 20 µL/well PBS, 0.1% Tween. The remaining solution was then removed and replaced with 20 µL/well blocking solution containing the secondary antibody (Alexa Fluor 594 donkey anti-rabbit IgG, 1:1000 dilution) and 1.6 µM Hoechst. After a 1-h incubation at room temperature on the shaker, the cells were washed three times with 20 µL/well PBS, 0.1% Tween. The plates were then sealed.
IN Cell 2000 high-content imaging protocol and algorithms
The assay plates were imaged and analyzed using the Developer software on the IN Cell 2000 (GE Healthcare, Piscataway, NJ) using a 4× objective, which enabled the image from a whole 384-well to be acquired. The filter combinations (excitation/emission) of DAPI/DAPI, YFP/YFP, and Texas Red/Texas Red were used to acquire images for Hoechst 33342, YFP, and Alexa Fluor 594, respectively. The image analysis algorithm was designed to detect YFP-associated nuclear staining intensity to identify cells expressing JMJD3. The intensity of nuclear staining associated with the anti-H3K27(Me)3 antibody was then analyzed in the JMJD3-YFP-expressing cell population. Restoration of global H3K27(Me)3 levels in the presence of test compound was expressed relative to DMSO-treated cells (0%) and cells treated with 100 µM of the hydroxamic acid, suberoylanilide hydroxamic acid (SAHA; 100%). To eliminate the potential for compound toxicity to skew the data, only wells containing >300 JMJD3 YFP-positive cells were used for subsequent analysis.
Focused compound set composition
It was decided to conduct this screen as a medium-throughput diversity screen of a subset of the GlaxoSmithKline (GSK) screening collection biased toward cell-permeable compounds. The size of the set was fixed at 87 500, a number that could be profiled in a reasonable timeframe while giving good coverage of chemical diversity. A 78 000–compound set was selected covering structural chemical diversity, good predicted membrane permeability, and mainly possessing lead-like chemical properties (molecular weight [MW] <400, cLogP <4). The insilico model used to bias the selection of compounds to be included in this set was a model to predict permeability though MDCK2 membranes, which was good at removing compounds expected to have low cell permeability. In total, 8500 compounds that had previously shown biological activity in cellular assays at GSK were added. A small 1000-compound set was also included based on a pharmacophore model of the JMJD3 crystal structure.
Expression of JMJD3 protein
JMJD3 DNA encoding the catalytic domain (residues 1141–1682) and lacking residues 1637–1675 (a splice variant deletion; GenBank BC009994) was amplified by PCR to add an N-terminal Flag tag and cloned into the pFB-HTb vector (Invitrogen, cat. #10584027) using BamHI and XhoI restriction sites, which encodes for a protein with an N-terminal 6His tag followed by a TEV-protease cleavage site and Flag tag. pFB1-HTb-Flag-JMJD3 1141-1682 (del 1637–1675) was transposed into the baculovirus genome using the Bac-to-Bac technology (Invitrogen). Bacmid DNA was transfected into Spodoptera frugiperda (Sf9) cells using Cellfectin II (Invitrogen, cat. #10362100), and expression was performed at 80L scale using Excell 420 media (SAFC Biosciences, cat. #14420) in a 100-L working volume wave reactor (Wave Biotech, Somerset, NJ). The culture, at a cell concentration of 3.6 × e6 cells/mL, was infected with P2 recombinant baculovirus at a nominal MOI of 6 and incubated for 48 h. The cells were removed from the media by continuous centrifugation at 2500 g with an 80-L/h flow rate using a ViaFuge Pilot (Carr Centritech Separation Systems, Kent, UK), and the cell pellet was frozen for subsequent purification. The pellet from the baculovirus culture was resuspended in buffer A (20 mM Tris [pH 8.0], 300 mM NaCl, 10% glycerol, and 1 µL/mL Protease Inhibitor Cocktail Set III [539134; Calbiochem, San Diego, CA]). Cells were lysed by Dounce homogenization, on ice, and centrifuged at 100 000 g for 90 min at 4 °C. The 100 000 g supernatant was applied to a HisTRAP HP Column (GE Healthcare, 17-5248-02). The column was washed with 10 column volumes of buffer A, followed by 10 column volumes of buffer A containing 20 mM imidazole. Bound protein was eluted from the column using a linear gradient of 20 to 250 mM imidazole over 20 column volumes. The JMJD3 protein was eluted between 100 and 200 mM imidazole. Eluted JMJD3 protein from the HisTRAP column was concentrated threefold (Amicon [Houston, TX] Ultrafree-15 30 kDa, Millipore UFC903024) and loaded onto a HiLoad 26/20 Superdex 200 prep grade size exclusion column (GE Healthcare, 17-1071-01), equilibrated with buffer B (20 mM Tris [pH 8.0], 150 mM NaCl, and 5% glycerol). Fractions containing JMJD3 were pooled and concentrated (Ultrafree-15 30 kDa). Protein identity was confirmed by peptide mass fingerprinting and predicted molecular weight confirmed by mass spectrometry.
RapidFire mass spectrometry assay
For dose–response curves, threefold serial dilutions were prepared from 10-mM compound solutions in DMSO, and 100-nL volumes were transferred to V base assay plates (Greiner Bio-one), giving a final assay concentration range between 100 µM and 1.7 nM in the presence of 1% DMSO (10 µL assay volume). Uninhibited (high) and fully inhibited (low) control wells contained 100 nL DMSO, with enzyme activity in the low controls inhibited by addition of 30 µL of 0.5% TFA (v/v), prior to enzyme addition. Assays were performed by dispensing 5 µL of 2× enzyme solution containing 0.3 µM JMjD3, 0.5 mg/mL BSA, 200 µM CHAPS, and 50 mM HEPES, pH 7.0. Plates were incubated for 10 min at ambient temperature prior to the addition of 5 µL of 2× substrate solution containing 100 µM ascorbate, 100 µM Fe(NH2)2(SO4)2, 20 µM α-ketoglutarate, 60 µM H3K27(Me)3 peptide, and 50 mM HEPES, pH 7.0. Assays were incubated at room temperature for 6 min prior to quenching with 30 µL of a 0.5% (v/v) TFA solution.
Assay plates were transferred onto a high-throughput RapidFire200 integrated autosampler/solid-phase extraction (SPE) system (Agilent Technologies, Wakefield, MA) coupled to an API4000 triple quadrupole mass spectrometer (Applied Biosystems). Solvent A was water containing 0.01% (v/v) TFA and 0.09% (v/v) formic acid; solvent B was acetonitrile/water (8:2, v/v) containing 0.01% (v/v) TFA and 0.09% (v/v) formic acid. A C4 SPE cartridge was used. The mass spectrometer was operated in positive electrospray multiple-reaction monitoring (MRM) demethylated mode, monitoring the H3K27(Me)3 substrate and the demethylated product (H3K27(Me)2) using the following transitions (Q1/Q3): H3K27(Me)3 (418.3/414.8) and H3K27(Me)2 (519.7/515.0), with a dwell time of 50 ms for all transitions. The peaks detected by mass spectrometry were approximately 1.2 s wide at half-height and were integrated and processed using the RapidFire peak integration software.
Enzymatic conversion of H3K27(Me)3 substrate to the H3K27(Me)2 product was expressed as percent conversion as shown below:
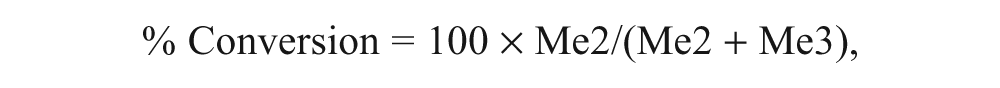
where Me2 and Me3 represent the integrated peak areas of the H3K27(Me)2 and H3K27(Me)3 peptides, respectively. Further data analysis was performed using the Activity Base Suite (ID Business Solutions Ltd, Surrey, UK).
Results
Assessment of histone modification antibody specificity
It is well known that many commercially available histone modification antibodies have substantial problems of specificity and utility. 25 Therefore, we set out to assess the specificity of a number of histone modification antibodies by employing a peptide array on the Meso-Scale Discovery platform. Representative specificity data from our preferred H3K27(Me)3-specific antibody, which was employed for the focused screen, are shown in Figure 1 .
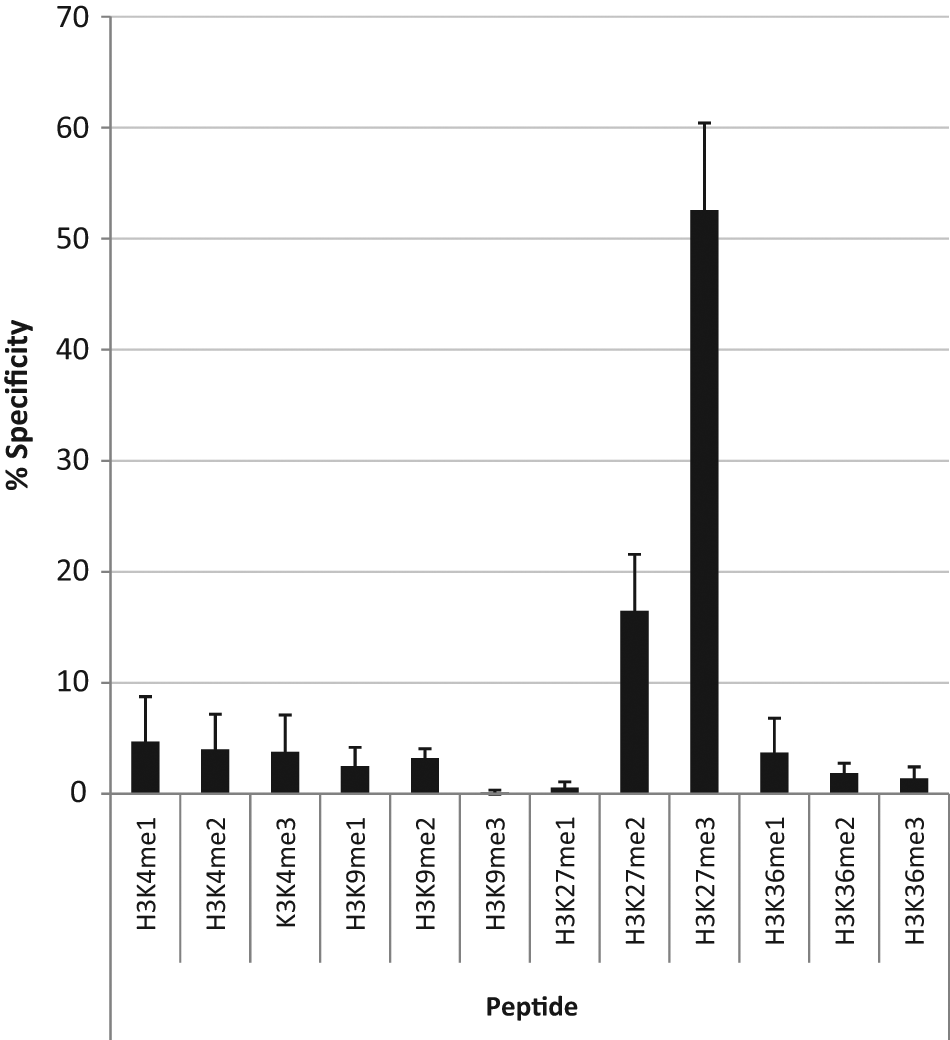
Relative specificity of the preferred H3K27(Me)3 antibody (Millipore, #07-449). In total, 15 ng of single modified peptides was spotted per well of an MSD high bind plate. Binding of the methylation-specific antibody was detected using an MSD ECL detection antibody. Data are provided as % specificity relative to total ECL counts generated by all modified peptides, normalized to background. Data shown are an average from three independent experiments.
JMJD3 demethylates histone H3 at lysine 27
Expression constructs encoding truncated, Flag-tagged JMJD3 were transfected into HEK293 MSRII cells, and the effect on histone lysine methylation was examined by immunocytochemistry. Cell nuclei were visualized by Hoechst staining, and the expression of the exogenous JMJD3 was visualized by immunostaining against the Flag epitope. Ectopic expression of wild-type Flag-tagged JMJD3 resulted in significant reduction in staining for H3K27(Me)3, in contrast to strong staining of H3K27(Me)3 in adjacent nonexpressing cells ( Fig. 2 ). Some reduction of staining was also observed for H3K27(Me)2, although to a lesser extent than H3K27(Me)3 ( Fig. 2 ). Little or no difference was observed in H3K27(Me)1 staining intensities between the wild-type Flag-tagged JMJD3 transfected and nontransfected cells ( Fig. 2 ). To determine whether the de-methylation of JMJD3 is specific for the residue of histone H3 lysine 27, similar immunocytochemistry experiments were carried out using specific antibodies against H3K9(Me)3 and H3K4(Me)2. Ectopic expression of wild-type Flag-tagged JMJD3 did not alter the immunostaining signal intensities of these antibodies ( Fig. 2 ), indicating that JMJD3 is not able to catalyze removal of methyl groups at lysine residues 4 or 9 of histone H3.
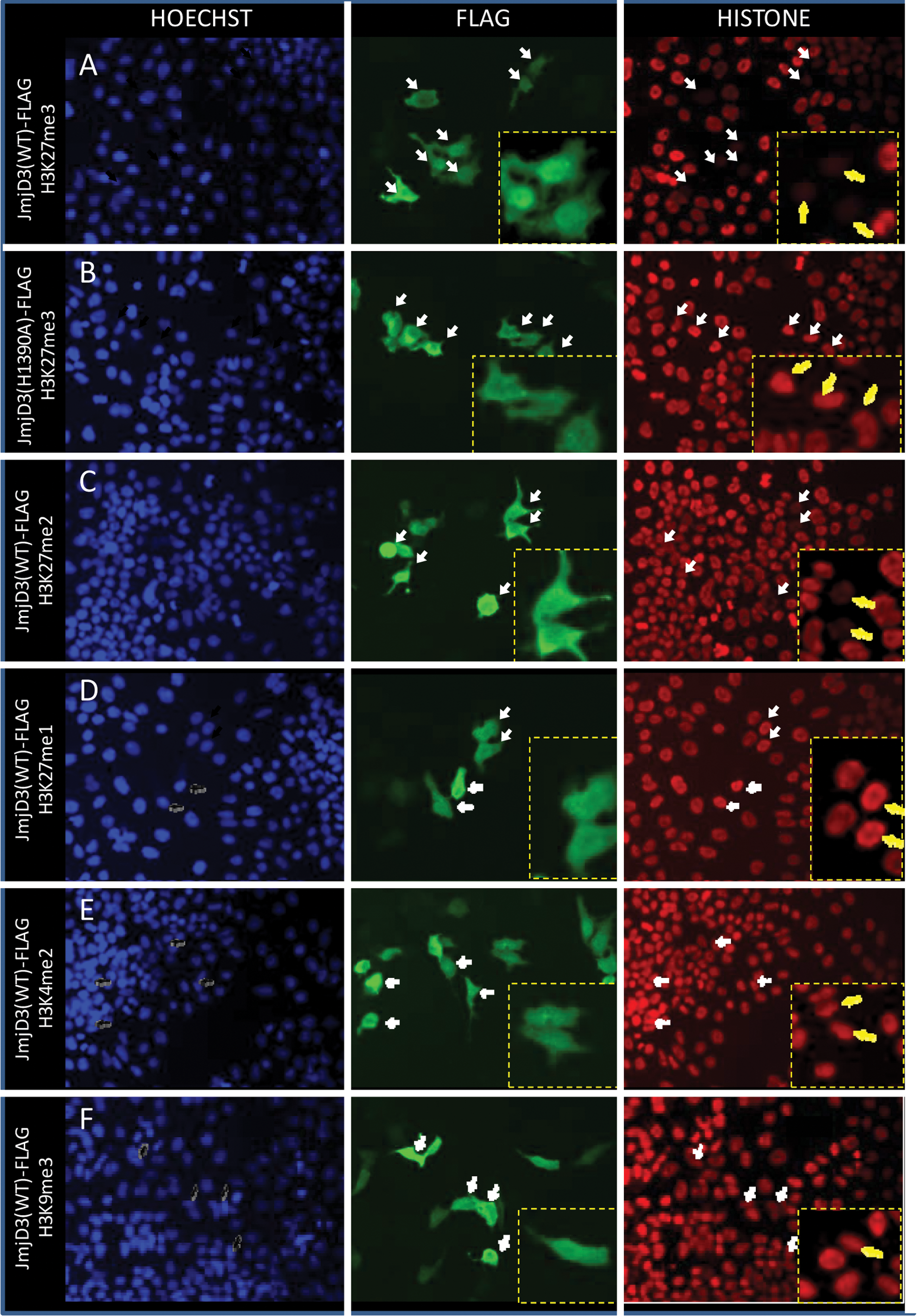
JMJD3 demethylates H3K27. HEK293 cells transiently transfected with JMJD3(WT/H1390A)-Flag were immunostained with specific antibodies against distinctively methylated lysine residues. Left panels are Hoechst staining, middle panels are Flag staining, and panels on the right are methylated lysine staining represented by (
To investigate whether demethylase activity of JMJD3 is directly responsible for demethylation of H3K27, we mutated histidine 1390 to alanine (H1390A), thus ablating its ability to chelate iron and thereby rendering it enzymatically dead. Following transfection of the mutated expression plasmid, expressing cells maintained similar immunostaining signals of H3K27(Me)3 as the adjacent nonexpressing cells, confirming that JMJD3 directly catalyzes demethylation at this residue ( Fig. 3 ).
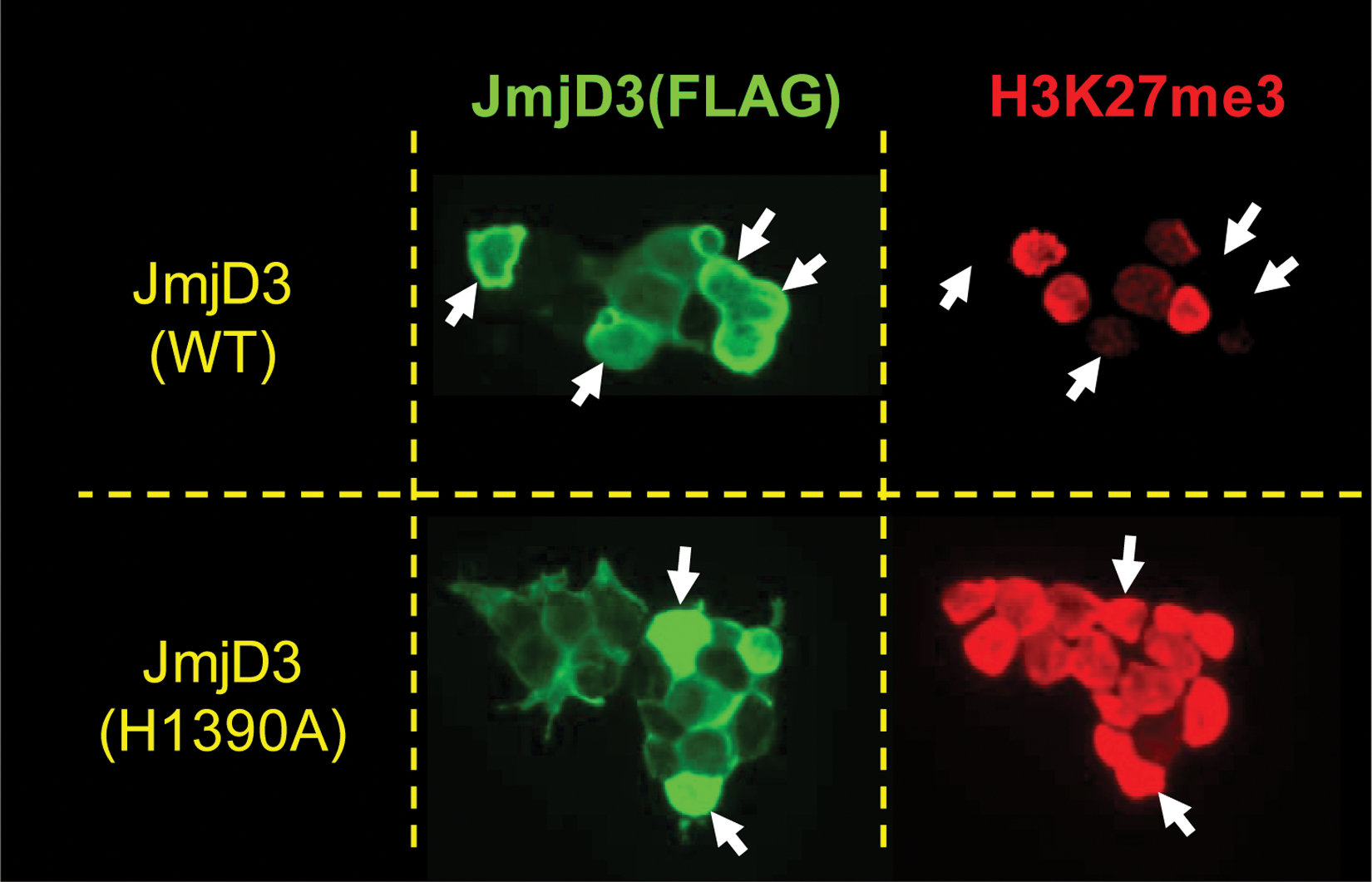
Global demethylation of H3K27(Me)3 by truncated JMJD3-expressing cells. Immunocytochemistry showing JMJD3 wild-type (WT) and JMJD3 demethylase dead (JMJD3 H1390A) expressing HEKMSRII cells stained using an anti-Flag primary and FITC-conjugated secondary antibodies. Also shown is H3K27(Me)3 staining using Millipore #07-449 and Alexa 594 conjugated secondary antibodies. Arrows indicate cells with high levels of JMJD3 expression.
Altogether, these data confirm previous published data, demonstrating that JMJD3 is a histone demethylase that is able to specifically catalyze the removal of trimethyl and dimethyl groups from H3K27.5,26
Due to the varying expression levels of JMJD3 in the transfected cells, we sought to investigate the impact of segmentation of the JMJD3-expressing cells into discrete bins of increasing expression levels on the signal-to-background (S/B) ratio, Z′, and potency of tool compounds. To segregate the expressing and nonexpressing cells, the total numbers of cells were identified on the imager by using the Hoechst channel prior to identifying the JMJD3-expressing cells in the FITC channel ( Fig. 4A ). Changes in the intensity of the H3K27(Me)3 mark were restricted only to the JMJD3-expressing cells. Quantification of the H3K27(Me)3 mark on the Cellomics ArrayScan demonstrated reduced nuclear intensity of the H3K27(Me)3 as one increases the JMJD3 expression level ( Fig. 4B ). Consequently, a concomitant increase in the S/B ratio and Z′ was also observed ( Fig. 4C , D ). In contrast, expression analysis of the cells expressing the JMJD3 demethylase dead enzyme (JMJD3 H1390A) did not display a similar trend to that observed with the wild-type enzyme ( Fig. 4B ). Pharmacological quantification of compounds of interest, at varying JMJD3 expression levels, demonstrated that the cellular system could be manipulated to either potentiate or desensitize the system to compound inhibition, depending on the expression threshold analysis employed. This provides the end user with the flexibility to modulate the cellular system to reflect the required compound pharmacological profile for a given situation. Ideally, the ectopic expression threshold selected would be as close as possible to the endogenous expression levels.
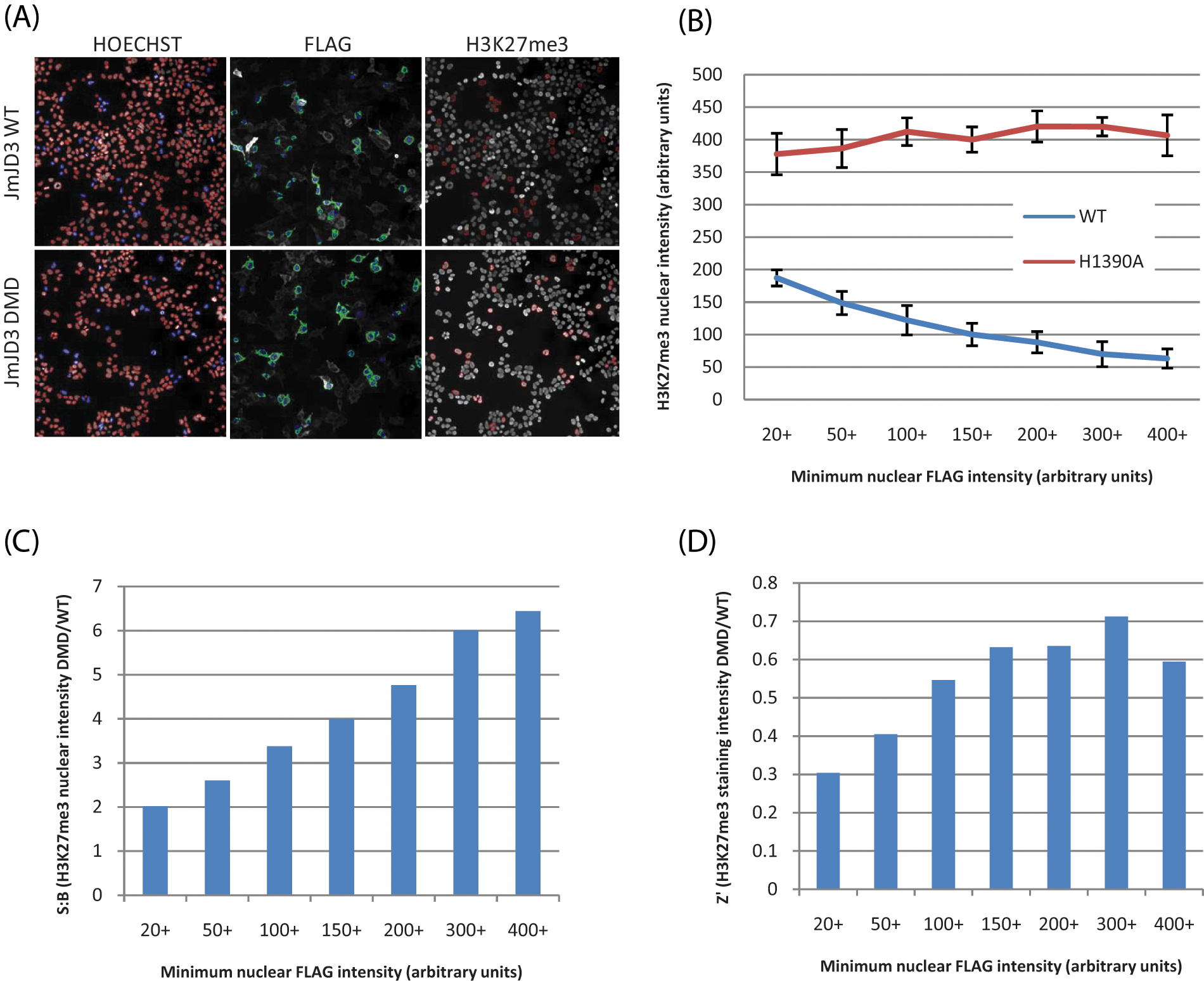
Global demethylation of H3K27(Me)3 by truncated JMJD3-expressing cells. (
Assay validation
For the focused campaign, we generated a full-length YFP-tagged JMJD3 BacMam virus to identify the potential full repertoire of direct and indirect modulators of this demethylase. Consistent with the data obtained for the truncated enzyme, we were able to demonstrate loss of the H3K27(Me)3 mark in cells expressing exogenous YFP-tagged JMJD3 (data not shown). In addition to the active site Fe(II) metal that is used by the Jumonji demethylases, it has been shown that the JmjD2 subfamily of enzymes also contains a zinc binding site, which is located close to the active site entrance. Subsequently, it has been demonstrated that a number of hydroxamic acids, including trichostatin A (TSA) and SAHA, inhibit JmjD2e. 20 Moreover, JmjD2a can also be inhibited by zinc ejecting compounds such as ebselen. 27 We therefore explored the utility of these compounds as potential inhibitors of JMJD3 and were able to demonstrate that SAHA inhibited JMJD3 with a potency of 10 µM, which is lower than the reported intracellular HDAC activity of 100 nM 28 but comparable to the 14 µM reported for JmjD2e. 20 By employing SAHA as our nominal inhibitor, we were able to show that we can restore levels of the H3K27(Me)3 mark in cells expressing YFP-tagged JMJD3 up to fourfold above that observed in the presence of 1% DMSO. This S/B ratio provided the most robust assay for compound profiling that could also identify weakly active compounds.
A set of 1408 structurally diverse compounds was used to generate a preliminary assessment of assay statistics prior to initiating the focused campaign. All compounds were screened at a single final assay concentration of 10 µM with the percent inhibition of each compound calculated (based on SAHA [100%] and DMSO-only [0%] wells within each plate). This compound set was screened on three independent test occasions to assess reproducibility and quality, with robust Z′ values 29 0.71 ± 0.1 and signal to basal ~5 (n = 12). A number of false-positive data points were observed due to reduced cell numbers, suggestive of compound toxicity ( Fig. 5 ). However, exclusion of these wells significantly improved the quality of the data. Standard compound (SAHA) pharmacology reported pIC50 5.21 ± 0.08, n = 12. Assay statistics generated a hit rate of 3.4% (±0.99; n = 3) using a robust cutoff of 6.4% (±0.94; n = 3) (robust cutoff = sample mean + 3× SD). The reproducibly low cutoff meant that we could potentially quantify JMJD3 inhibitors with intrinsically low efficacies in this cellular system, which is not always the case with cellular assay formats. The data showed good assay reproducibility and assay quality (Z′, S/B) throughout, and thus having fulfilled all performance criteria, they were considered fit for screening the 87 500–compound file at a final assay concentration of 10 µM.
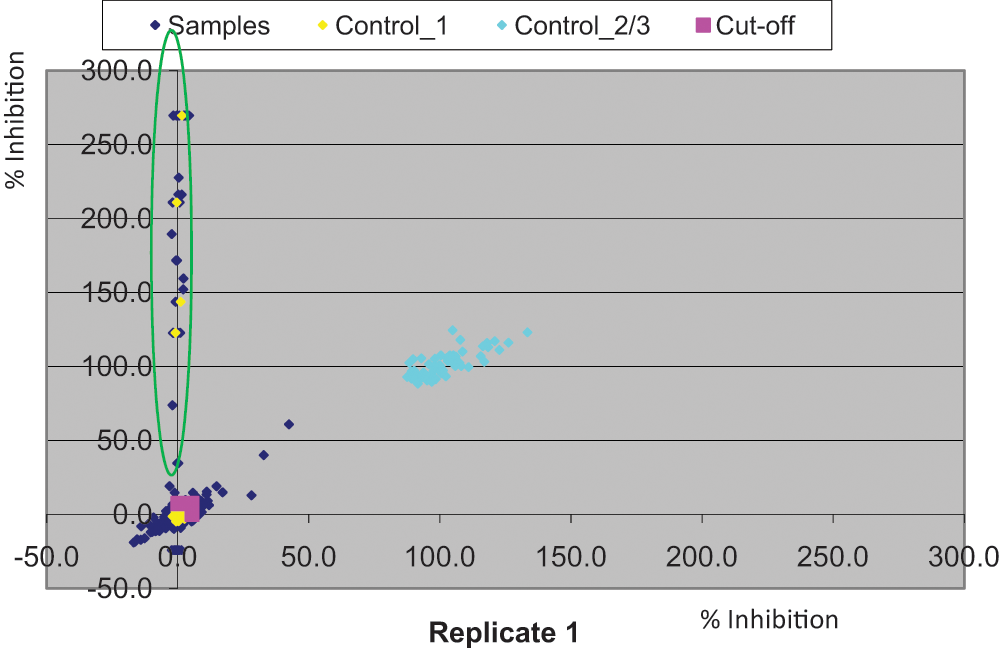
Correlation analysis of single-shot robustness data generated from a set of 1408 compounds tested on two independent occasions. Highlighted compounds on the y-axis were identified as false positives due to reduced number of counts in the respective compound wells potentially due to cellular toxicity. DMSO and 50 µM suberoylanilide hydroxamic acid (SAHA) are indicated in yellow and turquoise, respectively.
Analysis of focused set output
Assay reproducibility was consistent for the duration of the focused campaign, as evidenced by the Z′ (median ~0.70) and reproducible SAHA potency. A statistical hit rate of 4% was observed for the campaign, resulting in 3524 compounds being selected for full-curve analysis. An array of hits was identified with a range of inhibition responses, including a number of supra-maximal compounds exhibiting percent inhibition responses of JMJD3, which were twice as much as is normally observed with the nominal control compound, SAHA. In addition, the subset of compounds predicted to possess membrane permeability and mainly possessing lead-like chemical properties (MW <400, cLogP <4) provided the bulk of the hits (1828 compounds) for full-curve follow-up (see
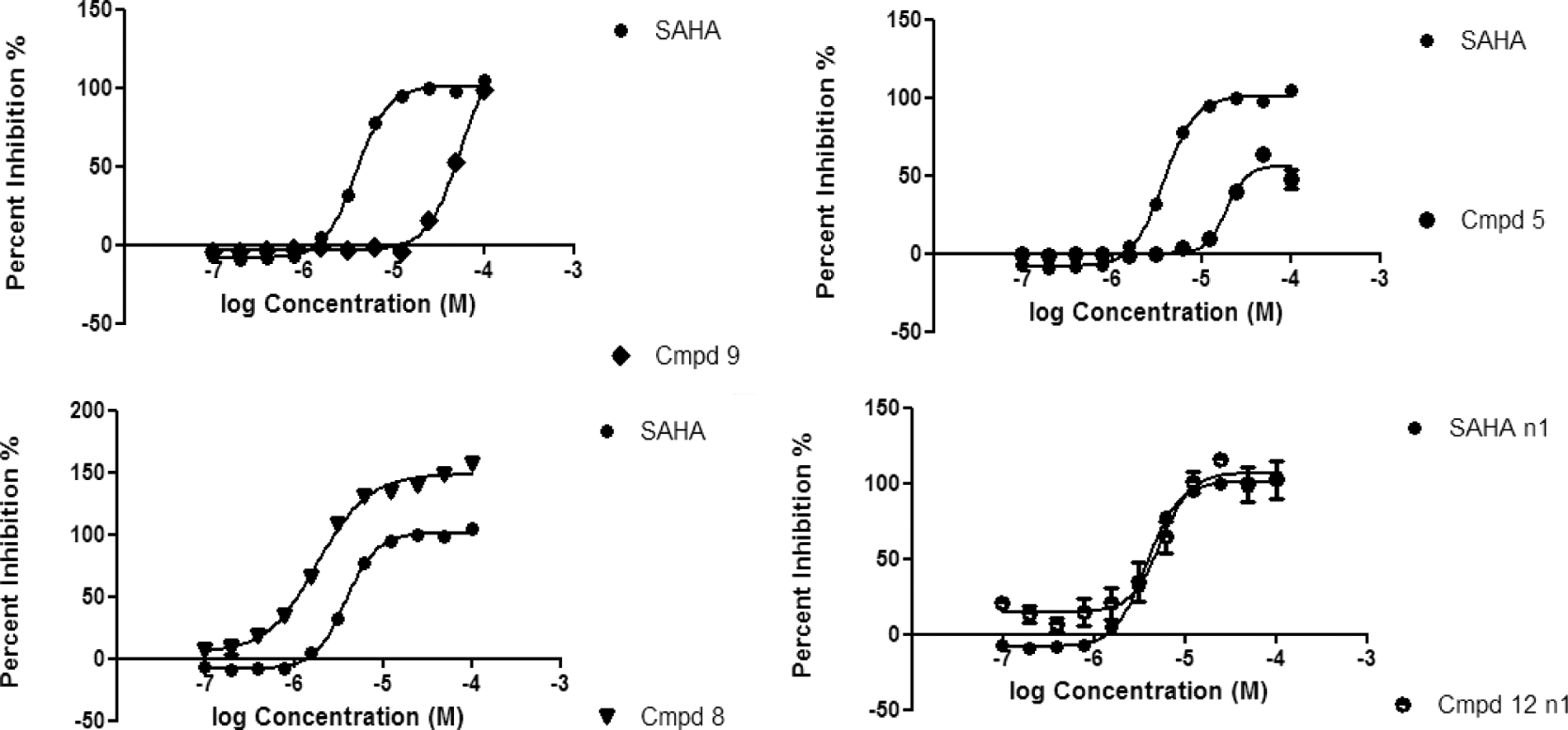
Pharmacological exemplars of JMJD3 compounds identified from the focused hit identification campaign. Responses are plotted as % inhibition relative to suberoylanilide hydroxamic acid (SAHA; average ± SD for n = 2). Example of SAHA concentration–response curve shown as reference against compound 9 (weak active), compound 5 (partial inhibitor), compound 8 (potent, supra-maximal inhibitor), and compound 12 (hit approximately equipotent to SAHA).
Identification of tractable JMJD3 chemotypes
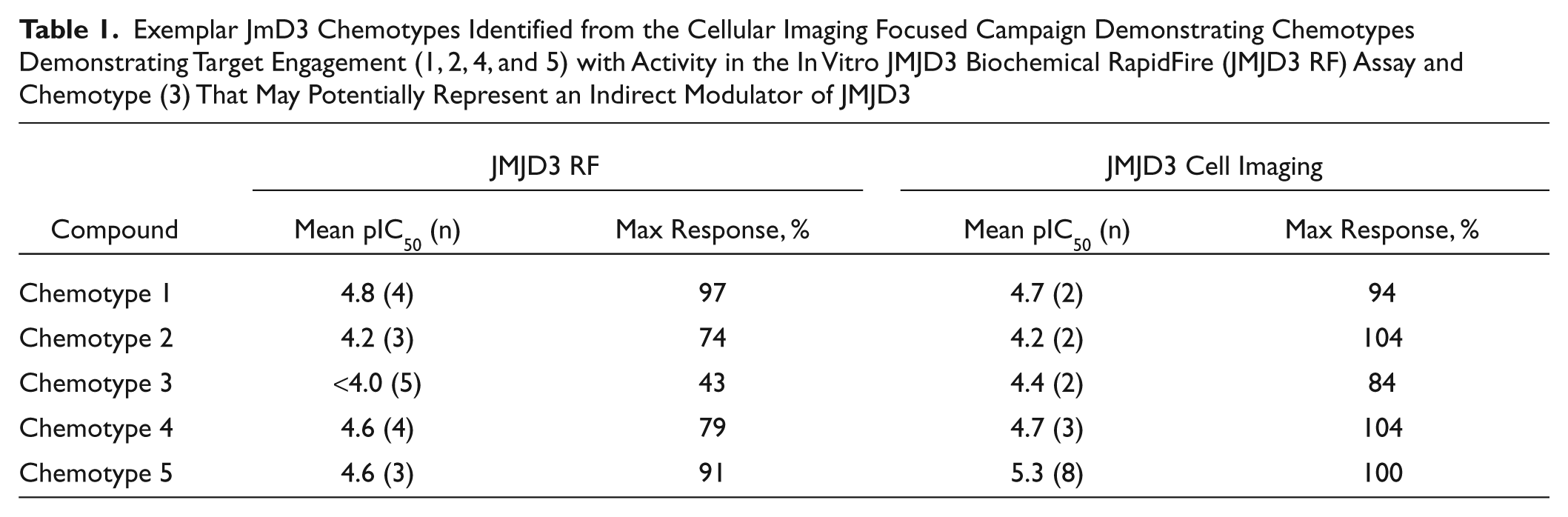
Ligand efficiency has emerged as an important parameter in drug discovery because smaller, more efficient ligands are predicted to have a greater chance of achieving the requisite bioavailability. A retrospective analysis of ligand efficiency (pIC50/HAC) versus lipophilic efficiencies (pIC50–cLogP) shows that there were some chemically tractable hits identified in the screen with a range of efficiencies. This suggests that this assay format possesses the ability to identify low molecular weight scaffolds in the required drug space as long as they possess good cellular permeability properties. Following hit identification, all selected putative JMJD3 hits were re-prepared and purified by HPLC to confirm that activity was driven by parent molecule and not trace impurities. Thus far, three diverse, low molecular weight chemotypes, 1 to 3, each giving robust inhibition in the cellular imaging assay (pIC50 4.2–4.7), have been identified from the focused screen and prioritized for optimization ( Table 1 ). Chemotypes 1 to 2 are likely to be active site inhibitors of the JMJD3 enzyme, as demonstrated by their ability to inhibit JMJD3 (pIC50 4.2 and 4.8) for chemotypes 1 and 2, respectively, in an in vitro RapidFire mass spectrometry assay format that uses truncated JMJD3 protein encompassing the catalytic region (amino acids 1141–1641). Chemotype 3 shows consistent activity in the imaging assay (pIC50 4.4) but is inactive in the RapidFire biochemical assay and may potentially represent an indirect modulator of JMJD3. Encouragingly, exemplars 4 to 5 from an alternative chemical series that was identified from a biochemical high-throughput screen also gave good inhibition in the imaging assay (pIC50 4.7–5.3).
Exemplar JmD3 Chemotypes Identified from the Cellular Imaging Focused Campaign Demonstrating Chemotypes Demonstrating Target Engagement (1, 2, 4, and 5) with Activity in the In Vitro JMJD3 Biochemical RapidFire (JMJD3 RF) Assay and Chemotype (3) That May Potentially Represent an Indirect Modulator of JMJD3
Discussion
The Jumonji histone demethylase family provides potentially exciting new opportunities for therapeutic intervention in a number of therapeutic areas, including inflammation, metabolism, and oncology. However, some key outstanding questions still need to be addressed to identify the targets that will provide the most optimal intervention points in these disease settings. First, understanding redundancy will be crucial for the design of treatment modalities because at least two demethylases have been identified for a given histone modification, implying that there may be overlapping functions. Second, despite the interesting studies on poised genes with dual modifications, 30 our understanding of the combinatorial impact of the possible histone modifications on gene expression is still at its infancy. Thus, it is still not clear whether targeting a specific methyl modification in isolation will result in an effective disease-modifying agent.
Moreover, it is widely presumed that the presence of a Jumonji histone demethylase at a specific gene locus is required to mediate an epigenetic change at the locus (i.e., an inheritable posttranslational modification that does not involve a change in the underlying DNA sequence). This is likely to be the case during development, where transition states are required to influence gene expression profiles and ultimately lineage commitment.31–36 However, there are likely to be situations where activity of the Jumonji histone demethylases at a specific locus does not result in an inheritable change. The identification of nonhistone demethylase substrates certainly suggests that some of these modifications may be transient. 37 There is also emerging evidence that the scaffolding role (i.e., noncatalytic function) of JMJD3 and its related H3K27(Me)3 demethylase, UTX, is required for chromatin remodeling of Infg in differentiated Th1 cells. 38 In addition, several point mutations in UTX, which have been associated with cancer, are located in both the catalytic and the noncatalytic region of this demethylase,39,40 implying both a demethylase dependent and independent role in carcinogenesis. Therefore, the complexity in biology will require the availability of exquisitely selective tool molecules to enable us to understand the overlapping and nonredundant functions and, more important, delineating the catalytic and chromatin remodeling functions of this enzyme class.
Traditional lead optimization campaigns of intracellular targets are often hindered by the lack of cellular permeability required to generate quality tool molecules. In this report, we outline a hit identification strategy, which has utility across the Jumonji demethylase enzyme family that armors the chemist with a selection of chemotypes with inherent cellular permeability and good physicochemical properties. This approach complements the other in vitro demethylase assay systems17,19,20 that are currently in use. This will assist in the identification of much-needed probe molecules for this enzyme class, which will enrich our understanding of their roles in development, differentiation, and pathogenesis of disease.
Footnotes
Acknowledgements
The authors sincerely thank Danuta Mossakowska, Martin Ruediger, Bill Cairns, David Wilson, Angela Worby, Steve Rees, Kevin Lee, and members of the JMJD3 program team for their support and input in enabling this focused campaign. In addition, they express their appreciation to the Sample Management Technology Group for assistance in compound sample management. They are grateful to the Molecular and Cellular Technology Group, particularly John Feild and Usman Shabon, for cloning and sequencing of the full-length JMJD3 cDNA construct and construction of the JMJD3-eYFP fusion expression vector.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.