
Abstract
In the past years, a lot of attention has been given to the identification and characterization of selective and potent inhibitors of chromatin-modifying enzymes to better understand their specific role in transcriptional regulation. As aberrant histone methylation is involved in different pathological processes, the search for methyltransferase and demethylase inhibitors has emerged as a crucial issue in current medicinal chemistry research. High-throughput in vitro assays are important tools for the identification of new methyltransferase or demethylase inhibitors. These usually use oligopeptide substrates derived from histone sequences, although in many cases, they are not good substrates for these enzymes. Here, the authors report about the setup and establishment of in vitro assays that use native core histones as substrates, enabling an assay environment that better resembles native conditions. They have applied these substrates for the known formaldehyde dehydrogenase assay for the histone demethylase LSD1 and have established two new antibody-based assays. For LSD1, a heterogeneous assay format was set up, and a homogeneous assay was used for the characterization of the arginine methyltransferase PRMT1. Validation of the system was achieved with reference inhibitors in each case.
Keywords
Introduction
Chromatin modifying enzymes
The chromatin of eukaryotic cells not only consists of DNA as the source of genetic information but also contains proteins, most prominently the histones that enable packaging of DNA into the nucleus. The nucleosomes, as the repetitive entities of chromatin, are composed of 146 bp of DNA and an octamer of four core histone proteins (H2A, H2B, H3, and H4). 1 The N-terminal tails of these core histones protrude from the nucleosome and are accessible to posttranslational modifications, such as acetylation, methylation, and phosphorylation.2,3 These covalent modifications lead to changes in chromatin structure and plasticity, which in turn lead to alterations in gene expression. Posttranslational histone modifications, together with the methylation of cytosine bases in DNA, are the biochemical mechanisms that underlie epigenetic gene regulation. The term epigenetics is defined as “heritable changes in gene expression that occur without changes in DNA sequence.” 4
Abnormal regulation of these processes leads to malfunction in gene expression and human disease. Aberrant histone methylation, for example, has in several studies been shown to be correlated with tumorigenesis.5–7 The steady state of histone lysine methylation is usually maintained by lysine methyltransferases and lysine demethylases. 8 In 2004, lysine-specific demethylase 1 (LSD1) was the first of these demethylases to be discovered.9,10 Since then, several members of the Jumonji domain–containing enzyme family (JmJC) have also been shown to possess lysine demethylase activity.11,12 Remarkably, the overexpression of LSD1 in prostate tissue correlates with the occurrence of prostate cancer and also with tumor recurrence during therapy. 13 The activity of this histone demethylase has also been demonstrated to be involved in the early cellular response to carcinogenic compounds 14 and seems to increase cell proliferation by promoting the G2/M transition. 15 Furthermore, for some histone demethylases from the JmJC family, elevated levels have been shown in different cancers. JARID1B and JMJD2C, for example, are overexpressed in testis and in breast cancer as well as in esophageal squamous carcinoma.16–18
Arginine methylation catalyzed by proteine arginine methyltransferases (PRMTs) is also involved in transcriptional regulation and the pathology of different diseases. One of the most prominent examples is the arginine methyltransferase PRMT1, which is an essential component of the mixed lineage leukemia (MLL) oncogenic transcriptional complex. Overexpressed PRMT1 enhances transcription and is thus critical for the development of leukemia in this case. 19 Another important role is the coactivation of nuclear receptors such as sexual hormone receptors.20–22 PRMT1 stimulates the estrogen receptor by interacting with members of the p160 family. 23 Thus, PRMTs are an interesting target for the therapy of hormone-dependent cancer. Methylation of arginines in other proteins is involved in cellular signaling. For example, methylation of a conserved arginine residue in the Igα subunit of the B cell receptor is catalyzed by PRMT1 and promotes B cell differentiation. 24 A detailed overview has recently been published. 6
These findings make histone demethylases and methyltransferases promising targets for novel treatment strategies in cancer as well as other diseases. Small molecules that selectively and potently inhibit these modifiers will serve as valuable tools to elucidate the functional importance of these enzymes in key cellular processes. Novel screening procedures for lead identification and optimization are important tools to that end.
In vitro assays for histone lysine demethylases and arginine methyltransferases
The activity of the histone-modifying enzymes that maintain the equilibrium of histone methylation can generally be determined by using antibodies that target these modifications. Usually, such antibody-based immunosorbent assays are performed with a secondary anti-IgG antibody that is labeled for detection purposes, which targets the primary antibody that recognizes the actual modification. By doing so, both the activity of enzymes introducing a posttranslational modification (methyltransferases) as well as those that remove the modification (demethylases) can be monitored. Nonspecific effects that result from binding of proteins to the immobilized substrate (and not from enzymatic activity) may falsely mimic removal of the modification. Hence, for such assay procedures, robust inhibitors are very helpful to differ between enzymatic turnover and such nonspecific binding effects.
Histone lysine demethylase activity can also be determined by monitoring the amount of small by-products that are released throughout the demethylation reaction. These by-products are formaldehyde in the case of LSD1 25 and the Jumonji domain–containing demethylases, 26 as well as hydrogen peroxide in the case of LSD1. 25 For various nonantibody detection methods, see Forneris et al., 25 Nash, 27 Lizcano et al., 28 Couture et al., 29 Wang et al., 30 Dorgan et al., 31 Collazo et al., 32 Nishioka and Reinberg, 33 and Wigle et al. 34
For the measurement of in vitro methyltransferase activity, heterogeneous assays using biotinylated histone peptides as substrates have been presented for arginine 35 as well as lysine 36 methyltransferases. The peptide is immobilized on streptavidin-coated microtiter plates, and the methylation is recognized by antibodies directed against the methyl mark transferred by the respective methyltransferase. Subsequent incubation with a europium-labeled secondary antibody allows the measurement of time-resolved fluorescence (TRF) to detect the methylation level. Alternatively, enzyme activity may be measured homogeneously using an AlphaScreen assay (the acronym Alpha means “amplified luminescent proximity homogeneous assay”) as published by Quinn et al. 37
In most published examples, synthetic oligopeptides are used as substrates instead of the native protein (i.e., core histones). In many cases, the use of such artificial peptide fragments does not influence substrate recognition by the corresponding enzyme. But for enzymes that show high substrate specificity, such as histone lysine demethylases, the use of proteins that differ from the native substrate may lead to a decrease or complete loss of substrate recognition and enzyme turnover. In addition, the substrates for immunosorbent assays need a tag for immobilization purposes that may have an impact on substrate recognition.
For these reasons, we decided to develop biochemical in vitro assays for reversible histone methylation that use core histones as substrates to mimic the native in vivo conditions more closely. As the first examples, assays for the histone demethylase LSD1 and the arginine methyltransferase PRMT1 are presented.
Materials and Methods
Proteins
As enzyme sources, GST-tagged LSD1 10 and recombinant GST-tagged hPRMT1 38 were used as previously described and purified according to standard procedures. Core histones (from calf thymus, type II-A) and formaldehyde dehydrogenase (from Pseudomonas putida) were purchased from Sigma-Aldrich (St. Louis, MO).
Inhibitors
The LSD1 reference inhibitor tranylcypromine was purchased from Sigma-Aldrich, and compound
Formaldehyde dehydrogenase–coupled assay for LSD1
The formaldehyde dehydrogenase–coupled assay was performed in 96-well plates according to the described procedure 28 but using native core histones instead of peptide substrates. Twenty µL LSD1 (0.1 µg/µL) was incubated with 5 µL formaldehyde dehydrogenase (FDH) (0.02 U/µL) and NAD+ (10 mM) for 5 min at 37 °C before the core histones (5 µg/µL) were added to start the demethylase reaction. The subsequent formation of NADH was quantified by the continuous measurement of fluorescence intensity (λEx = 330 nm; λEm = 460 nm) for 10 h. In case of inhibitor testing, DMSO solutions of the respective compound were added to the incubation batch in various concentrations. All tests were carried out in duplicates.
Heterogeneous antibody-based assay for LSD1
In the heterogeneous antibody-based assay, all tests were performed as duplicates in 96-well high-binding enzyme-linked immunosorbent assay (ELISA) plates (Greiner Bio-One, Monroe, NC). In a first step, the native core histones (0.1 µg/µL) were bound to the cavities of the plate in an incubation step in washing buffer (100 mM Tris, 150 mM NaCl, 0.1% Tween 20) with an overall volume of 100 µL for 60 min at 37 °C. After a washing step, 5 µL LSD1 (0.1 µg/µL) was added and incubated with or without inhibitor (DMSO solution in different concentrations) for 120 min at 37 °C in incubation buffer (100 mM Tris, 150 mM NaCl, 0.5% bovine serum albumin [BSA], 0.1% Tween 20), and after that time another washing step was performed. The demethylation reaction was followed by incubation with the primary antibody (α-H3K4me2, rabbit IgG; Millipore, Billerica, MA) in incubation buffer for 60 min at 37 °C. After that time and an additional washing step, the secondary antibody (anti–rabbit IgG, europium-labeled; PerkinElmer, Waltham, MA) was added in incubation buffer to each well. Subsequently, the microtiter plate was incubated for 60 min at 37 °C. In the final step, the Enhancement solution (PerkinElmer) was put in each well, and the plate was stirred gently at room temperature for 10 min. Finally, time-resolved fluorescence was measured (λEx = 340 nm; λEm = 615 nm; integration start: 400 µs, integration time: 400 µs).
Homogeneous antibody-based assay
The AlphaLISA assay was performed in 384-well plates (OptiPlate alpha; PerkinElmer), and all reactions were tested in duplicate. The incubation volume was 25 µL. First, antibody dilutions were prepared in buffer (50 mM Tris-HCl; 50 mM KCl; 5 mM MgCl2; anti-IgG 1:500, anti-H3: 1:25 000, anti–H4R3 (me)2: 1:1000). Antibodies, which have to be conjugated with the donor beads, were preincubated in the same buffer as above (1 µL of each dilution with 47 µL buffer) for 5 min at room temperature. Then, 1 µL of the commercially available donor bead suspension was added in the dark. The incubation of this donor bead mixture for 60 min at room temperature was also performed in the dark (by covering the test tube with aluminium foil). In parallel, the acceptor bead mixture was prepared and also incubated for 60 min at room temperature. Then, 1 µL of the respective antibody dilution and 1 µL of the acceptor bead suspension were added to 48 µL of the buffer described. For the methylation reaction, 5 µL PRMT1 diluted 1:5 in methyltransferase buffer (15 mM Tris-HCl; 10 mM NaCl; 0.25 mM Na2EDTA, 10% glycerol 85% [v/v], 1 mM 2-mercaptoethanol) was preincubated for 10 min at 30 °C with or without 0.75 µL of the respective inhibitor in a total volume of 10 µL. As substrates, native core histones were diluted with buffer to a concentration of 0.2 µg/µL. Then, 4 µL of this substrate dilution was added in the presence of the cofactor SAM (final concentration 40 µM) to all wells except the negative control, where 1 µL of methyltransferase buffer was added instead. All reaction mixtures were incubated for 60 min at 30 °C. The next incubation step consisted of the addition of 15 µL of the enzyme mixture and each 5 µL of the acceptor and donor bead mixtures and the transfer of the respective mixtures to the microtiter plate. This combination again took place in the dark just as well as the following incubation for 60 min at room temperature.
All incubation steps were performed with gentle shaking. Finally, the AlphaLISA signal was determined in a microplate reader (λEx = 680 nm; λEm = 615 nm; excitation time: 180 ms, total measurement time: 550 ms).
Results
Formaldehyde dehydrogenase–coupled assay for LSD1
At first it was important to test the ability of native core histones to serve as substrates for LSD1. Therefore, one of the assay protocols described previously 28 using FDH was modified to demonstrate the turnover of core histones by LSD1. LSD1 catalyzes the removal of methyl groups from mono- and dimethyl lysine 4 on histone 3 under formation of formaldehyde. This is detected in the FDH assay via an enzyme-coupled reaction (see Fig. 1 ). It is oxidized to formic acid by FDH that is added to the incubation mixture. The cofactor of this redox reaction is NAD+, which thereby is reduced to NADH. The formation of the reduced cofactor NADH can be quantified by the measurement of its absorbance (λ = 340 nm) or more sensitively using fluorescence intensity (λEx: 330 nm, λEm: 460 nm).
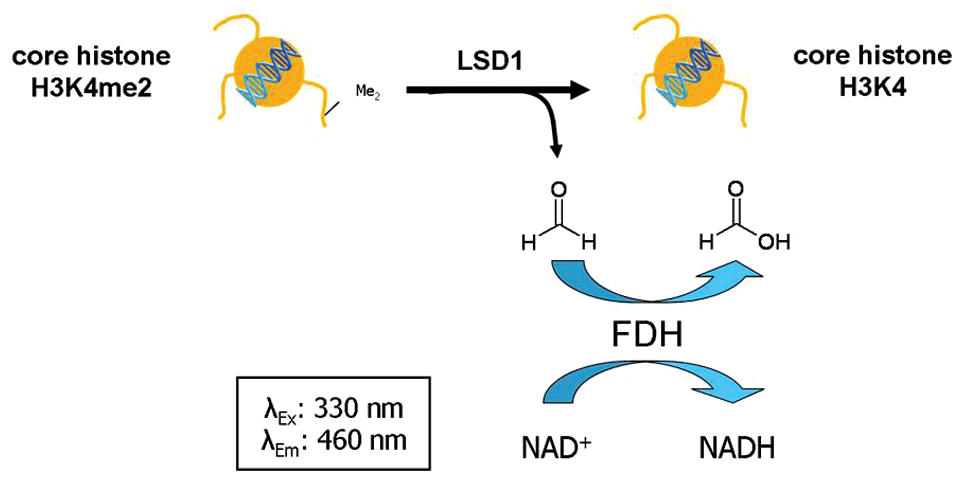
Detection principle of the formaldehyde dehydrogenase–coupled assay using native core histones as substrates. LSD1 demethylates H3K4-dimethylated core histones, leading to the generation of formaldehyde, which in turn is oxidized to formic acid by the activity of a formaldehyde dehydrogenase. Thereby, the cofactor NAD is reduced to NADH, which can finally be quantified by the measurement of fluorescence intensity (λEx: 330 nm, λEm: 460 nm).
To show the capability of LSD1 to accept core histones as substrates in this assay setup, the histones (2.5 µg/µL) were incubated with FDH, NAD+, and varying concentrations of LSD1. Figure 2 shows the dose-dependent conversion of the core histone substrates by LSD1.
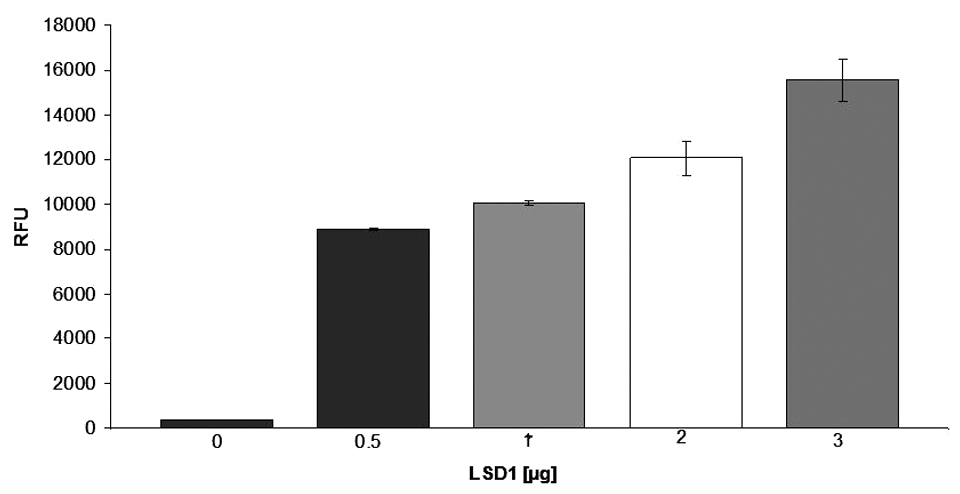
Conversion of core histones by varying amounts of LSD1, displayed as fluorescence intensity due to generation of NADH (n = 2).
To prove the linearity of this enzyme reaction, we performed the assay with varying concentrations of core histones (0.01–10 µg/µL). By doing so, we could show that in the initial enzyme reaction, there is a linear response over the whole range of histone concentrations. Therefore, we chose a core histone concentration of 2.5 µg/µL for the following experiments.
To further prove the feasibility of identifying inhibitors of LSD1 with this newly established protocol using native core histones as substrates, the inhibition by the known LSD1 inhibitor tranylcypromine
40
(TCP,
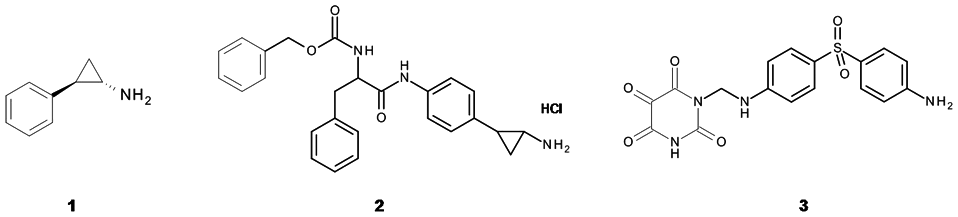
Chemical structures of the reference LSD1 inhibitors tranylcypromine (
Heterogeneous antibody-based assay for LSD1
We have shown successfully that the FDH assay protocol using histones is applicable for LSD1 testing. To be able to test compounds whose LSD1 inhibition cannot be determined in the FDH assay, we decided to use native substrates in another assay setup using a different detection reaction. This may be important for compounds that lead to quenching of the NADH fluorescence intensity or that show intrinsic fluorescence properties at the respective wavelength. So we established a heterogeneous, antibody-based assay that would allow us to avoid these disadvantages by quantifying the enzyme turnover by antibody-based recognition and ultimately via the measurement of time-resolved fluorescence. Compared to other commonly used fluorophores, the fluorescence intensity can be measured over a relatively long time interval (µs to ms), minimizing the interference with background fluorescence. Therefore, it is not surprising that this detection method has already been described for testing chromatin-modifying enzymes.35,41 The heterogeneous immunosorbent assays that have been reported so far use labeled (usually biotin) oligopeptide substrates that are immobilized to (streptavidin-) coated microtiter plates, commonly taking advantage of the strong biotin–streptavidin interactions. However, this has the disadvantage of using reagents that have to be prepared by solid-phase synthesis or purchased for a relatively high price.
To coat the walls of the reaction cavity with core histones, we used microtiter plates that show a high binding capacity for proteins due to hydrogen bond donors and other polar compounds incorporated into the plate raw material. This eliminates the need of a tag for immobilization purposes. After the immobilization of the protein substrate to the cavities of the high-binding plate, the reaction batch was incubated with LSD1, leading to H3K4 demethylation depending on the activity of the enzyme. In case of an inhibitor screen, the enzyme was preincubated with the corresponding compound. In the next incubation step, the added α-H3K4me2 antibody recognizes the remaining amount of dimethylated histone substrate before this primary antibody in turn is targeted by the secondary, labeled antibody in the subsequent incubation step. In each case, several washing steps are performed after every incubation step to remove the unbound antibody fraction. In a final detection step, Eu 3+ is released from this complex by the addition of an acidic enhancement solution. 42 The amount of europium that has been released correlates with the LSD1 activity and can be quantified by determination of time-resolved fluorescence (λ = 615 nm) after excitation at a wavelength of 340 nm. Figure 4a schematically recapitulates the described procedure.
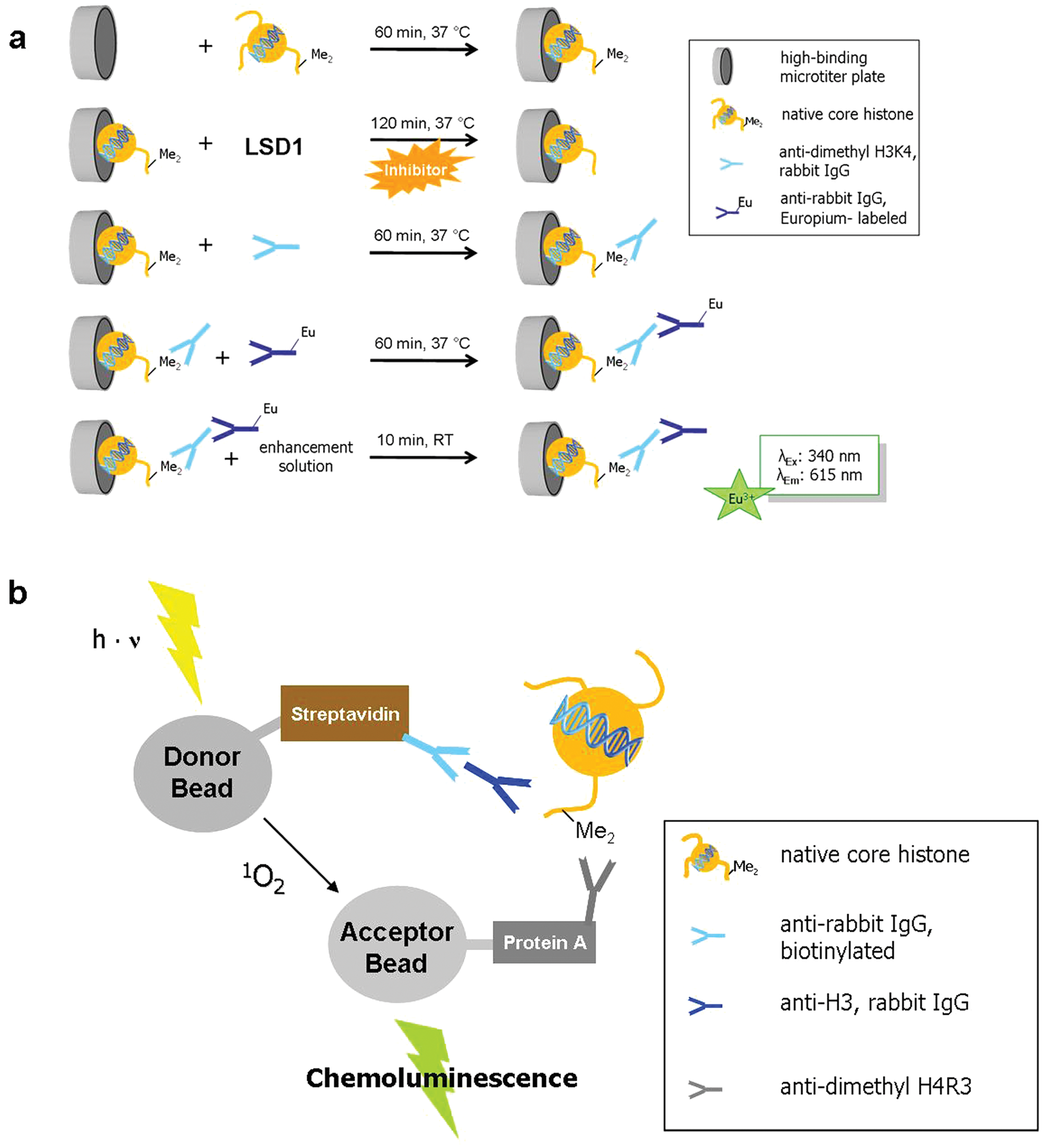
Schematic illustration of (
To verify the conversion of core histones in this assay setup, the histones were incubated with and without LSD1, and after adding the primary and secondary antibodies, time-resolved fluorescence was measured. Figure 5 shows the resulting fluorescence intensities.
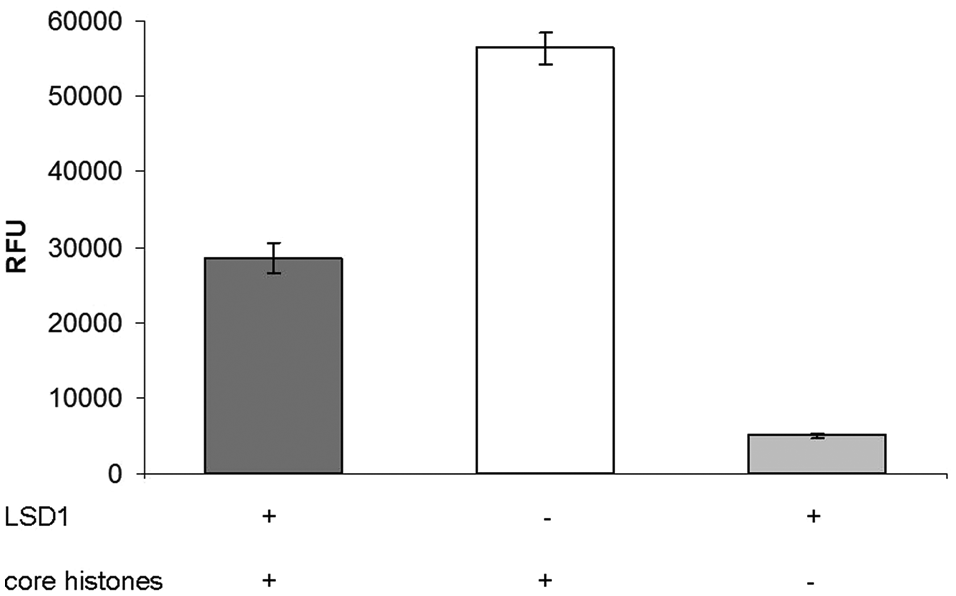
Conversion of core histones by LSD1 in the heterogeneous antibody-based assay, displayed as intensity of time-resolved fluorescence due to released Eu3+ (n = 2).
To show the capability of this assay to identify LSD1 inhibitors, again the reference inhibitor TCP (100 µM) and also the potent inhibitor
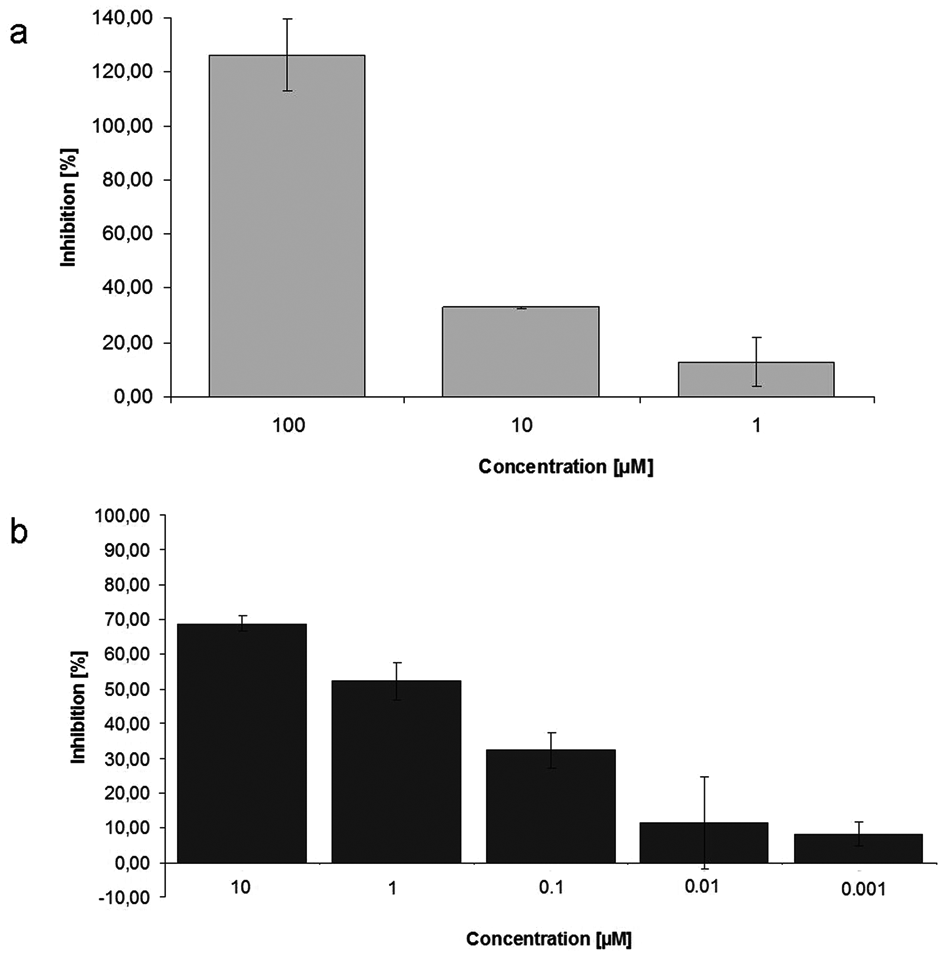
Dose-dependent inhibition of LSD1 activity by the reference inhibitors tranylcypromine (
With this protocol, a versatile in vitro assay for the determination of demethylase activity could be established. The use of native histones should allow the (only slightly modified) application for the testing of other chromatin-modifying enzymes as well, given that a respective primary antibody is available. In each instance, the same secondary antibody can then be used. However, one has to appreciate that validated antibodies do not exist for all histone marks and that combinatorial histone modifications may have an influence on antibody recognition. 43
Homogeneous antibody-based assay for in vitro testing of PRMT1
As a variation of the procedure for LSD1 and to combine the advantages of homogeneous assays in terms of a high throughput and the versatility of immunosorbent detection, we decided to establish an assay protocol that is based on the AlphaLISA technology and also uses native histones as substrates. We used the arginine methyltransferase PRMT1 for this example.
The acronym Alpha (amplified luminescent proximity homogeneous assay) describes an assay based on a luminescence signal that is produced upon the proximity of two latex-based beads, so-called donor and acceptor beads. The donor beads are conjugated with streptavidin and also contain a photosensitizer converting oxygen to singlet oxygen after excitation at a wavelength of 680 nm. Within its half-life of approximately 4 µs, the singlet oxygen is able to cover a distance of about 200 nm in solution. When the acceptor bead resides within this distance, the singlet oxygen is transferred to a thioxene derivative on the acceptor bead. 44 The resulting adduct rapidly decomposes under chemiluminescence. The energy is transferred to the lanthanide europium, which is also placed on the acceptor bead and can be read out at λ = 615 nm. A signal is produced only if donor and acceptor beads are in close proximity. Coated with streptavidin, the donor beads enable the binding of the substrate, usually biotinylated oligopeptides.
As we decided to use native histones as substrates, we employed streptavidin-coated donor beads, which target a biotinylated anti–rabbit IgG antibody. This biotin-tagged antibody in turn binds a second antibody that is directed against the C-terminus of histone H3. The acceptor beads are coated with protein A and are connected to an anti–dimethyl H4R3 antibody due to the strong interactions of protein A and the Fc part of IgG antibodies. Binding of these antibodies to the dimethylated H4R3 substrate leads to proximity of the donor and the acceptor beads, enabling quantification of methyltransferase turnover by measurement of the resulting chemiluminescence.
Figure 4b
illustrates this procedure schematically. We could show that the reference inhibitor allantodapsone (
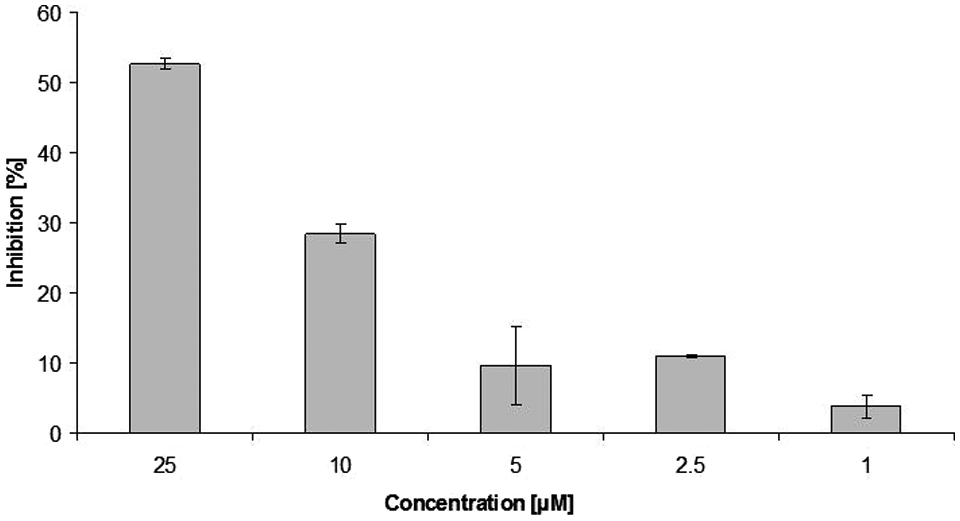
Dose dependent inhibition of PRMT1 activity by the reference inhibitor allantodapsone (n = 2).
Discussion
Biochemical in vitro assays are one of the most important tools in the identification and characterization of novel modifiers of epigenetic targets, not only for the development of future drugs but also to elucidate the specific biological role of the respective enzyme. Several assay protocols for the screening of inhibitors of lysine demethylases and arginine methyltransferases have been described so far. One of the problems with these in vitro assays is the use of artificial oligopeptide substrates that may lead to aberrant substrate recognition, especially in the case of enzymes that show high substrate specificity, as reported for some histone lysine demethylases. We were able to set up different in vitro assays that circumvent this problem by the use of core histones as native substrates but that are still amenable to high-throughput screening. We have shown that LSD1 accepts core histones as substrates under cell-free in vitro conditions and that the enzyme turnover can be inhibited by the known reference inhibitors tranylcypromine and compound
The assays discussed herein possess diverse readout procedures (absorbance, fluorescence, time-resolved fluorescence), making it possible to test a variety of compounds even if they show absorbance, fluorescence, or quenching properties under assay conditions themselves. The use of native histones mimics in vivo conditions, and the application of recombinant and then specifically modified histones would allow the setup of tailor-made assays. One such possibility is the targeted installation of methyl lysine analogs into recombinant histones. 45 The use of such tailor-made histones would enable the characterization of distinct modifications, especially in the context of interdependence with other modifications. We are well aware of the fact that not core histones but nucleosomes are the physiological substrates and that there also have been reports about aberrant substrate recognition when using histones instead of nucleosomes. 46 Still, they are of higher relevance than oligopeptides and, for some enzymes histone peptides, are very poor substrates. Thus, the use of histones in the in vitro screening assays should broaden the possibilities for epigenetic drug discovery.
Footnotes
Acknowledgements
The authors thank the Deutsche Krebshilfe (PRMT, Nr. 107898) and the Deutsche Forschungsgemeinschaft (LSD1, Ju 295/7-1, Schu 688/11-1) for funding.