
Abstract
Histone posttranslational modifications are among the epigenetic mechanisms that modulate chromatin structure and gene transcription. Histone methylation and demethylation are dynamic processes controlled respectively by histone methyltransferases (HMTs) and demethylases (HDMs). Several HMTs and HDMs have been implicated in cancer, inflammation, and diabetes, making them attractive targets for drug therapy. Hence, the discovery of small-molecule modulators for these two enzyme classes has drawn significant attention from the pharmaceutical industry. Herein, the authors describe the development and optimization of homogeneous LANCE Ultra and AlphaLISA antibody-based assays for measuring the catalytic activity of two epigenetic enzymes acting on lysine 4 of histone H3: SET7/9 methyltransferase and LSD1 demethylase. Both the SET7/9 and LSD1 assays were designed as signal-increase assays using biotinylated peptides derived from the N-terminus of histone H3. In addition, the SET7/9 assay was demonstrated using full-length histone H3 protein as substrate in the AlphaLISA format. Optimized assays in 384-well plates are robust (Z′ factors ≥0.7) and sensitive, requiring only nanomolar concentrations of enzyme and substrate. All assays allowed profiling of known SET7/9 and LSD1 inhibitors. The results demonstrate that the optimized LANCE Ultra and AlphaLISA assay formats provide a relevant biochemical screening approach toward the identification of small-molecule inhibitors of HMTs and HDMs that could lead to novel epigenetic therapies.
Introduction
Posttranslational modifications on histone proteins have been shown to play important roles in the regulation of chromatin structure and gene expression. 1 The reversible nature of histone covalent modifications or “marks,” 2 along with the deregulation of histone-modifying enzymes observed in diseased states such as cancer 3 and neuropathologies, 4 has promoted this enzyme group to an attractive class of drug targets. 5
SET7/9 (KMT7), SMYD3, and members of the MLL (KMT2) protein family are histone methyltransferases that, together with LSD1 demethylase (KDM1), are known to affect the methylation level of histone H3 on the lysine 4 (H3K4) residue (reviewed by Ruthenburg et al. 6 ). Although methylation of H3K4 by SET7/9 and MLL has been linked to active transcription,7–9 demethylation elicited by LSD1 in association with the CoREST complex was suggested to repress gene expression. 10 SET7/9 contains an evolutionary conserved SET-domain that uses S-adenosyl-L-methionine (SAM, or AdoMet) as a co-substrate to catalyze the methylation of lysine.7,8 Unlike most other SET proteins that catalyze the addition of up to three methyl groups, SET7/9 catalyzes the transfer of a single methyl group to the unmodified H3K4 residue and is thus referred to as a histone lysine mono-methyltransferase. 11 On the other hand, LSD1 belongs to the flavin adenine dinucleotide (FAD)–dependent amine oxidase superfamily and specifically catalyzes the demethylation of mono- or dimethylated H3K4 residues with the concomitant formation of hydrogen peroxide and formaldehyde. 10 In addition to being a repressor of transcription when associated with CoREST, LSD1 can also act as a transcriptional coactivator when complexed with the androgen receptor by demethylating mono- or dimethylated histone H3 lysine 9 residues. 12 Both SET7/9 and LSD1 have been associated with disease: SET7/9 with inflammatory disorders and diabetes 13 and LSD1 with various types of cancer, 14 making these two enzymes potential targets for therapeutic intervention.
Several methodological approaches have been used for quantifying the activity of histone methyltransferases (HMTs) and histone demethylases (HDMs), including radiometric assays,8,15,16 enzyme-linked immunoassays, 17 mass spectrometry, 10 and enzyme-coupled detection of reaction by-products such as S-adenosyl-homocysteine (SAH, or AdoHcy), 18 formaldehyde, 10 or hydrogen peroxide. 19 These methods suffer from drawbacks such as low throughput, lack of selectivity, high enzyme and peptide consumption, multiple wash steps, generation of hazardous waste, requirement for expensive equipment, or artifacts associated with the use of enzyme-coupled assays. The nonradioactive homogeneous LANCE Ultra and Alpha (AlphaScreen and AlphaLISA) platforms ( Fig. 1 ), based respectively on time-resolved fluorescence resonance energy transfer (TR-FRET) 20 and luminescent oxygen channeling assays, 21 do not share these limitations and are now firmly established for the development and validation of high-throughput screening (HTS) campaigns.22,23 In this regard, the AlphaScreen technology has been recently employed with success in the development of HTS assays for two epigenetic enzymes, G9a methyltransferase 24 and JMJD2E demethylase. 25
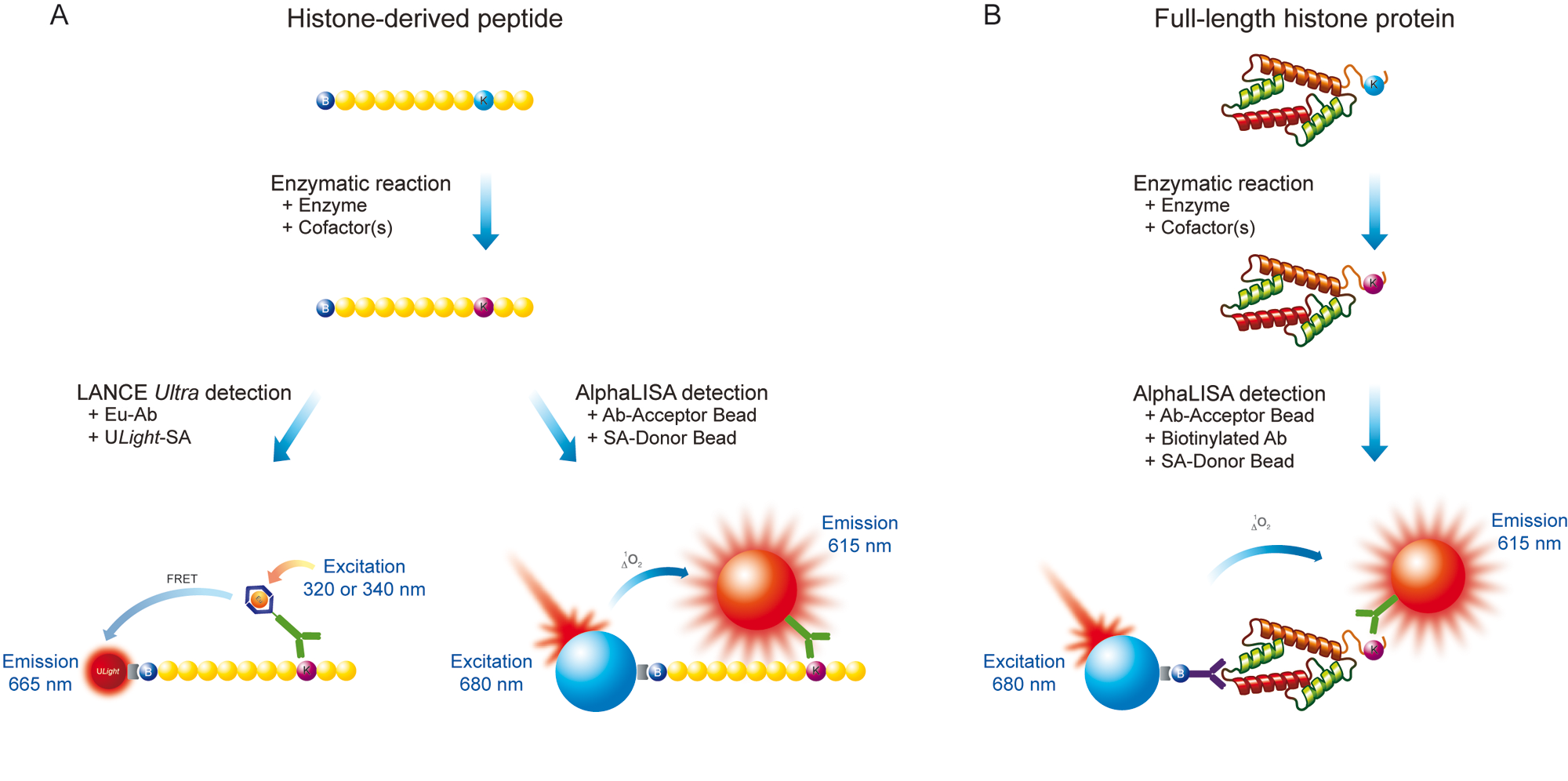
Schematic representation of LANCE Ultra and AlphaLISA epigenetic assays. (
Using LANCE Ultra and AlphaLISA technologies, we undertook the development of sensitive and selective biochemical assays for measuring the activity of SET7/9 and LSD1 enzymes in a signal-increase mode. With both technologies, synthetic biotinylated peptides derived from the N-terminus of histone H3 (residues 1–21) were used as substrates ( Fig. 1A ). Furthermore, taking advantage of the AlphaLISA platform flexibility, we developed a SET7/9 assay using a full-length histone H3 protein substrate ( Fig. 1B ). In all cases, the methylation state of histone H3K4 residues was interrogated through the use of specific antibodies detecting either methylated or unmethylated H3K4 residues.
Materials and Methods
All biotinylated peptides, including substrate peptides histone H3 (1-21) (cat. 61702) and Lys4(me1)-histone H3 (1–21) (cat. 64355), were purchased from AnaSpec (Fremont, CA), whereas recombinant full-length histone H3 substrate (C110A) (cat. 31207) and modified positive controls were from Active Motif (Carlsbad, CA). S-(5′-adenosyl)-L-methionine chloride (cat. A7007), S-(5′-adenosyl)-L-homocysteine (cat. A9384), sinefungin (cat. S7559), trans-2-phenylcyclopropylamine (tranylcypromine; cat. P8511), and poly-L-lysine (cat. P1399) were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant human SET7/9 methyltransferase (cat. ALX-201-178-C100) and LSD1 demethylase (cat. 50100) were purchased from Enzo Life Sciences International, Inc. (Plymouth Meeting, PA) and BPS Bioscience, Inc. (San Diego, CA), respectively. All other reagents were from PerkinElmer (Waltham, MA) unless otherwise stated. For LANCE Ultra assays, these included europium (Eu)–labeled anti-H3K4me1-2 (cat. TRF0402) and anti-unmodified H3K4 (cat. TRF0404) antibodies, ULight-labeled Streptavidin (ULight-SA; cat. TRF0102), and 10× LANCE Detection Buffer (cat. CR97-100). For AlphaLISA assays, reagents included anti-H3K4me1-2 (cat. AL116) and anti-unmodified H3K4 (cat. AL119) acceptor beads, streptavidin (SA) donor beads (cat. 6760002), biotinylated anti-H3 (C-ter) antibody (cat. AL118), and 5× Epigenetics Buffer 1 Kit (cat. AL008). Assays for both technologies were performed in white opaque OptiPlate 384-well microplates (cat. 6007290) using TopSeal-A sealing film (cat. 6005185). All steps using streptavidin donor beads were performed under subdued laboratory lighting (<100 lux) using green filters (Roscolux filters #389) from Rosco (Stamford, CT) to avoid photo-depleting the beads. Plates were read on an EnVision Multilabel Plate Reader (PerkinElmer) in TR-FRET or Alpha mode according to the technology used.
LANCE Ultra enzymatic assays
LANCE Ultra SET7/9 and LSD1 assays were developed using a biotinylated histone H3-derived peptide (residues 1–21) as substrate. SET7/9 methyltransferase reactions were carried out in 50 mM Tris-HCl (pH 8.8) supplemented with 5 mM MgCl2, 1 mM dithiothreitol (DTT), and 0.01% Tween-20 and were initiated by the addition of a mix of biotinylated histone H3 peptide and SAM cofactor. LSD1 demethylase reactions were carried out in 50 mM Tris-HCl (pH 9.0) supplemented with 50 mM NaCl, 1 mM DTT, and 0.01% Tween-20 and were initiated by the addition of biotinylated H3K4me1 peptide substrate. All enzymatic reactions were performed for a fixed period of time at room temperature in a 10-µL volume (see Table 1 for details on experimental conditions).
Optimized Conditions for SET7/9 Methyltransferase and LSD1 Demethylase Assays

SAM, S-adenosyl-L-methionine.
SET7/9 methyltransferase reactions were stopped by adding a mixture of Eu-labeled anti-H3K4me1-2 antibody and ULight-SA prepared in 1× LANCE Detection Buffer. LSD1 demethylase reactions were stopped by the addition of 300 µM tranylcypromine prepared in 1× LANCE Detection Buffer and incubated for 5 min before the addition of a mixture of Eu-labeled anti-unmodified H3K4 antibody and ULight-SA. For both assays, Eu-labeled antibody and ULight-SA were used at final concentrations of 2 nM and 50 nM, respectively (total assay volume of 20 µL). Capture of the reaction products by the Eu-labeled antibodies and ULight-SA was allowed to proceed for 60 min at room temperature before measuring TR-FRET signal (excitation at 320 nm, emission at 615 and 665 nm, delay time 50 µs, window time 100 µs).
AlphaLISA enzymatic assays
AlphaLISA SET7/9 assays were developed using two different substrates: a biotinylated histone H3-derived peptide (residues 1–21) and a recombinant full-length histone H3 protein substrate. The AlphaLISA LSD1 assay was developed using a biotinylated histone H3K4me1 (1–21) peptide substrate.
SET7/9 and LSD1 assays with biotinylated histone H3 peptide substrates
AlphaLISA SET7/9 methyltransferase and LSD1 demethylase reactions were run at room temperature in a 10-µL volume under conditions identical to those described for the LANCE Ultra assays. The enzymatic reactions were stopped by the addition of antibody-acceptor beads diluted in 1× Epigenetics Buffer 1. Anti-H3K4me1-2 acceptor beads were added to the SET7/9 reactions and anti-unmodified H3K4 acceptor beads to the LSD1 reactions. After 60 min at room temperature, SA donor beads prepared in 1× Epigenetics Buffer 1 were added, and the capture of the reaction product was allowed to proceed for an additional 30 min at room temperature before detecting the Alpha signal. For both assays, acceptor and SA donor beads were used at a final concentration of 20 µg/mL (total assay volume of 25 µL).
SET7/9 assay using a recombinant histone H3 protein substrate
SET7/9 reactions using a recombinant full-length histone H3 protein as substrate were conducted at room temperature in a 10-µL volume under the conditions described before for the biotinylated peptide substrate.
Methyltransferase reactions were stopped by the addition of 5 µL of high salt buffer consisting of 50 mM Tris-HCl (pH 7.4), 1 M NaCl, 0.1% Tween-20, and 0.3% poly-L-lysine. After 15 min at room temperature, capture of the reaction product was initiated by the addition of a mixture of anti-H3K4me1-2 acceptor beads and biotinylated antihistone H3 (C-ter) antibody. SA donor beads were added 60 min later, and reactions were incubated for an additional 30 min before detection of the Alpha signal. Alpha beads and biotinylated antihistone H3 (C-ter) antibody were used at a final concentration of 20 µg/mL and 1 nM, respectively (total assay volume of 25 µL). All detection reagents were prepared in a detection buffer containing 50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 0.1% Tween-20, and 0.001% poly-L-lysine.
Data analysis
Data analysis was conducted using the GraphPad Prism 5 software package (GraphPad Software, San Diego, CA). Enzymatic progress curves were fitted using a one-site binding hyperbola equation. SAM and inhibitor concentration–response curves were generated by fitting the data using a four-parameter logistic equation and used for the determination of EC50 (apparent K m) and IC50 values. Values are expressed as mean of triplicates ± standard deviation. Assay performance and robustness were evaluated using the Z′ factor equation. 26
Results
Assay development workflow
Developing LANCE Ultra and AlphaLISA reagents for the immunodetection of defined epigenetic marks requires the careful selection of appropriate antibodies.
27
These should allow the sensitive and specific detection of the mark(s) of interest while at the same time minimizing assay background due to cross-reactivity to unmodified substrate molecules. To this end, antibodies recognizing either H3K4me1-2 or unmodified H3K4 were selected and labeled with the LANCE europium chelate W1024 or covalently linked to AlphaLISA acceptor beads using standard protocols. The specificity of the resulting conjugates was characterized in titration experiments using histone H3-derived peptides and full-length histone H3 proteins bearing appropriate epigenetic marks (
The goal of our enzymatic assay development process was to develop and optimize robust and sensitive assays that can be used for the identification of compounds modulating enzyme activity. The first step of assay development consisted of selecting a substrate concentration between the EC60 and EC90 values taken from substrate titration curves. This allowed maximizing enzyme velocity while eliminating the risk of “hooking” on the substrate. A “hook” effect is usually observed in three-component assays when substrate concentrations exceed the binding capacity of the system. 28 Following determination of optimal substrate concentration, enzyme progress curves were generated at increasing enzyme concentrations in the presence of saturating cofactor (when required). An enzyme concentration and reaction time providing a linear signal increase over time were selected and used for all subsequent experiments. For methyltransferase assays, SAM titration curves were performed next, at the selected enzyme, substrate, and incubation time conditions. SAM concentrations near the apparent K m values were further selected in order for the assays to be sensitive to SAM-competitive inhibitors. Assay linearity was then reconfirmed prior to assessing DMSO tolerance and performing inhibitor concentration–response curves, as well as Z′ factor determination.
SET7/9 methyltransferase assay using a histone H3 peptide substrate
Using a biotinylated histone H3 (1–21) peptide substrate, LANCE Ultra and AlphaLISA SET7/9 methyltransferase assays were optimized following the steps described above. Based on peptide titration data (not shown), 200 nM and 50 nM peptide substrate were selected for the LANCE Ultra and AlphaLISA assays, respectively. Enzyme progress curves were generated ( Fig. 2A , B ), and a 60-min reaction time using 5 nM SET7/9 was selected for LANCE Ultra, whereas a 30-min reaction time using 1 nM enzyme was chosen for AlphaLISA experiments. The effect of SAM concentration on the methyltransferase activity was then investigated, where both LANCE Ultra and AlphaLISA assays showed equivalent apparent K m concentrations for SAM, being 75 and 79 nM, respectively ( Fig. 2C , D ). These values are significantly lower than what has been reported in the literature for SET7/9 assays conducted with H3-derived peptides (17 µM; see Ibañez et al. 29 ). However, it should be noted that in the Ibañez et al. 29 assay protocol, much higher SET7/9 enzyme and peptide substrate concentrations (200- to 1000-fold) were used, a difference that might account for this discrepancy.
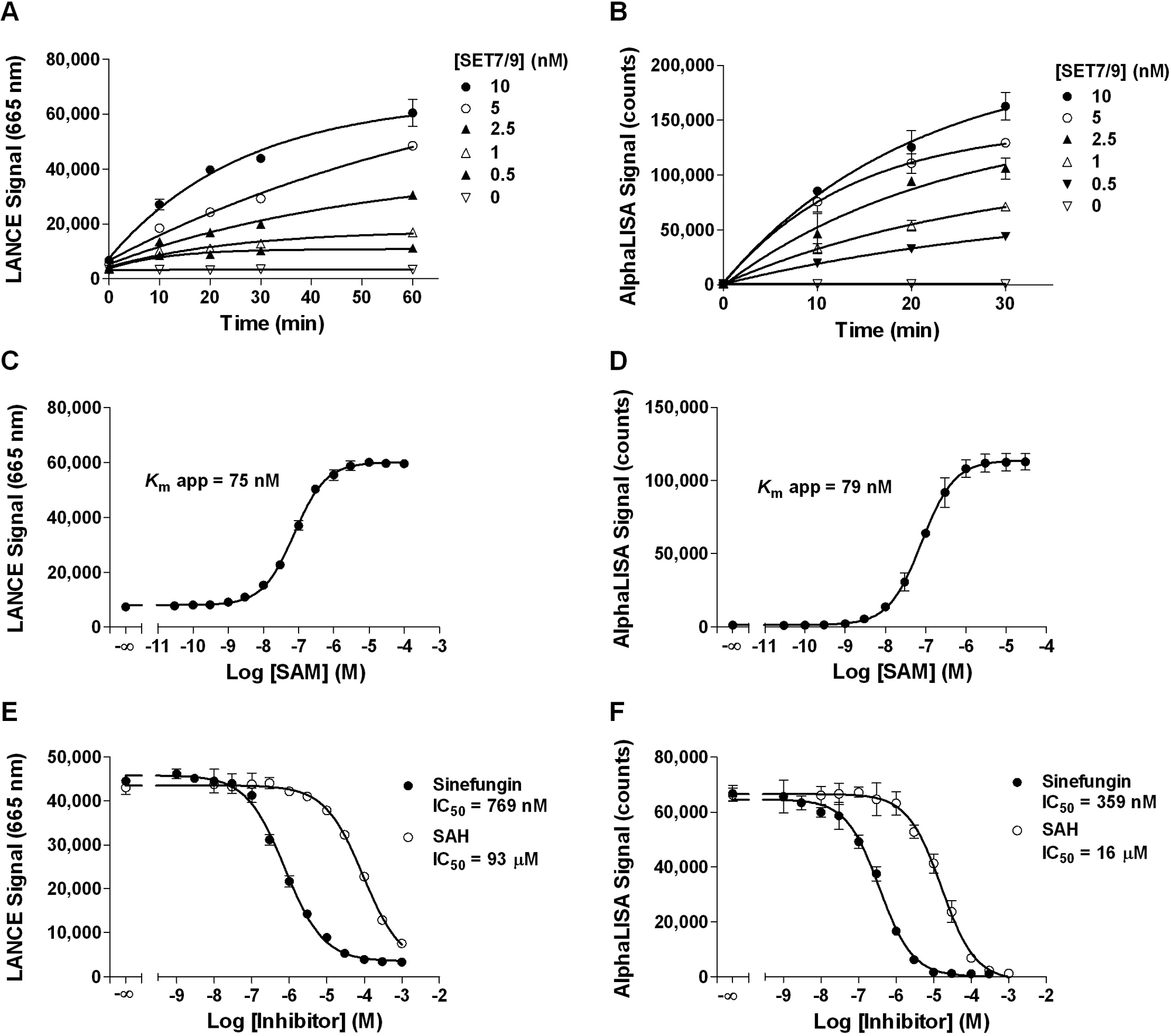
LANCE Ultra and AlphaLISA SET7/9 methyltransferase assay optimization using a biotinylated histone H3 peptide substrate. (
A concentration of 100 nM SAM (i.e., near the apparent Km) was selected for the AlphaLISA assay, whereas 300 nM (~4× apparent Km) was chosen for LANCE Ultra to obtain a larger assay window. Under the selected conditions (summarized in Table 1 ), the LANCE Ultra assay was linear for up to 60 min (r2 = 0.925) with a signal to background (S/B) ratio of 7.3 at 60 min, whereas the AlphaLISA assay was linear for up to 30 min (r2 = 0.980) with an S/B ratio of 60 at 30 min (data not shown).
To assess the sensitivity of the LANCE Ultra and AlphaLISA assays toward small-molecule modulators, two nonselective SAM-competitive HMT inhibitors were tested: sinefungin, a structural analog of SAM isolated from Streptomyces, and SAH, a by-product of all SAM-promoted methyltransferase reactions. Both compounds inhibited SET7/9 activity in a concentration-dependent manner with IC50 values of 769 and 359 nM for sinefungin and 93 and 16 µM for SAH in the LANCE Ultra and AlphaLISA assays, respectively ( Fig. 2E , F ). The lower IC50 values obtained with AlphaLISA are consistent with the threefold lower SAM concentration used in the enzymatic reaction. Our data indicate that, under these assay conditions, sinefungin behaves as a more potent inhibitor of SET7/9 than SAH.
Robustness of the optimized LANCE Ultra and AlphaLISA assays was determined by evaluating their respective Z′ factor values. Inhibited signal was measured using sinefungin at 300 µM in LANCE Ultra and 100 µM for the AlphaLISA assay. Calculated Z′ factor values were above 0.80 with both technologies ( Table 2 ), with S/B ratios of 9.4 in LANCE Ultra and 36 in AlphaLISA. Assay stability was demonstrated for both platforms, with Z′ factor values higher than 0.70 following overnight incubation. Taken together, these results demonstrate the robustness of the optimized SET7/9 methyltransferase peptide assays on both detection platforms and their suitability for HTS.
Statistical Evaluation of Assay Quality
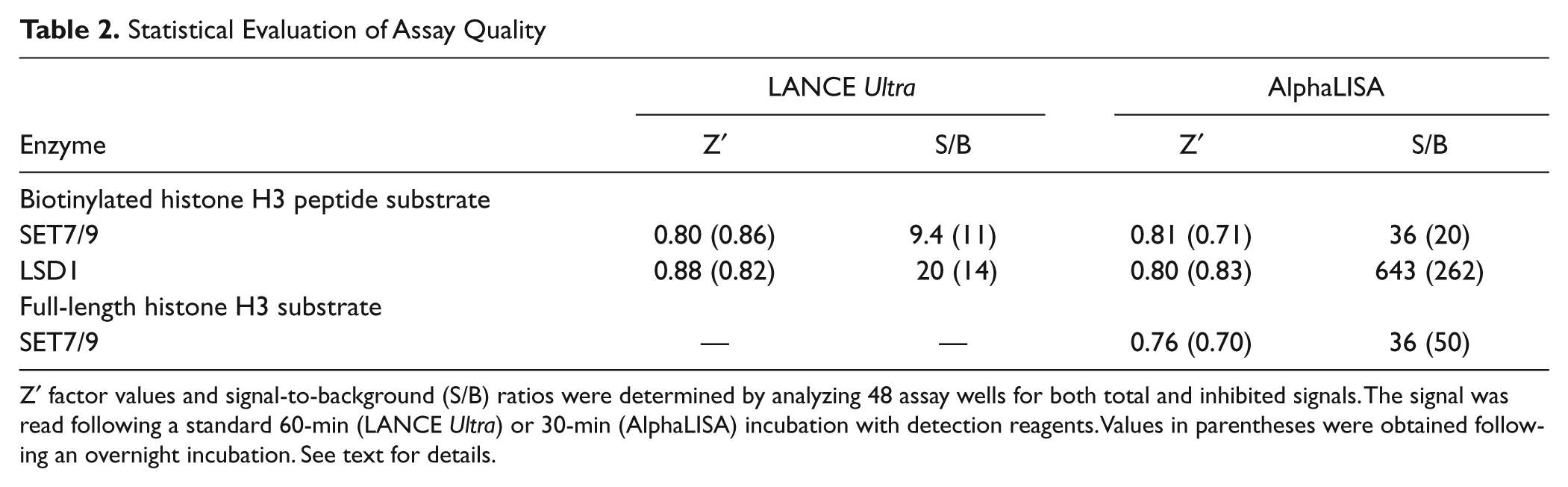
Z′ factor values and signal-to-background (S/B) ratios were determined by analyzing 48 assay wells for both total and inhibited signals. The signal was read following a standard 60-min (LANCE Ultra) or 30-min (AlphaLISA) incubation with detection reagents. Values in parentheses were obtained following an overnight incubation. See text for details.
SET7/9 methyltransferase assay using a full-length histone H3 substrate
In contrast to TR-FRET, the AlphaLISA technology theoretically allows measuring interactions between binding partners over distances up to 250 nm. 21 This characteristic makes the Alpha technology a highly suitable tool for developing assays aimed at measuring protein–peptide30,31 and protein–protein 32 interactions, as well as enabling the detection of full-length proteins bearing specific posttranslational modifications.23,32 The AlphaLISA platform was thus selected for developing a SET7/9 methyltransferase assay using recombinant full-length histone H3 as substrate. The use of detection buffers optimized for short histone H3-derived peptide substrates led to high background levels and a concomitant loss of antibody specificity. Further buffer optimization experiments (not shown) proved that high salt concentration was essential to minimize assay background, whereas the addition of a higher concentration of poly-L-lysine was required to stop the enzymatic reaction. In this regard, the exact mechanism of action of poly-L-lysine as an inhibitor of SET7/9 is not known and was not further studied. However, it could be hypothesized that the addition of a large amount of nonmodified lysine residues will successfully outcompete the ones present on a given substrate, hence bringing its enzymatic modification to a halt. A buffer solution combining both 1 M NaCl and 0.3% poly-L-lysine was thus used to stop the enzymatic reactions prior to the addition of acceptor beads and biotinylated antihistone H3 (C-ter) antibody. With these protocol adjustments in place, development and optimization of the SET7/9 protein assay were resumed through experiments similar to those performed for the SET7/9 assay using biotinylated peptide substrate.
To determine the optimal substrate concentration to be used in the assay, full-length histone H3 titration experiments were performed and a concentration of 300 nM histone H3 protein was selected for further assay optimization (data not shown). Enzyme progress curves were then performed at different SET7/9 concentrations ( Fig. 3A ). These curves showed a decline of SET7/9 activity over time, with almost no residual activity after 15 min. On the basis of these results, we selected a reaction time of 10 min, using an enzyme concentration of 0.3 nM.
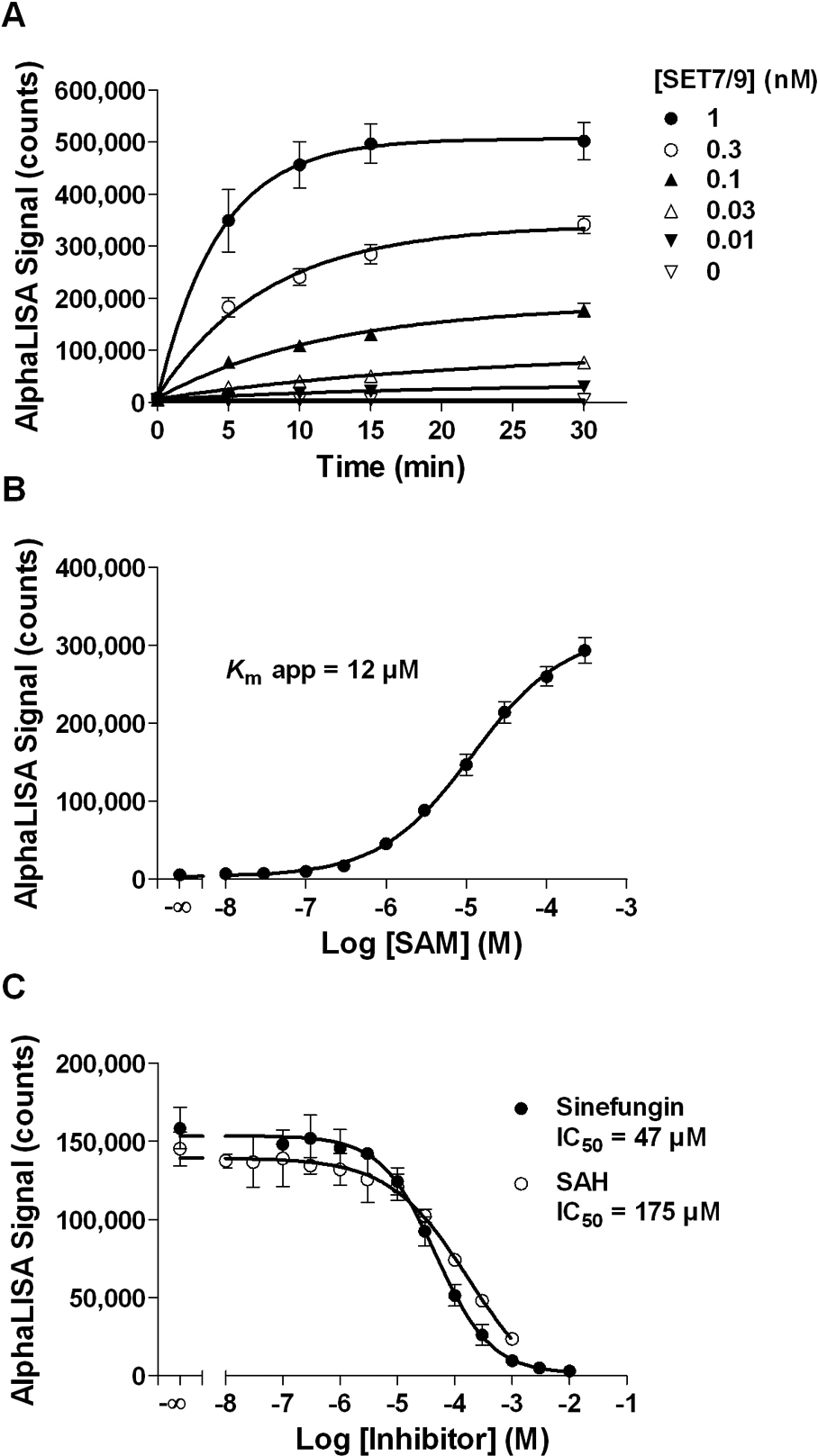
AlphaLISA SET7/9 methyltransferase assay optimization using full-length histone H3 protein substrate. (
The apparent Km value for SAM was then evaluated at the selected enzyme concentration and incubation time ( Fig. 3B ). A value of 12 µM was obtained, which is in agreement with literature data (6 µM, Trievel et al. 15 ; 3.5 µM, Dirk et al. 33 ). A concentration of 10 µM SAM was selected for the optimized protein assay. Interestingly, this apparent Km value is considerably higher than the one obtained for the AlphaLISA peptide assay (79 nM), a fact that could be attributable to the different nature of the substrates. Linearity of the optimized enzymatic reaction as a function of time was then confirmed for up to 10 min (r2 = 0.941) with an S/B ratio of 29 (data not shown). Optimized assay conditions are summarized in Table 1 .
Concentration–response curves for sinefungin and SAH were performed, and both compounds inhibited SET7/9 activity with IC50 values of 47 µM for sinefungin and 175 µM for SAH ( Fig. 3C ). As expected, IC50 values were higher than those obtained in the SET7/9 methyltransferase peptide assay because a higher SAM concentration was used for the protein assay. However, the same rank order of potency was obtained for the two compounds regardless of the substrate used ( Figs. 2F and 3C ). A Z′ factor value of 0.76 with an S/B ratio of 36 were obtained using 10 mM sinefungin for inhibited samples and, importantly, performance was maintained after an overnight incubation ( Table 2 ). These results confirm the AlphaLISA SET7/9 methyltransferase protein assay robustness as well as its suitability for HTS.
LSD1 demethylase assay using a H3K4me1 peptide substrate
The availability of LANCE Ultra and AlphaLISA reagents specifically detecting unmethylated lysine 4 allowed developing LSD1 demethylase signal increase assays measuring the appearance of the demethylated product. Because LSD1 has been reported to demethylate monomethyl-lysine 4 on histone H3-derived peptides at least 20 residues long, 19 a biotinylated H3K4me1 peptide (residues 1–21) was selected as substrate for these assays. Following titration of the H3K4me1 peptide, a concentration of 200 nM was chosen for LANCE Ultra and 80 nM for AlphaLISA assays (data not shown). Enzyme progress curves showed that signal increased linearly with time for up to 2 h when 2 nM LSD1 was used ( Fig. 4A , B ). On the basis of these results, we selected a 60-min reaction time and a 2-nM LSD1 enzyme concentration for the subsequent experiments with both technologies ( Table 1 ). Under these conditions, S/B ratios were equal to 18 and 292 for LANCE Ultra and AlphaLISA assays, respectively. The redox cofactor FAD is essential for LSD1 activity and is strongly embedded in the amine oxidase domain. As such, FAD addition to the enzyme reaction buffer is not necessary for optimal enzymatic activity as it is already present in recombinant LSD1 enzyme preparations. 10
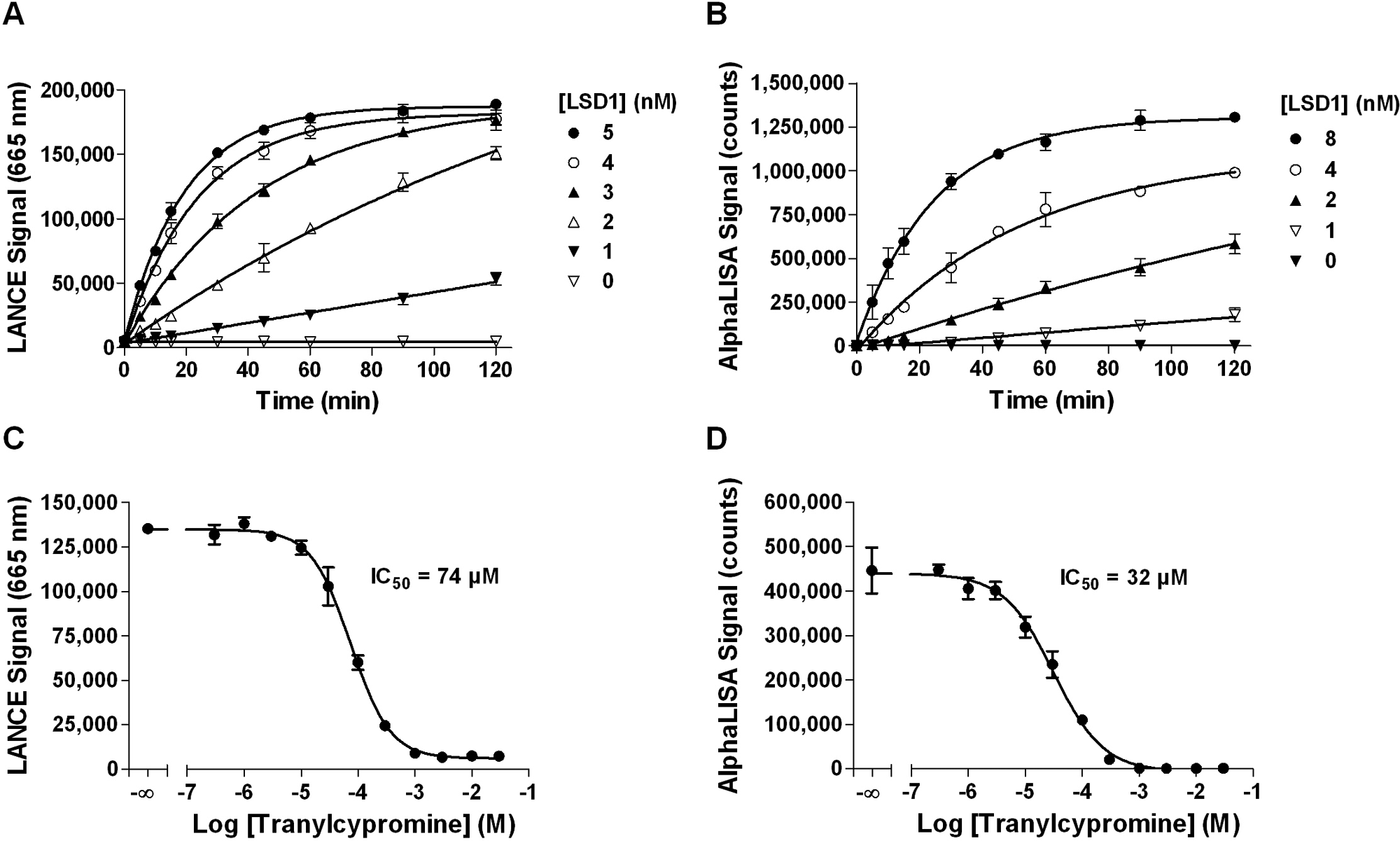
LANCE Ultra and AlphaLISA LSD1 demethylase assay optimization using biotinylated H3K4me1 peptide substrate. (
The antidepressant drug tranylcypromine is a monoamine oxidase inhibitor reported to inhibit the activity of LSD1 demethylase. 34 Data in Figure 4C , D indicate that tranylcypromine inhibits LSD1 enzyme activity in a concentration-dependent manner with similar IC50 values of 74 and 32 µM for the LANCE Ultra and AlphaLISA assays, respectively. Given that tranylcypromine is an irreversible covalent inhibitor of enzyme activity, comparison of IC50 values reported herein with those published in the literature has not been performed due to significant differences in assay conditions. Finally, assay robustness was assessed by Z′ factor determination, where inhibited signal was measured using 3 mM tranylcypromine. Values of 0.88 and 0.80 were obtained, with S/B ratios of 20 and 643 for LANCE Ultra and AlphaLISA assays, respectively, demonstrating their suitability for HTS ( Table 2 ).
Discussion
In this study, we presented the development and optimization of LANCE Ultra and AlphaLISA assays for two epigenetic enzymes controlling the methylation state of histone H3 lysine 4 residues. Optimized assays were highly sensitive, requiring low enzyme and substrate concentrations, which confers a significant advantage in terms of cost over less sensitive methods as liquid chromatography/mass spectrometry or enzyme-coupled assays. Importantly, the use of low nanomolar enzyme concentrations can enable structure–activity relationship studies where the use of very low concentrations of small-molecule compounds is required. Our assays also allowed inhibitor profiling and were highly robust, as highlighted by Z′ factor values equal or above 0.7. Notably, assay quality was maintained following an overnight incubation, allowing both online and off-line plate reading.
Although the presented data focused on the development of SET7/9 and LSD1 assays, the same antimark specific detection reagents could also be used for studying other enzymes involved in the modification of the H3K4 residue. For instance, a signal increase SIRT1 assay measuring the deacetylation of H3K4 residues has been optimized using reagents detecting unmodified H3K4 (unpublished data). Similarly, demethylation of an H3K4 trimethylated substrate could also be monitored using either the H3K4me1-2 or unmodified H3K4 detection reagents. In brief, the assay development approach described here is broadly applicable to other epigenetic marks and in fact has been used for developing LANCE Ultra and AlphaLISA reagents detecting H3K9me2, H3K9ac, H3K27ac, H3K27me1-2, H3K27me3, and H3K36me2 (unpublished data). This could aid in the identification of novel small-molecule compounds of pharmacological value targeting important epigenetic enzymes such as EZH2, the Jumonji domain–containing family of demethylases, acetyltransferases such as p300 and pCAF, and deacetylases from the HDAC and sirtuin classes.
Footnotes
All authors are employees of PerkinElmer. None of the authors or PerkinElmer received at any time payment or services from a third party for any aspect of the submitted work. There are no other relationships/conditions/circumstances that present a potential conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.