
Abstract
Aberrant expression of chromatin-modifying enzymes (CMEs) is associated with a range of human diseases, including cancer. CMEs are now an important target area in drug discovery. Although the role that histone and protein (lysine) methyltransferases (PMTs) play in the regulation of transcription and cell growth is increasingly recognized, few small-molecule inhibitors of this class of enzyme have been reported. Here we describe an assay suitable for primary compound screening for the identification of PMT inhibitors. The assay followed the methylation of histones in the presence of the PMT SET7/9 and the radioactive cofactor S-adenosyl-methionine using scintillating microplates (FlashPlate) and was used to screen approximately 65 000 compounds (% coefficient of variation = 10%; Z′ = 0.6). The hits identified from a library of more than 63 000 diverse small molecules included a series of rhodanine compounds with micromolar activity. A screen of the National CancerInstitute Diversity Set (2000 compounds) identified an orsein derivative that inhibited SET7/9 (~20 µM) and showed modest growth inhibition associated with the expected cellular phenotype of reduced histone methylation in a human tumor cell line. The assay represents a useful tool for the identification of inhibitors of PMT activity.
Introduction
The two dominating forms of chromatin structure, transcriptionally repressive heterochromatin and transcriptionally permissive euchromatin, are regulated by several mechanisms, including a range of posttranslational histone modifications carried out by chromatin-modifying enzymes (CMEs). These modifications, including methylation of lysine and arginine, acetylation of lysine, and phosphorylation of serine and threonine, mostly occur on the N-terminal tails of histone proteins and are critically involved in the control of gene transcription, replication, and DNA damage repair. The enzymes that covalently modify histones and a range of other signaling proteins are thus key mediators of transcriptional regulation. Several CMEs, including histone acetyltransferases (HATs), histone deacetylases (HDACs), and histone protein kinases, have been implicated in a variety of human cancers and as such have been identified as targets for therapeutic intervention in cancer and other diseases.
Protein (lysine K) methyltransferases (PMTs) are essential enzymes in the regulation of transcriptional events through histone and nonhistone protein methylation. Many PMTs contain a highly homologous catalytically active SET (Su(var)3-9, Enhancer of Zeste, Trithorax) domain that uses the cofactor S-adenosylmethionine (SAM) as a methyl group donor. PMTs have been implicated in several human diseases, particularly cancer,1,2 including mixed-lineage leukemia through translocation and resultant fusion proteins of the MLL gene, an H3K4 PMT; prostate breast and ovarian cancer involving upregulation of EZH2, an H3K27 PMT; and colorectal and hepatocellular carcinoma through overexpression of SMYD3, another H3K4 PMT. Recently, an H3K9 and K27 methyltransferase, G9a, was shown to have a role in the invasion and metastasis of lung cancer. 3
On the basis of the mounting evidence implicating SET domain-containing PMT enzymes in cancer, we commenced a project to identify small-molecule inhibitors of SET domain-catalytic activity by high-throughput screening (HTS). At the time this work commenced, it was decided to use the PMT enzyme SET7/9 for the primary screen as a model for SET domain-containing proteins. SET7/9 monomethylates H3K4, leading to transcriptional activation of a number of genes.4,5 However, evidence linking SET7/9 to cell growth in human disease remains weak. It was hypothesized that inhibitors of SET7/9 would provide possible chemical tools to explore the role of SET domain-containing enzymes in disease, as well as being potential starting points for drug discovery. In addition, unlike other enzymes in this class at the time, large quantities of catalytically active SET7/9 were available. In addition, co-crystal structures of SET7/9 with an H3 peptide substrate and the cofactor reaction product S-adenosylhomocysteine (SAH) had been solved,6,7 permitting crystallographic and in silico approaches to hit confirmation and analogue design. Over the past few years, several PMT inhibitors1,8 have been disclosed but none have progressed to clinical evaluation. These include the SAM-competitive mycotoxin chaetocin and the quinazoline BIX-01294, a micromolar inhibitor of G9a. Both compounds were identified by screening campaigns and were shown to modulate methylation at appropriate histone methylation sites. Recently, a more potent analogue of BIX-01294, UNC0224, has been reported. 9 Here we describe the optimization of a radiometric biochemical assay suitable for primary screening, the outcome of an HTS campaign and some of the properties of the SET7/9 inhibitors identified.
Materials and Methods
Materials
The SET7/9 cofactor SAM, S-adenosylornithine (SO), and SAH (A2408, S8559, and A9384 respectively; Sigma-Aldrich, Gillingham, Dorset, UK) were obtained as powdered stocks and stored at 4 °C. The 10-mM stock solutions were prepared in water and stored aliquoted at −20 °C. SAHA was obtained from BioCat GmbH (Heidelberg, Germany). The screening library (63 226 compounds) included compounds from Maybridge (Thermo Fisher Scientific, Tintagel, Cornwall, UK), Chemical Diversity Laboratories (ChemDiv, San Diego, CA), Bionet Research (Key Organics Ltd, Camelford, Cornwall, UK), SPECS and BioSPECS B.V. (Delft, the Netherlands), and the Cancer Research UK Compound Collection. The National Cancer Institute (NCI) Diversity Set (~2000 compounds from the Development Therapeutics Program, NCI, Bethesda, MD; http://dtp.nci.nih.gov) was also screened. All compounds were stored as 10-mM stocks in 100% DMSO in 96-well polypropylene plates at −20 °C. Compounds identified as hits in the primary screen were cherry-picked from these stocks. Daughter plates (200 µM in 2% DMSO in 384-well plates) were removed from −20 °C storage and stored at room temperature for 24 h before screening. Tritiated SAM (3H-SAM); NET155H was obtained from PerkinElmer Life Sciences. GST-SET7/9 enzyme (aa52-366) was kindly provided by Dr. S. Gamblin (NIMR, Mill Hill, UK), and crude histone extracted from calf thymus (Cat No. 223-665) was obtained from Roche, Burgess Hill, West Sussex, UK. DELFIA reagents were obtained from PerkinElmer Life Sciences (Seer Green, Bucks, UK). All other chemicals were obtained from Sigma-Aldrich.
Optimized FlashPlate Assay for HTS
Compounds (3 µL of 200 µM in 2% DMSO), DMSO control (2%), or adenosylornithine (3 µL of 53.7 µM stock) were added to the wells of a basic 384-well FlashPlate (cat. no. SMP400001PK; PerkinElmer Life Sciences). GST-SET7/9 (5 µL of 600 nM) diluted in assay buffer (50 mM Tris, 1 mM EDTA, 100 mM NaCl, 10% glycerol, 0.01% Triton X-100, 4 mM dithiothreitol [DTT], made up in distilled water, pH 8.5) was added to compound and total wells, and assay buffer was added to blank wells. Last, 12 µL of substrate mix (5.66 µCi/mL 3H-SAM, 4.2 µM SAM, 0.0167 mg/mL crude histone extract diluted in assay buffer) was then added to all wells. Final concentrations in the reaction (20 µL volume) were 150 nM GST-SET7/9, 0.068 µCi 3H-SAM, 2.5 µM SAM, and 0.01 mg/mL crude histone extract. The reaction was incubated for 30 min at room temperature and stopped by the addition of 70 µL 0.5% perchloric acid (PCA). The plate was sealed, incubated (2 h at room temperature), and read for 30 s per well on a Topcount NXT (PerkinElmer Life Sciences). Results from the screen were analyzed using robust statistics using Excel (Microsoft Corp., Redmond, WA) and OMMM Enterprise 2.0 software (Oxford Molecular Group, Oxford, UK), and all data were later migrated to the data manager ActivityBase (IDBS, Guildford, UK). Compounds were identified as hits if they gave ≥50% inhibition of enzyme activity. The potency of hit compounds (IC50) was determined in GraphPad Prism 4 (GraphPad Software, La Jolla, CA), using the built-in analysis for nonlinear regression (curve fit) and the sigmoidal dose-response (variable slope) equation after the % inhibition for each compound was calculated and compound concentrations converted into Log10 values.
DELFIA Assay for SET7/9 Activity
DMSO control or compound (3 µL) was added as before to the wells of a polypropylene, round-bottomed, 96-well plate. GST-SET7/9 (5 µL of 400 nM diluted in assay buffer as described above) was added to the total activity and compound wells and assay buffer (5 µL) added to the enzyme blank wells. SAM (7 µL of 71.4 µM) and substrate peptide (10 µL of 1.88 µM; sequence ARTKQTA RKSTGGKAPRKQLA-biotin) were added to give a total of 25 µL reaction volume. Final reagent concentrations were 80 nM GST-SET7/9, 20 µM SAM, and 750 nM peptide. The enzyme reaction was allowed to proceed at room temperature for 1 h and then stopped by the addition of 100 µL 0.5% PCA. Reaction mixture (100 µL) was transferred to a 96-well, NeutrAvidin-coated plate (Perbio Science UK Ltd, Northumberland, UK) and incubated at room temperature for 1 h with gentle agitation. The plate was washed (Wellwash 5000; Thermo Fisher Sciences, Basingstoke, UK) four times in wash buffer (25 mM Tris, 150 mM NaCl, and 0.1% Tween-20, pH 8.0) and 100 µL primary antibody (1 nM monomethyl H3K4 antibody, ab8895; Abcam Ltd., Cambridge, UK) diluted in DELFIA buffer added and incubated for 1 h with agitation, then washed 4× in wash buffer. Europium-labeled secondary antibody (Eu-N1 antirabbit antibody at 0.15 µg/mL diluted in DELFIA buffer) was added for a further hour with agitation. The plate was washed as before and enhancement solution (100 µL) added to each well. The plate was shaken for 5 min and the time-resolved fluorescent signal measured on a Victor II plate reader (PerkinElmer Life Sciences).
Growth inhibition assay
HCT116 human colorectal tumor cells were obtained from American Type Culture Collection (ATCC), supplied by LGC Promochem (Teddington, Middlesex, UK) and routinely tested for mycoplasma contamination. Cells were seeded at 1000 cells per well (200 µL 5 × 103 cell/mL) in 96-well tissue culture plates, leaving an empty row of cells as a no-stain blank control. After 24 h growth, compounds and DMSO control were added at a range of concentrations and the cells incubated for a further 72 h. Cell number was measured using sulforhodamine B, and growth inhibition (GI50) was calculated using GraphPad Prism 4.0 software.
Fixed-Cell Enzyme-Linked Immunosorbent Assay (TRF-Cellisa)
HCT116 human colorectal tumor cells were seeded at 4000 cells/well in 200 µL supplemented Dulbecco’s modified Eagle’s medium (DMEM; 2 × 104 cells/mL) into 96-well tissue culture plates (Falcon cat. no. F3072 from Marathon Laboratory Supplies, London, UK) and cultured for 48 h. Cells were treated with compound or DMSO control (typically 10 µL in 200 µL of media) for 24 and 48 h and fixed by the addition of 200 µL fixing solution (3% formaldehyde, 0.25% glutaraldehyde, 0.25% Tween-20) for 30 min at 37 °C. The fixed cell layer was washed once in phosphate-buffered saline and the plates stored at 4 °C until ready to be processed. The wells were blocked in 5% dried milk (Marvel, Premier International Foods, Spalding, Lincs., UK)–Tris-buffered saline (TBS) solution (100 µL) for 30 min at 37 °C and washed once in 0.1% Tween-20 solution. Primary rabbit antibodies (100 µL) to monomethyl H3K4 (ab8895; Abcam) or acetylated H3 (06-599; UpState, Dundee, UK) diluted in TBS were added and the plate incubated overnight at room temperature. The plate was washed three times in 0.1% Tween-20 and incubated with europium-labeled antirabbit secondary antibody (AD0105) diluted in DELFIA buffer for a further 1 h at 37 °C. Wells were washed as before prior to the addition of 100 µL enhancement solution and plates shaken for 5 min and read as above. Plates were washed once in PBS and protein measured by the bicinchoninic acid (BCA) assay (Perbio UK Ltd.) and absorbance read at 570 nm. DELFIA counts were normalized by dividing europium counts by BCA reading minus the BCA background. Results were expressed as percent DMSO controls.
Results and Discussion
Optimization of PMT Activity Assay
Several methods have been used for measuring PMT catalytic activity. 10 Recently, homogeneous nonradioactive antibody-based assays (LANCE Ultra and AlphaLISA) have been described. 11 During assay development, we investigated the use of a similar peptide-based AlphaScreen method. However, the screen described here, based on a nonwash radiometric FlashPlate assay previously described for HAT activity, 12 provided a greater signal window. The histone substrate used in the assay is thought to passively coat the surface of the wells and, when methylated in the presence of enzyme and 3H-SAM, produces a signal detectable by proximity counting. A similar assay for PMT activity that uses a coated peptide substrate has been published. 13
Preliminary results in a filter-based assay (see supplemental information) using GST-SET7/9 in the presence of tritiated SAM (1.0 µCi) confirmed histone extract as a substrate for this enzyme. An enzyme titration in the presence of 0.55 µCi tritiated SAM, and 10 µM SAM allowed the selection of conditions (200 nM GST-SET7/9 for 30-min incubation) for plate-based optimization experiments.
Initial reagent titrations were carried out in 96-well FlashPlates. An acceptable signal window (Z′ = 0.66) was obtained with 0.137 µCi 3H-SAM, 200 nM SET7/9, and a 30-min reaction time. Further assay optimization was carried out in a 384-well plate format. As a dilution step (3.5-fold) with assay buffer was found to be inadequate as a means of stopping the enzyme reaction, trichloroacetic acid (TCA 10%) and PCA (0.5%–8%) were tested as stopping solutions. Although the enzyme reaction was effectively stopped by both acids over a 20-h period, PCA (0.5%) was selected for future work as it resulted in a higher signal (2301 ± 775 cpm) than TCA (1587 ± 95) (
The reagent concentrations were finally optimized in the presence of 0.2% DMSO to replicate screening conditions. The enzyme reaction was approximately linear under these optimized conditions (2.5 µM SAM, 0.068 µCi 3H-SAM, and 0.01 mg/mL crude histone) (
Screening Results
Compounds were screened at a final concentration of 30 µM in 11 batches of 10 to 20 plates. For each batch, the assay components were made up as four solutions: The positive control was manually pipetted into the assay plate, the DMSO compound solution or DMSO controls were removed directly from daughter plates (200 µM; 2% DMSO), the substrate mix ( 3 H-SAM, SAM, and crude histone extract diluted in assay buffer), and finally enzyme solution. The last three solutions were dispensed using a Minitrak V (PerkinElmer Life Sciences) from reservoirs on the workstation. Compound plates were rescreened if the performance criteria were unacceptable (Z′ < 0.5 and CV greater than ~10%).
The assay performed well during the screen ( Fig. 1 ). Assay parameters were consistent between plates and batches, resulting in a wide signal window (signal to background [S/B] = 38.4 ± 7.9). The mean Z′ for the entire screen was 0.6 ± 0.05 with a CV of 9.1%. In total, only four plates (~2%) failed to meet the criteria set to pass a plate, and these were rescreened. No false positives were identified on any of the DMSO-only plates included in the screen.
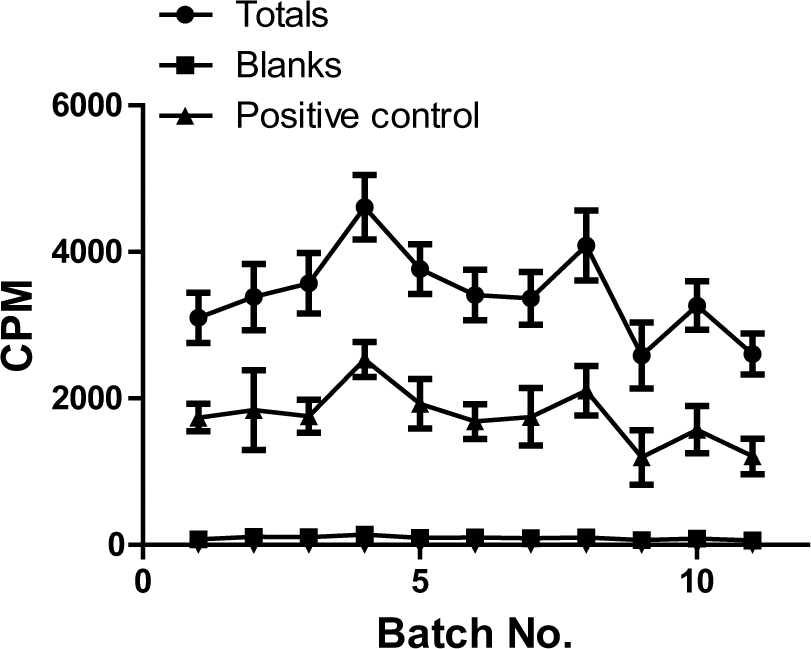
Summary (mean ± SD) of overall screen performance (~63 226 compounds) in 11 batches. Overall total activity counts were 3434 ± 591, and no-enzyme blank counts were 95 ± 22.4 (the error bars are within the symbol). Mean inhibition by the positive control S-adenosylornithine (SO) was 47.3% ± 5.0%. CPM, count per minute.
Hit Compounds
The activity of 10 hits was confirmed by reassay (n = 5) in the FlashPlate assay. On the basis of their pharmaceutical properties, we selected five compounds that fell into two structural classes ( Table 1 ) for further evaluation. A signal quenching assay was established to check for optical interference. This assay was run in the normal way except that compounds were added after the enzyme reaction had been stopped. Where there was no interference, the signal obtained would be similar to that observed in the enzyme total activity wells. These results were compared with a standard assay where the compounds were present for the duration of the enzyme reaction, allowing inhibition of the enzyme. Four of the compounds did not cause optical interference, although CCT021895 caused interference equivalent to an apparent inhibition of enzyme activity of approximately 25% at 200 µM.
Chemical Structures and SET7/9 Inhibition of Five Hits Selected for Evaluation
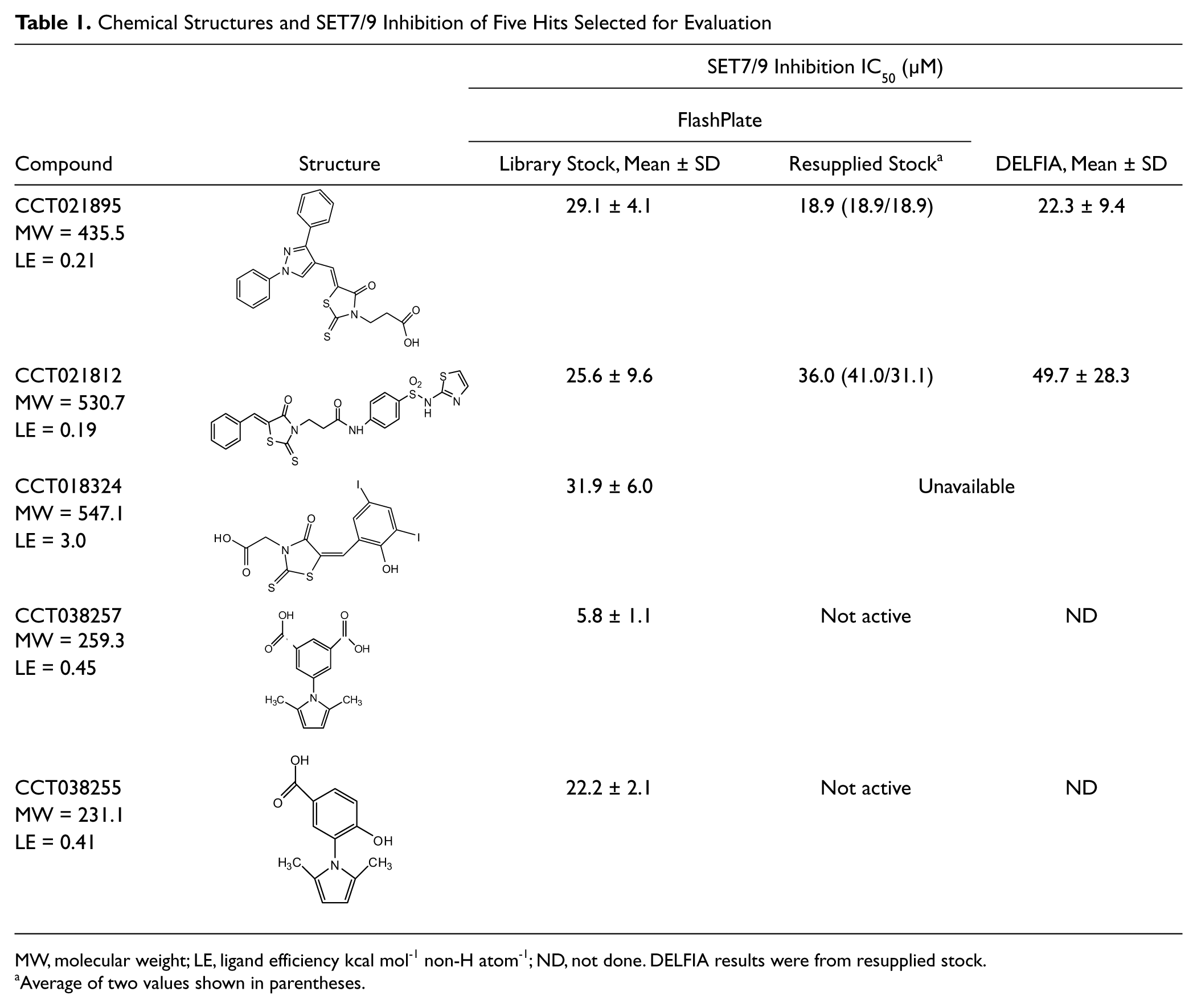
MW, molecular weight; LE, ligand efficiency kcal mol-1 non-H atom-1; ND, not done. DELFIA results were from resupplied stock.
Average of two values shown in parentheses.
The activity of the two N-aryl pyrrole compounds (CCT038257 and CCT038255; Table 1 ) could not be reproduced in freshly prepared solutions of resupplied compound. Subsequent mass spectrometry did not demonstrate the presence of the parent compound in library master stocks, and this hit series was classified as chemically unstable.
Two of the three rhodanine compounds (CCT021812 and CCT021895) were available for repurchase, and fresh stocks of compound inhibited SET7/9 with comparable potency to that determined for the library compounds (
Table 1
). The activity of these two hits was confirmed (
Table 1
) in a biochemical DELFIA assay using a primary antibody to monomethylated H3 K4 (
Although a relatively high compound concentration (30 µM) was used in the screen, the confirmed hit rate was low (0.02%) compared with that seen in screens of other targets using the same assay format. 12 Chemical diversity within the compound collection may have been insufficient to find inhibitors of this novel class of enzymes. Alternatively, the low hit rate could have been due to the fact that, as revealed by the crystal structure of SET7/9,7,14 the substrate and cofactor binding sites lie on either side of a narrow channel in the protein active site formed by the folding of the C-flanking domain. There may be increased flexibility in the protein structure in the absence of the cofactor and/or substrate peptide. The terminal amine of the lysine residue has access to the methyl donor through a tunnel in the protein linking the two binding domains. This feature of the active site may result in SET7/9 being less drugable than other PMTs or some other target classes.
Nevertheless, our screen identified two structurally different chemotypes that inhibited PMT activity. The micromolar activity of the two rhodanine compounds was confirmed in two different assay formats. Compounds similar to the structure of CCT021812 have been shown to be inhibitors of the phosphatase JSP-1 15 and their activity ascribed to the ability of the C=C bond to act as a Michael acceptor. This does not seem to be the case with CCT021812 at least, since its activity was approximately the same in the absence and presence of DTT. However, due to the large number of related structures identified by substructure searching, their low ligand efficiency against SET7/9, substantial prior art as inhibitors in various diseases (including cancer), and their potential for chemical reactivity, the series did not progress further. The N-arylpyrrole compounds proved to be chemically unstable, and this series has been subsequently classified as frequent hitters.
NCI Diversity Set
In the interest of finding additional PMT inhibitors, the NCI Diversity Set was screened using the FlashPlate format. Overall assay performance was satisfactory (Z′ = 0.55; CV = 11.2%). Thirteen compounds were identified as hits, and these were sourced as powdered stocks for further study. Because of poor potency, it was possible to calculate the IC50 values of only 6 of the 13 compounds. The growth inhibitory effects of these 6 compounds were determined using HCT116 human colorectal tumor cells. Compounds were not investigated further if cellular potency was greater than that against SET7/9 as this suggested non-mechanism-based, off-target cytotoxicity. Two compounds (119913-X and 610930-N) were selected. The biochemical inhibition (IC50) of 119913-X and 610930-N was confirmed using the DELFIA format (2.3 ± 0.7 µM and 22.1 ± 8.1 µM, respectively). Growth inhibition potency (GI50) for these two compounds was ~20- to 3-fold, respectively, less than their enzyme inhibition ( Table 2 ).
Chemical Structures, SET7/9 Inhibition (IC50), and HCT116 Human Colorectal Tumor Cell Growth Inhibition (GI50) by the Two Hits from the National Cancer Institute Diversity Set
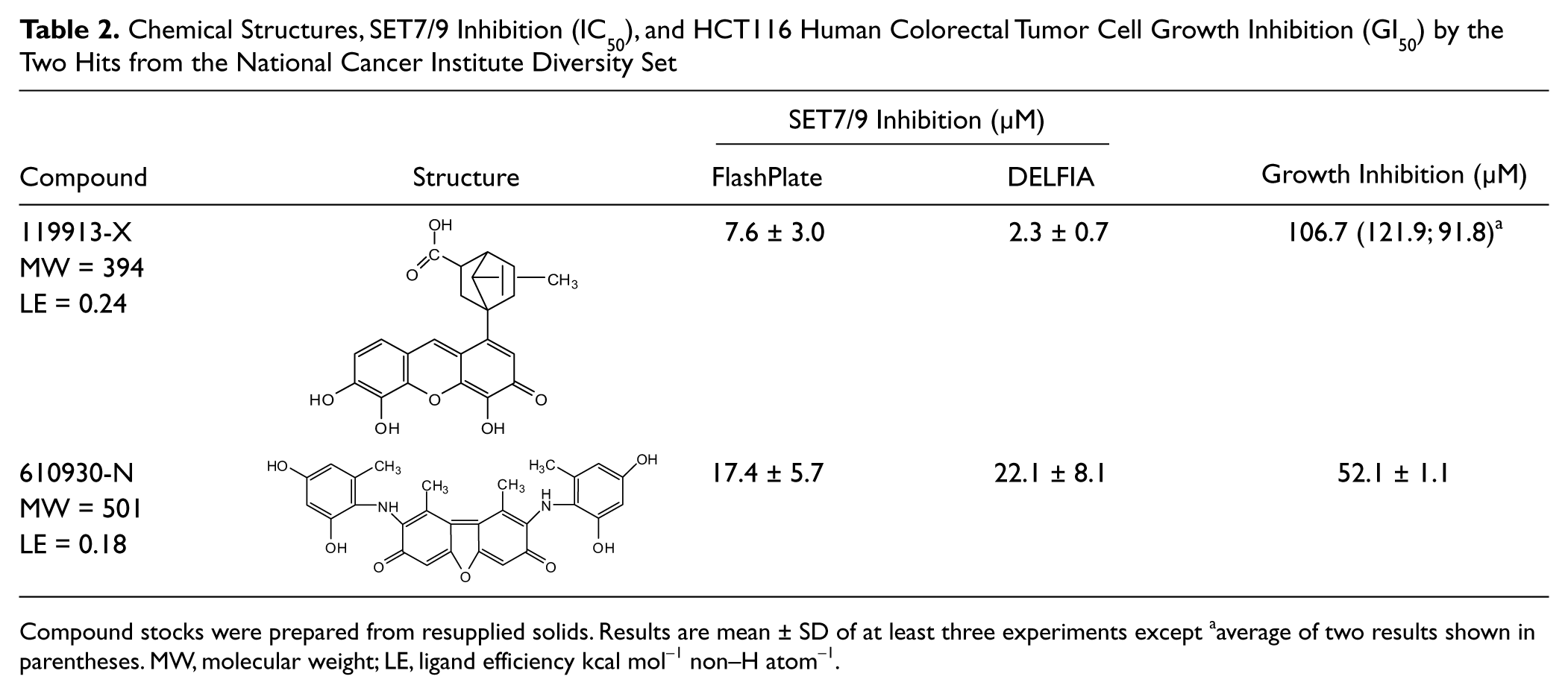
Compound stocks were prepared from resupplied solids. Results are mean ± SD of at least three experiments except aaverage of two results shown in parentheses. MW, molecular weight; LE, ligand efficiency kcal mol−1 non–H atom−1.
Mechanistic Assay
The effects of 119913-X and 610930-N on histone methylation were investigated in a fixed-cell enzyme-linked immunosorbent assay (ELISA; TRF-Cellisa) developed to monitor the cellular effects of PMT inhibitors using an antibody to monomethylated H3K4. It was hypothesized that inhibition of SET7/9 in cells would cause decreased monomethylation of H3K4, a known SET7/9 (and other PMT) methylation site.4,5 Increased acetylation on histone H3, particularly H3K9, as occurs with HDAC inhibition, encourages methylation at H3K4 as both these marks denote euchromatic domains. The HDAC inhibitor SAHA and the demethylase inhibitor pargylline had previously been used to validate this fixed-cell ELISA (data not shown).
Compound 119913-X had no statistically significant effects in this assay ( Fig. 2a ), which may be due to a high enzyme to growth inhibition (IC50/GI50) ratio limiting cellular uptake and intracellular activity. In contrast, the IC50/GI50 ratio for 610930-N was lower (~3), building some confidence that growth inhibition may not be due to off-target effects. The effects on H3K4 monomethylation were greater than for 119913-X. Although at 24 h, methylation appeared to be reduced (p < 0.5) by 610930-N at the lowest concentration used (52 µM), this effect was not observed at 104 µM. In contrast and more encouragingly, monomethylation was reduced in a concentration-dependent manner at 48 h, with this effect reaching significance (p < 0.05) at 104 µM. As expected, an increase in acetylation at lysine H3K9 and methylation at H3K4 was observed in response to treatment with SAHA, which was used as a positive control for the assay ( Fig. 2b ).
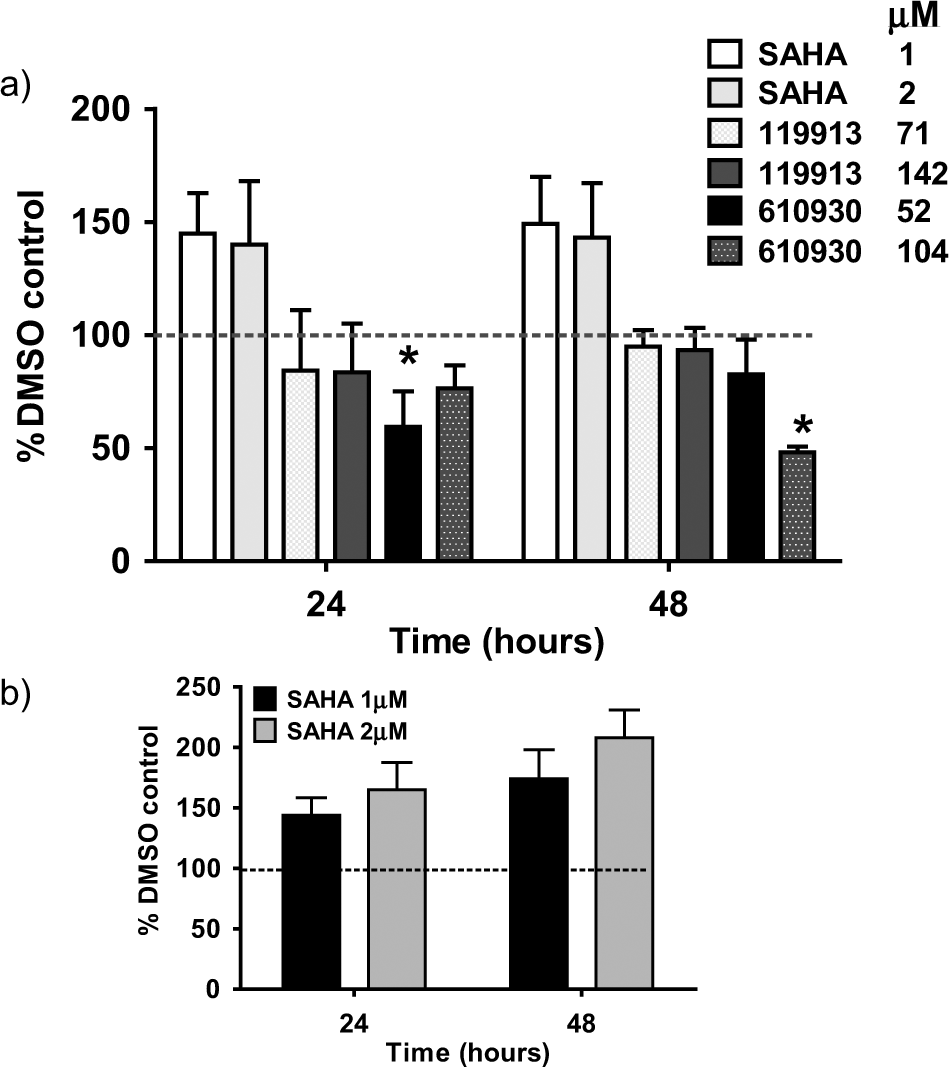
TRF-Cellisa mechanistic assay: (
Thus, 610930-N (2,8-bis(2,4-dihydroxy-6-methylphen ylamino)-1,9-dimethyl dibenzo[b,d]furan-3,7-dione) had antiproliferative activity and was associated with the mechanism expected of PMT inhibition in a cellular context (i.e., reduced H3K4 monomethylation). This compound, known as orsein, is one of a series of natural products used as nuclear counterstains (e.g., Sejben et al. 16 ) and food dyes and was shown to be modestly active across the cell line panels used by the NCI (http://dtp.nci.nih.gov/docs/dtp_search.html) in growth inhibition assays.
In summary, the PMT SET7/9 has been used for screening in high-throughput approximately 65 000 compounds that included the NCI Diversity Set. The assay was subsequently used by our collaborators to screen an additional 112 000 compounds with similarly acceptable assay performance and hit rates (0.03%). None of the hit matter identified in these screens was suitable for progression due to poor potency, unfavorable chemical properties, or lack of directional structure-activity relationship (SAR). However, it is of note that the assay identified inhibitors of different chemical classes, albeit of low potency. One compound identified from the NCI Diversity Set was modestly antiproliferative and showed the expected mechanistic effect on H3K4 methylation consistent with PMT inhibition.
Several screening assays, including that presented here, have been described to meet the increased drug discovery interest in PMT inhibitors as novel therapeutic agents. Alternative approaches to finding drug-like inhibitors of PMT enzymes would be to use in silico screens of large virtual libraries or biophysical screens of fragment libraries. Alternatively, because PMTs are known to form and to be active in multiprotein complexes in vitro, phenotypic cell-based screens, based on the DELFIA assay described here or an analogous high-content assay, to identify modulators of cellular or histone methylation could also be useful primary screening tools. With the aim of reversing aberrant transcriptional histone methylation marks, it is likely that attractive hit compounds that modulate these increasingly important classes of enzymes will be discovered by using the full range of contemporary screening strategies.
Footnotes
Acknowledgements
We are grateful to Professor Steve Gamblin for the supply of SET7/9 protein and to Dr. Jon Wilson, The Institute of Cancer Research, and colleagues at Chroma Therapeutics for advice and useful discussions. We thank Johannes Reynisson for molecular modeling.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: WA has acted as consultant and PW as Founder and Board Member for Chroma Therapeutics.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support was provided by Cancer Research UK (Programme Grants C309/A8274 and C309/A8365) and NHS funding to the NIHR Biomedical Research Centre. N-JF was awarded a PhD studentship funded by The Institute of Cancer Research.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.