
Abstract
PML is a multi-functional protein with roles in tumor suppression and host defense against viruses. When active, PML localizes to subnuclear structures named PML oncogenic domains (PODs) or PML nuclear bodies (PML-NBs), whereas inactive PML is located diffusely throughout the nucleus of cells. The objective of the current study was to develop a high content screening (HCS) assay for the identification of chemical activators of PML. We describe methods for automated analysis of POD formation using high throughput microscopy (HTM) to localize PML immunofluorescence in conjunction with image analysis software for POD quantification. Using this HCS assay in 384 well format, we performed pilot screens of a small synthetic chemical library and mixture-based combinatorial libraries, demonstrating the robust performance of the assay. HCS counter-screening assays were also developed for hit characterization, based on immunofluorescence analyses of the subcellular location of phosphorylated H2AX or phosphorylated CHK1, which increase in a punctate nuclear pattern in response to DNA damage. Thus, the HCS assay devised here represents a high throughput screen that can be utilized to discover POD-inducing compounds that may restore the tumor suppressor activity of PML in cancers or possibly promote anti-viral states.
Introduction
P
PML plays an essential role in both caspase-dependent and caspase-independent cell death. 2 pml–/– mice are resistant to apoptosis induced by numerous stimuli and have an increased tumor incidence. 4,5 PML contributes to cell death induced by γ-radiation, the primary treatment modality for a wide variety of tumors. 6 Interferons and arsenicals (e.g., As2O3) increase the number and size of PODs per cell and sensitize and/or induce apoptosis in a variety of tumor cell types. 7,8 Interestingly, PML confers direct resistance to many viruses, and numerous viruses have evolved mechanisms for disrupting POD formation. 9
Among the proteins that localize to PODs are p53, Daxx, and RecQ DNA helicase. The tumor suppressor p53 requires acetylation by CBP/p300 at PODs for the induction of apoptosis. 2,10 Daxx localization to PODs increases PTEN nuclear localization and PTEN tumor suppressor activity by inhibiting HAUSP-mediated PTEN deubiquitinylation. 11 PTEN nuclear exclusion is associated with cancer progression, and HAUSP overexpression coincides with PTEN nuclear exclusion in prostate cancer. 11 Daxx also represses the transcription of various antiapoptotic Rel-B-associated genes, including cIAP2, cFLIP, and Bfl-1 (A1), via histone deacetylase (HDAC) and DNA methyltransferase binding and recruitment. 12,13 PODs are required for the maintenance of genomic stability in combination with proteins such as RecQ DNA. 14 Notably, PML also suppresses the anchorage-independent growth of transformed cells, is required for hypophosphorylated Rb-mediated cell cycle arrest, and inhibits neoangiogenesis in human and mouse tumors. 1,15
The fundamental roles of PODs and POD-related proteins in tumor suppression validate targeting PODs for drug discovery. POD formation is essential in interferon and arsenical cancer therapy, especially for leukemias and multiple myeloma. 2,16,17 Interferons and arsenicals, however, induce a plethora of toxic effects, limiting their effectiveness. 17,18 The objective of the current study was to develop a high-content screening (HCS) assay for the high-throughput identification of chemical activators of PODs.
Materials and Methods
Cell culture
HeLa cells were originally obtained from ATCC (Manassas, VA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA), with 10% fetal bovine serum (FBS; Clontech, Mountain View, CA) and penicillin-streptomycin (diluted according to the manufacturer’s specifications; Invitrogen) at 37 °C, 5% CO2. PPC-1 cells were cultured similarly but with RPMI 1640 (Invitrogen) instead of DMEM.
Compounds
The LOPAC1280 collection of 1280 pharmacologically active single compounds was obtained from Sigma-Aldrich (St. Louis, MO). The Torrey Pines Institute for Molecular Studies Combinatorial Libraries are mixture-based libraries in positional scanning format, and they were dissolved in dimethylformamide (DMF). 19-21
Immunofluorescence assay
In total, 3150 cells/well (50-µL/well volume) were seeded onto 384-well clear-bottom plates (Greiner Bio-One, Monroe, NC) using the Matrix WellMate liquid dispenser (Thermo Fisher Scientific, Hudson, NH) and incubated at 37 °C (5% CO2). After 24 h, the Biomek FX Laboratory Automation Workstation (Beckman Coulter, Fullerton, CA) was used to add interferon-γ (IFN-γ; 4 U/µL with either 0.1% DMSO or DMF; R&D Systems, Minneapolis, MN), DMSO (0.1% final concentration; Sigma-Aldrich), DMF (0.1% final concentration; Sigma-Aldrich), or compounds. After 12 h, cells were washed with phosphate-buffered saline (PBS), fixed with 4% formaldehyde (Sigma-Aldrich) for 15 min, washed, permeabilized with 0.5% Triton X-100 (Sigma-Aldrich) for 5 min, washed, incubated with the primary antibody diluted to 0.5 µg/mL in 5% bovine serum albumin (BSA; Thermo Fisher Scientific) for 1 h, washed, incubated with the secondary antibody diluted to 5 µg/mL in 5% BSA for 1 h, washed, and placed in a 100-ng/mL DAPI-PBS solution (Invitrogen) overnight. Each wash step was a multiple fluid change using the Titertek MAP-C II Microplate Washer and Stacker (aspiration to 10 µL, 50 µL PBS addition, repeated a total of 3 times; Titertek, Huntsville, AL).
For PML immunostaining, mouse monoclonal antihuman PML primary antibody (PG-M3, Santa Cruz Biotechnology, Santa Cruz, CA) was used with an Alexa Fluor 488 chicken antimouse IgG secondary antibody (Invitrogen). For phospho-H2AX (Ser139; p-H2AX or γ-H2AX) or phospho-Chk1 (Ser317) immunostaining, rabbit polyclonal antihuman phosphohistone H2A.X (Ser139; diluted 1:400; Cell Signaling Technology, Danvers, MA) or phospho-Chk1 (Ser317; diluted 1:400; Cell Signaling Technology) primary antibodies were used, respectively, with an Alexa Fluor 568 goat antirabbit IgG secondary antibody (Invitrogen).
High-content imaging
Plates were imaged using the Beckman Coulter Cell Lab IC-100 Image Cytometer with a 40× 0.6NA ELWD Plan Fluor dry (air) objective (6 images/well). The images (>200 cells/well) were analyzed using the POD detection algorithm, which was developed based on CytoShop (Beckman Coulter) and MATLAB (MathWorks, Natick, MA) software.
HCS was performed at the Conrad Prebys Center for Chemical Genomics at the Sanford-Burnham Medical Research Institute (La Jolla, CA).
Statistical analyses
HCS performance was characterized using the following equation: Z′ factor = 1 − (3σpositive + 3σnegative)/│(µpositive − µnegative)│, where σpositive is the standard deviation of the positive control, σnegative is the standard deviation of the negative control, µpositive is the mean of the positive control, and µnegative is the mean of the negative control. 22
Results
Development and optimization of the PML-POD localization assay
Immunostaining conditions were optimized for detection of PML using a commercially available mouse monoclonal antibody. HeLa cells were seeded in 384-well plates, treated with either DMSO or IFN-γ for 24 h, and immunofluorescently stained for PML to confirm IFN-γ-induced PML localization into PODs ( Fig. 1A ). IFN-γ-induced localization of PML into PODs is accompanied by the localization of various other proteins to PODs, such as Daxx. 23 Thus, simultaneous immunofluorescence detection of both PML and Daxx in HeLa cells confirmed co-localization/formation of PODs and validated IFN-γ as a positive control ( Fig. 1B ). IFN-γ-induced PML and Daxx co-localization to PODs was also confirmed to occur in PPC-1 cells (observed by immunofluorescence; data not shown).
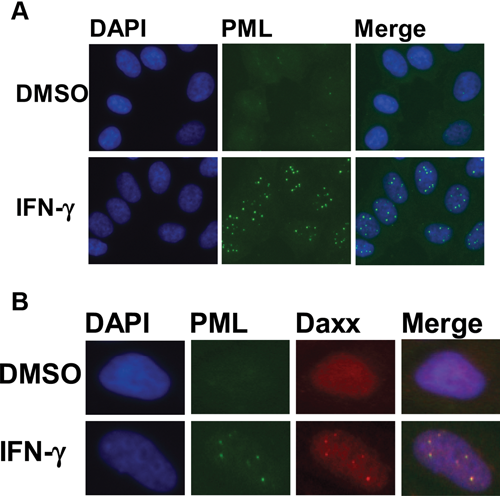
PML and Daxx localize to PML oncogenic domains (PODs) after interferon-γ (IFN-γ) treatment. (
To quantify the extent of POD formation (i.e., the number of PODs per cell, the intensity of PML localization, and the fraction of cells per well with extensive numbers of PODs) in an automated fashion, the “POD detection algorithm” was developed using Beckman Coulter CytoShop and MathWorks MATLAB software ( Fig. 2A ). First, the nuclear image (DAPI stain) was used to produce a “nuclear mask,” which identified all nuclei in an image. This nuclear mask was applied to the PML (“green”) image, and all green pixels outside of this nuclear mask were eliminated. Next, PODs were outlined based on the identification of green pixels with higher intensities than their surrounding pixels (using CytoShop’s “Aggregate Detection”), with the minimum size of a POD defined based on IFN-γ control wells (CytoShop’s “Object Scale”). This number of detected PODs was then reported on a per nucleus basis and used to determine the percentage of nuclei per image that were “POD positive.” The value for the number of detected PODs above which a nucleus is considered POD positive was determined to be 4.0 by iteratively setting increasing threshold values and determining the Z′ factor for each threshold (on control plates).
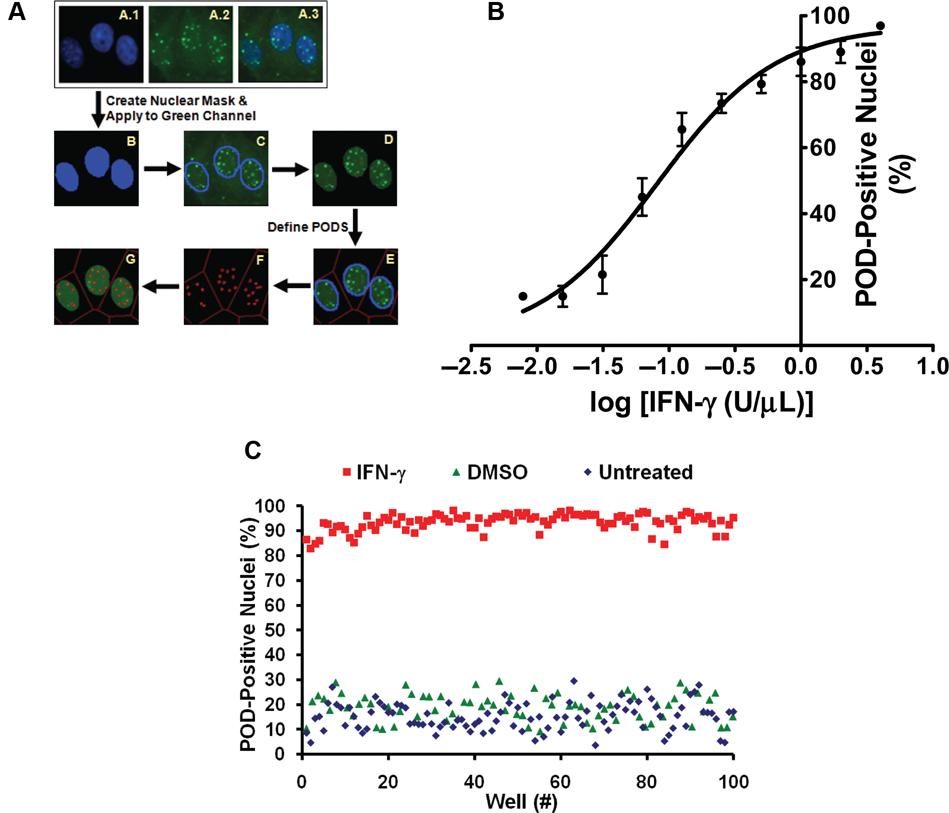
High-content screen (HCS) development and optimization. (
To both further validate the algorithm, HeLa cells were treated with increasing concentrations of IFN-γ, stained with anti-PML antibody, and imaged, and the percentage of POD-positive cells was determined (
Fig. 2B
). Using this automated method, IFN-γ was determined to induce concentration-dependent POD formation in HeLa cells. Manual counting of POD-positive cells confirmed the algorithm-defined quantification (data not shown). Examples of whole-field images from single wells (multiple images from single wells) are shown in Supplemental Figure S1 (
To characterize the reproducibility of the assay, multiple replicates were prepared (n > 80 per condition), where cells were seeded into a 384-well plate using the Matrix Wellmate bulk liquid dispenser (3150 cells/well); treated with IFN-γ (4 U/µL), DMSO (0.1%), or nothing for 12 h; and immunostained for PML, imaged, and analyzed. The Z′ factor was determined to be 0.64 or 0.65 using DMSO or untreated cells, respectively, as the negative control ( Fig. 2C ).
High-content screening
The HCS assay was used to screen the LOPAC1280 library of pharmacologically active compounds for PML activators ( Fig. 3A ). Compounds that increased the number of POD-positive nuclei by at least 50% (relative to the controls), as measured by the POD detection algorithm, were considered hits. No hits were identified when the library was screened at 5 µM. Moreover, dose-response curves (serial dilutions performed from 250 µM to 0.25 µM) were generated for the 5 LOPAC1280 compounds inducing the most POD positivity, and the highest value observed was less than 40% POD-positive nuclei (data not shown). Thus, the HCS assay is not promiscuous with respect to hit rate.
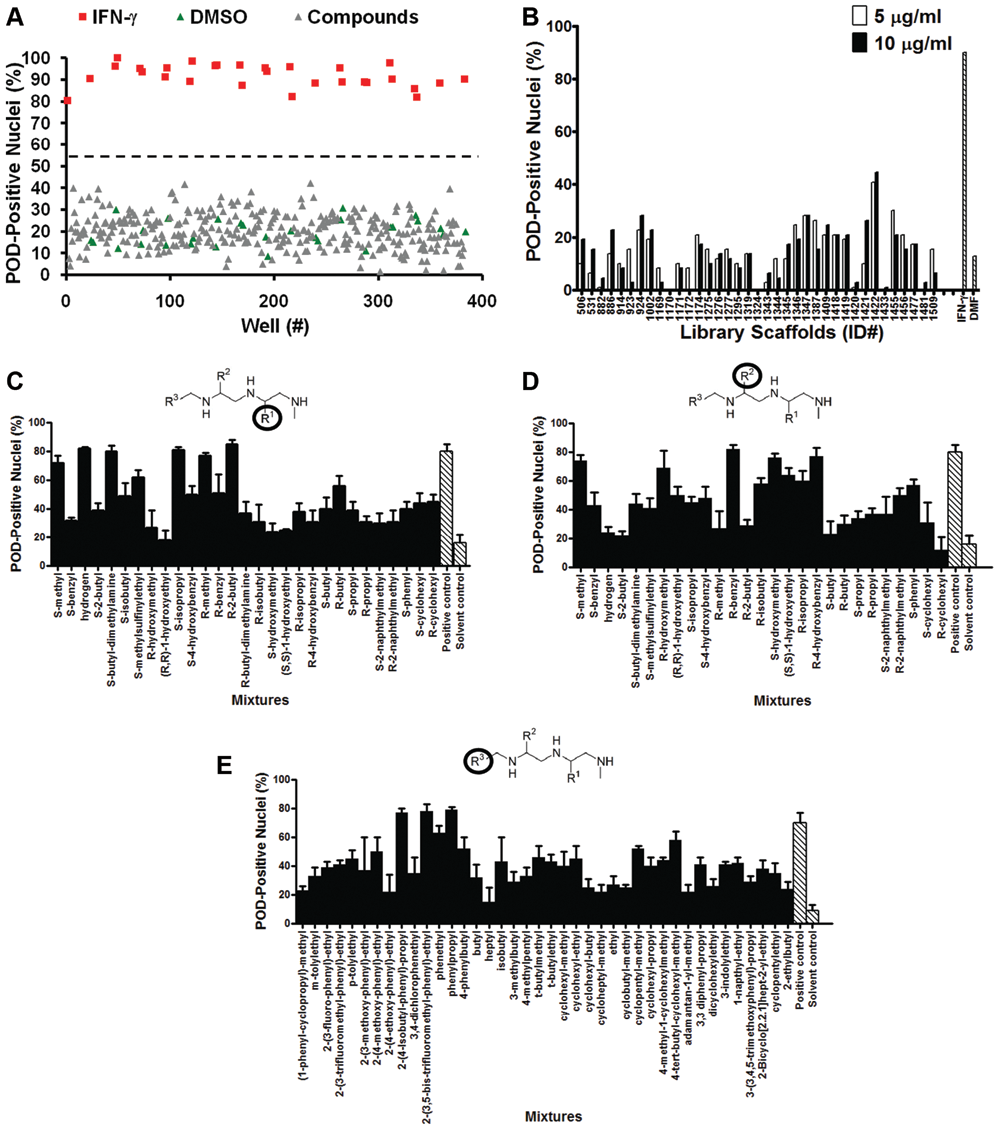
High-content screening (HCS) for chemical activators of PML oncogenic domains (PODs). (
The TPIMS mixture-based combinatorial libraries were formatted and plated as a “scaffold ranking library,” which was subsequently screened to determine the most active chemical scaffolds before further screening/deconvolution of mixtures. 24 The scaffold ranking library was formatted as 38 mixtures (dissolved in DMF), grouped according to scaffold, and represented 5,287,896 compounds (and several million peptides). Each mixture was present in 2 different wells at 2 different concentrations. Use of mixtures makes it readily feasible to screen large collections of compounds. This library was screened at 5 µg/mL (~10 µM for average molecular weight [MW] ≈500 g/mol) and 10 µg/mL (~20 µM for average MW ≈500 g/mol; Fig. 3B ). The most active mixture was “1422,” which induced >40% POD-positive cells.
The 1422 library contains compounds with an N-methyl triamine scaffold in which chemical diversity was created at 3 positions, R1, R2, and R3. Analysis of a collection of N-methyl triamines, at which one of these diversity positions was fixed, allowing the others to vary as a mixture of all possibilities built into the combinational library, revealed substituents at R1, R2, or R3 that induced >50% POD-positive cells when tested at 4 µg/mL (~8 µM; Fig. 3C - E ). Successful implementation of the HCS assay for POD inducers for structure-activity relation (SAR) analysis of the N-methyl triamine combinatorial library demonstrates the robust performance of the assay.
POD-inducing specificity
To complement the HCS assay for POD activation, we also devised 2 HCS counterscreen assays using immunostaining for the DNA damage/repair–related proteins, H2AX and Chk1. H2AX is phosphorylated primarily by ATM, which “senses” double-stranded DNA breaks, whereas Chk1 is phosphorylated primarily by ATR, which “senses” single-stranded DNA breaks (a common intermediate found at sites of DNA damage detection and repair pathways). 25 Immunofluorescence staining for phospho-H2AX (Ser139) (γ-H2AX) and phospho-Chk1 (Ser317), which are visible as nuclear aggregates, was used as a counterscreen to test the specificity of the most active 1422 compounds. The similarity in antigen redistribution for H2AX and Chk1 compared to PML provides the basis for counterscreens that eliminate false-positive compounds that might affect PML or alter immunostaining patterns in a nonspecific manner. In addition, because some types of DNA-damaging agents can stimulate POD formation, 25-29 the phospho-H2AX and phospho-Chk1 immunofluorescence counterscreens serve to eliminate compounds that operate as DNA-damaging agents and thus eliminate these from further consideration.
For these counterscreen HCS assays, CytoShop algorithms were used to quantify the intensity of nuclear phospho-H2AX (Ser139) (γ-H2AX) or phospho-Chk1 (Ser317) immunofluorescence. As shown in
Figure 4
(and
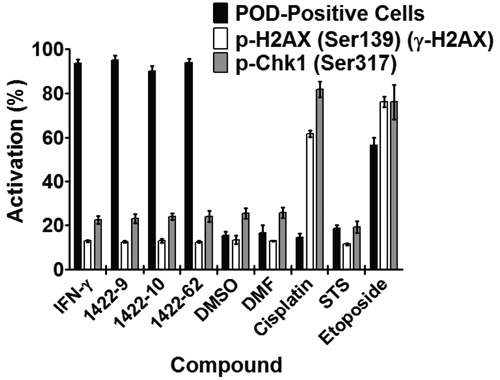
PML oncogenic domain (POD)–inducing N-methyl triamines do not induce DNA damage. HeLa cells were treated for 12 h with interferon-γ (IFN-γ; 4 U/µL), DMSO (0.1%), dimethylformamide (DMF; 0.1%), cisplatin (25 µM), staurosporine (STS; 25 nM), etoposide (25 µM), or 1422 compounds (N-methyl triamines; 10 µM). Cells were then immunostained for PODs (mouse monoclonal antihuman PML, Alexa Fluor 488 chicken antimouse antibodies), phospho-H2AX (rabbit polyclonal antihuman phospho-H2AX-Ser139, Alexa Fluor 568 goat antirabbit antibodies), or phospho-Chk1 (rabbit polyclonal antihuman phospho-Chk1-Ser317, Alexa Fluor 568 goat antirabbit antibodies). POD-positive nuclei (%) were quantified as previously described. p-H2AX and p-Chk1 nuclear staining intensity was quantified using CytoShop. Mean and standard deviation are shown (n = 4 wells/condition, >200 cells imaged and quantified per well).
Discussion
The current study describes an HCS assay for compounds that induce PML to localize to PODs, subnuclear structures involved in a variety of tumor-suppressive pathways, and host defense against some types of viruses. After developing algorithms for automated analyses of POD formation, the LOPAC1280 and TPIMS combinatorial libraries were screened. No hits were identified among the 1280 pharmacologically active LOPAC1280 compounds, possibly providing evidence that the HCS assay is highly specific and does not suffer from promiscuous reactivity. Screening of the TPIMS combinatorial libraries, which consist of mixtures representing 5,287,896 compounds and several million peptides, revealed a bioactive N-methyl triamine mixture. Further SAR evaluations where library deconvolution was performed by the positional scanning method revealed a clear SAR and thus validated the HCS assay for detection of POD-inducing compounds. Furthermore, N-methyl triamines that were active in the POD assay did not affect the distribution of control proteins, H2AX and Chk1, demonstrating specificity.
Some types of DNA damage have been shown to induce the formation of PML-type bodies that are thought to be functionally different from the tumor-suppressive and antiviral PODs mentioned in the current study. 25-29 Thus, the HCS counterscreen assays described here eliminate DNA-damaging compounds from further consideration.
Active compounds such as the N-methyl triamines could potentially induce PODs via several different mechanisms. First, IFN-α, -β, and -γ induce POD formation by upregulating PML and various POD-associated proteins. 9 Thus, compounds might induce interferon production, a mechanism that can be readily determined by measuring interferon elaboration into culture supernatants (e.g., using immunoassays) or by testing activation of interferon-inducible reporter genes (e.g., ISGE-luciferase). Second, arsenicals have been shown to activate PML by directly conjugating cysteines within the zinc fingers of the PML RBCC domain, 30 and thus compounds that covalently modify these sites define an additional mechanism for POD activation. Third, conjugation of PML by the ubiquitin-like protein SUMO is required for POD formation. 31 Hence, compounds that affect SUMOylation represent another potential mechanism, which might include, for instance, inhibitors of the proteases responsible for de-SUMOylation. Finally, PML binding proteins such as Daxx are regulated by phosphorylation. 32 The protein kinase ZIPK, for example, has been shown to be required for interferon to induce POD formation. 28 Thus, compounds that influence the relevant kinases or phosphatases represent another potential class of POD activators that might be revealed by our HCS assay.
Because of the role of PODs in tumor suppression and host defense against viruses, POD-inducing compounds may have relevance to a wide variety of cancers and infectious diseases. Arsenicals are already being used to treat leukemias (such as APL) and multiple myeloma. 2,16 In addition, adenoviruses, herpes simplex virus–1, human cytomegalovirus, Epstein-Barr virus, papillomavirus, hepatitis D virus, human T-lymphotropic virus–1, lymphocytic choriomeningitis, and rabies viruses disrupt PODs, 9 suggesting that POD-inducing compounds may also promote antiviral states that could be therapeutically useful. The HCS assay described here enables high-throughput screening (HTS) for compounds with potential medicinal activity based on induction of PODs, thus providing a route to chemical modulators of PML that accommodates the diversity of cellular mechanisms responsible for POD regulation in a manner unachievable with standard biochemical HTS assays.
Footnotes
Acknowledgements
We thank Tessa Siegfried and Melanie Hanaii for assistance with manuscript preparation, as well as Drs. Paul Diaz and Satoshi Ogasawara for technical assistance. Support by DoD grant W81XWH-08-0574 and NIH grant CA-55164.