
Abstract
Autophagy is an evolutionarily conserved process for catabolizing damaged proteins and organelles in a lysosome-dependent manner. Dysregulation of autophagy may cause various diseases, such as cancer and neurodegeneration. However, the relevance of autophagy to diseases remains controversial because of the limited availability of chemical modulators. Herein, the authors developed a fluorescence-based assay for measuring activity of the autophagy protease, autophagin-1(Atg4B). The assay employs a novel reporter substrate of Atg4B composed of a natural substrate (LC3B) fused to an assayable enzyme (PLA2) that becomes active upon cleavage by this cysteine protease. A high-throughput screening (HTS) assay was validated with excellent Z′ factor (>0.7), remaining robust for more than 5 h and suitable for screening of large chemical libraries. The HTS assay was validated by performing pilot screens with 2 small collections of compounds enriched in bioactive molecules (n = 1280 for Lopac™ and 2000 for Spectrum™ library), yielding confirmed hit rates of 0.23% and 0.70%, respectively. As counterscreens, PLA2 and caspase-3 assays were employed to eliminate nonspecific inhibitors. In conclusion, the LC3B-PLA2 reporter assay provides a platform for compound library screening for identification and characterization of Atg4B-specific inhibitors that may be useful as tools for interrogating the role of autophagy in disease models.
Introduction
A
Roles for autophagy have been suggested in either causation or prevention of disease. For example, autophagy has been implicated in eliminating senescent and insoluble proteins that are not degraded via proteasome-based mechanisms, representing an important “housekeeping” function for some types of cells, such as neurons, where it may be helpful for preventing protein inclusion body diseases associated with neurodegeneration. 7,8 Autophagy also plays helpful roles in the context of host-pathogen interactions, where autophagic vesicles help to surround intracellular virus particles and bacteria, targeting them for lysosomal destruction. 9,10 In contrast, autophagy may promote the pathogenesis and progression of tumors, allowing cancer cells to survive nutrient-poor and hypoxic environments—such as those found in the centers of rapidly growing malignant lesions that have outstripped their vascular supply. 11 Conversely, some components of the autophagy machinery have also been reported to be required for nonapoptotic cell death, suggesting the concept of autophagic cell death. 12 Altogether, the role of autophagy in disease remains largely controversial.
Chemical inhibitors of autophagy are essentially nonexistent, with the few agents available for aiding research. The compound 3-methyladenine (3-MA) has been touted as an autophagy inhibitor but requires millimolar concentrations to inhibit class III phosphatidyl inositol kinases (PI3Ks) involved in autophagy. 13 Bafilomycin A1 is also commonly employed for suppressing autophagic flux due to its inhibition of the vacuolar type H+-ATPase (V-ATPase), blocking acidification of lysosomes and endosomes. However, long-term treatment (>4 h) of cells with bafilomycin A1 also interferes the trafficking of proteosomes, endosomes, and other cellular processes. 14,15 A need therefore exists for chemicals that target specific components of the autophagy machinery for use as research tools for addressing questions about autophagy mechanisms and investigating the role of autophagy in diseases.
Autophagins are a class of cytosolic cysteine proteases required for autophagy. 16 Autophagins cleave Atg8 to promote its conjugation with phosphatidylethanolamine (PE) in the membranes of autophagic vesicles by a ubiquitin-like system, which is required for autophagosome formation and participates in targeting these vesicles to lysosomes for fusion and degradation of their contents. 17 Autophagins also promote deconjugation of Atg8-PE to liberate Atg8 from membrane at or before the final stage of fusion between autophagosome and lysosomes, suggesting that deconjugation of Atg8-PE is required for the fusion. 18 The human genome contains 4 independent genes encoding the Atg4 orthologs (termed autophagins), including Atg4A/autophagin-2, Atg4B/autophagin-1, 4C/autophagin-3, and 4D/autophagin-4. 16 Atg4B cleaves most of human Atg8 homologs in vitro, including (1) microtubule-associated light chain 3 (LC3; which has A, B, and C isoforms), (2) GABA(A) receptor–associated protein-like 1 (GABARAP-L1), (3) GABA(A) receptor–associated protein (GABARAP), and (4) Golgi-associated ATPase enhancer of 16 kDa (GATE-16, also known as GABA(A) receptor–associated protein-like 2). 19-21 In contrast, Atg4A has activity mainly for GATE-16 in vitro. 22 Atg4B knockout mice show reduction of proteolysis of all Atg8 homologs except GATE-16, suggesting that Atg4A compensates for cleavage of GATE-16 in Atg4B knockout mice. 21 By comparison, Atg4C and Atg4D show minor protease activity or require posttranslational modification (caspase-3 cleavage) for activation, 20,23 indicating Atg4B is functionally dominant in the regulation of autophagy.
In this study, we have selected autophagin-1/Atg4B as a target for developing novel inhibitors of autophagy. We devised a Atg4B high-throughput screening (HTS) assay using a LC3B-PLA2 reporter substrate (Shu, 2010 #16880) in 384-well format that has excellent performance characteristics (Z′ factor >0.7) and verified by pilot library screening and counterscreens that the assay is suitable for HTS of chemical libraries.
Experimental Procedures
Plasmid constructions
Plasmids for bacterial expression of autophagin-1 (Atg4B) and substrate protein LC3B were constructed as previously described. 19 Briefly, PCR products were cloned into bacterial expression vector pETDuet-1 (Novagen, San Diego, CA) in frame with an N-terminal His tag. The mature human phospholipase A2 (PLA2) group X G10 (amino acids 43-165) was amplified from a human fetal brain cDNA library (Open Biosystems, San Diego, CA) and fused with LC3B in pETDuet-1. Site-directed mutagenesis of Atg4B C74A or LC3B G120A was carried out by the QuickChange kit according to the manufacturer’s instructions (Stratagene, La Jolla, CA).
Protein expression and purification in Escherichia coli
Atg4B or catalytic mutant Atg4B C74A expression plasmid was transformed into Escherichia coli BL21 (DE3; Invitrogen, San Diego, CA). Protein expression was induced by 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG; Invitrogen) at 15°C for 6 h. His6-tagged proteins were purified using Ni-NTA-agarose (Qiagen, Valencia, CA) and eluted with a 20- to 250-mM imidazole gradient in 20 mM Tris (pH 8.0), 150 mM NaCl, and 10 mM β-mercaptoethanol.
His6-tagged LC3B-PLA2 protein was induced by 1 mM IPTG at 37°C for 4 h to obtain inclusion bodies. The inclusion bodies were solubilized with 20 mM Tris buffer (pH 8.0) containing 0.5 M NaCl, 5 mM imidazole, 6 M guanidine hydrochloride, and 1 mM β-mercaptoethanol and loaded into the Ni-NTA column. The denatured proteins were refolded with refolding buffer (20 mM Tris [pH 8.0], 0.5 M NaCl, 20 mM imidazole, 1 mM β-mercaptoethanol) in the presence of linear 6-0 M gradient urea. The refolded proteins were eluted with a 20- to 500-mM gradient imidazole in 20 mM Tris (pH 8.0), 150 mM NaCl, and 10 mM β-mercaptoethanol.
For production of active PLA2 protein, LC3B-PLA2 fusion protein (2.5 µM) was incubated with Atg4B (100 nM) in PLA2 reaction buffer (20 mM Tris-HCl [pH 8.0], 2 mM CaCl2, and 1 mM dithiothreitol [DTT]) at room temperate for 16 h. The cleaved PLA2 protein was purified with Ni-NTA column (Qiagen) to remove His6-tagged LC3B and Atg4B. Caspase-3 protein was purified as described previously. 24,25
Atg4B cleavage of synthetic protein substrates
Atg4B (wild-type [WT]) or catalytic mutant Atg4B C74A was incubated with 400 nM protein substrates in a 50-µL reaction buffer containing 50 mM Tris (pH 8.0), 150 mM NaCl, and 1 mM DTT at 37°C for 1 h. Reactions were stopped by addition of 50 µL of 2× Laemmli sodium dodecyl sulfate (SDS) sample and subjected to 13% SDS–polyacrylamide gel electrophoresis (PAGE) followed by GelCode Blue staining (Thermo Fisher Scientific, Asheville, NC) or transfer to nitrocellulose membranes (Sigma-Aldrich, St. Louis, MO) for immunoblotting analysis using anti-myc antibody (Roche, Indianapolis, IN). Proteins were detected using peroxidase-conjugated antimouse IgG secondary antibody by Super Signal and chemiluminescence substrate (Pierce, Rockford, IL) with exposure to X-ray film.
Atg4B activity measured using LC3B-PLA2 substrate
The assay was modified from a previous report. 26 Briefly, recombinant Atg4B was mixed with LC3B-PLA2 fusion protein to a final volume of 20 µL PLA2 reaction buffer (20 mM Tris-HCl [pH 8.0], 2 mM CaCl2, and 1 mM DTT) containing 20 µM 2-(6-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino) hexanoyl-1-hexadecanoyl-sn-glycero-3-phosphocholine (NBD-C6-HPC) (Invitrogen) in wells of black-walled 384-well plates (Greiner Bio-One, Frickenhausen, Germany). Fluorescence intensity was measured for various times using an Analyst™ HT (Molecular Devices, Sunnyvale, CA) at room temperature with excitation and emission wavelengths of 485 and 530, respectively. Data were analyzed by GraphPad Prism software (GraphPad Software, San Diego, CA). To determine robustness of the assay, Z′ factor was calculated as a defined equation: 1 – 3(SD+Atg4B + SD−Atg4B)/mean+Atg4B – mean−Atg4B, where SD is the standard derivation of signal for both positive (+Atg4B) and negative (−Atg4B) controls. 27
High-throughput screening
For HTS, 20 nL of each compound (10 mM in DMSO) of 1280 compounds from the LOPAC™ library (Sigma-Aldrich) or 2000 compounds from the Spectrum™ library (Spectrum Chemicals & Laboratory Products, Gardena, CA) were plated into black 384-well, round-well plates using a Labcyte Echo 550 acoustic pipetter (Agilent Technologies, Wilmington, DE). Recombinant Atg4B (0.2 nM, 10 µL) was dispensed to each well using ThermoScientific Matrix WellMate bulk dispenser (Thermo Fisher Scientific). LC3B-PLA2 fusion protein (50 nM, 10 µL) diluted in 2× PLA2 reaction buffer (40 mM Tris-HCl [pH 8.0], 4 mM CaCl2, and 2 mM DTT) containing 40 µM NBD-C6-HPC (Invitrogen) was then added to each well. Fluorescence intensity was measured within 30 to 60 min using an Analyst™ HT (Molecular Devices) at room temperature with excitation and emission wavelengths of 485 and 530, respectively. The inhibition of compounds was recorded as percentage compared to DMSO control. Twofold serial dilutions (maximal final concentration 100 µM) of compounds were used for determination of dose-responsive curves. IC50 values were calculated by nonlinear regression analysis employing GraphPad Prism software (GraphPad Software).
Counterscreens for PLA2 and caspase-3 assay
For the PLA2 assay, 20 nL of each hit (10 mM in DMSO) from LOPAC™ or Spectrum™ libraries was plated into black 384-well, round-well plates using a Labcyte Echo 550 acoustic pipetter (Agilent Technologies). Active PLA2 (500 nM, 10 µL) was dispensed to each well, then mixed with NBD-C6-HPC (Invitrogen; 20 µM, 10 µL) in PLA2 reaction buffer at room temperature for 2 h. Fluorescence intensity was measured with excitation and emission wavelengths of 485 and 530, respectively. Caspase-3 assay was determined using methods modified from a previous report. 25 Briefly, His6-tagged caspase-3 (1 nM, 10 µL) was mixed with acetyl-Asp-Glu-Val-Asp-AFC (200 µM, 10 µL) in assay buffer (50 mM HEPES [pH 7.4], 10% sucrose, 1 mM EDTA, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate [CHAPS], 100 mM NaCl, and 1 mM DTT) in wells of white 384-well plates containing 20 nL hit compound (10 mM in DMSO). Fluorogenic AFC product was measured at 37°C for 1 h with excitation and emission wavelengths of 405 and 510, respectively.
Results
Development of Atg4B assay based on LC3B-PLA2 fusion protein substrate
To develop a primary HTS assay for autophagins, we generated an expression plasmid for production of LC3B-PLA2 fusion protein (substrate) in bacteria. The enzyme phospholipase A2 (PLA2) is inactive when expressed as a fusion protein with polypeptide appendages added to its N-terminus (
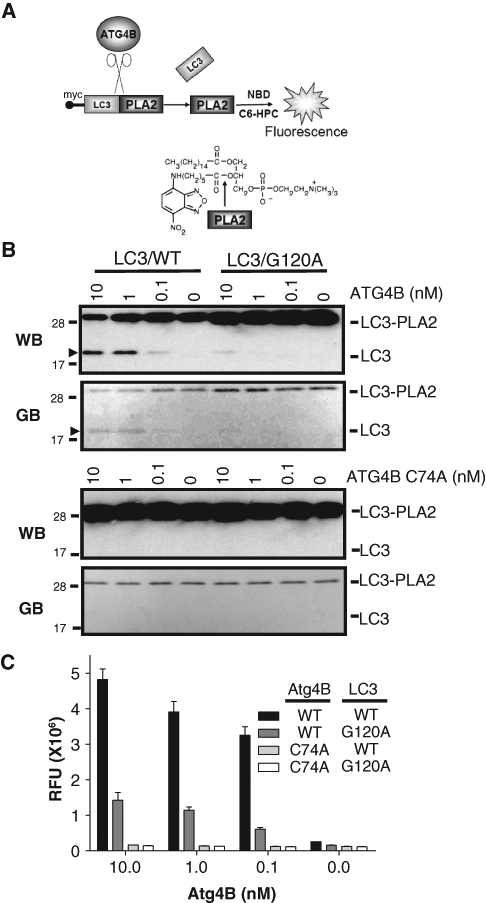
Generation of Atg4B assay using LC3B-PLA2 substrate. (
We also explored the reactivity of Atg4B on a mutant of LC3B-PLA2 in which the glycine residues at which LC3B cleavage normally occurs was converted to alanine. Atg4B only slightly cleaved the LC3B-PLA2 (Gly/Ala) mutant as measured by generation of PLA2 activity (
To determine the optimal concentration of Atg4B and LC3B-PLA2 for the assay, the concentration of Atg4B was titrated by 2-fold serial dilution from 1250 to 0 pM (
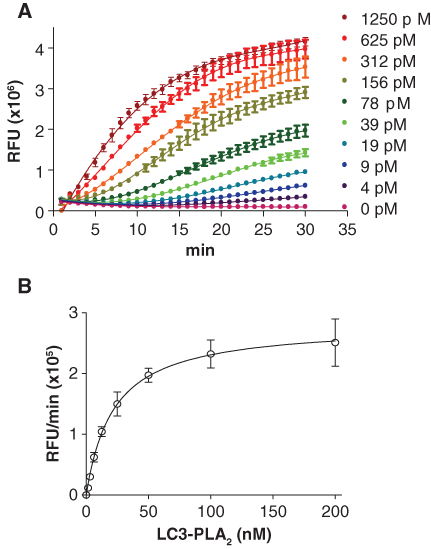
Determination of optimal concentrations of Atg4B and LC3B-PLA2 for the high-throughput screening assay. (
HTS implementation of the LC3B-PLA2 reporter assay for Atg4B
To study assay performance for the HTS environment, the Atg4B assay was tested in 384-well plates using 96 wells each of (a) the negative control (without Atg4B); (b) the positive control (Atg4B without inhibitors); (c) a control for protease inhibition (Atg4B with general cysteine protease inhibitor N-ethylmaleimide (10 mM), which covalently modifies the active site cysteine of proteases; and (d) EGTA (5 mM), a chelator of calcium, which is essential for PLA2 activity (
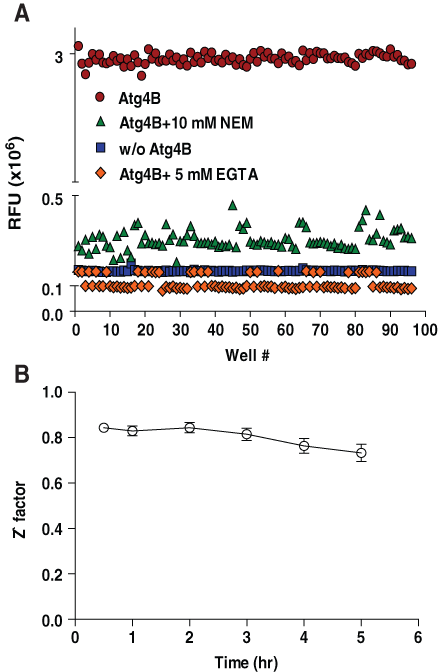
Characterization of LC3B-PLA2 reporter assay for high-throughput screening. (A) LC3B-PLA2 (25 nM) was incubated without (squares) or with (circles) Atg4B (0.1 nM) in the absence or presence of calcium chelator, EGTA (5 mM) (diamonds), or general cysteine protease inhibitor, N-ethylmaleimide (10 mM) (triangles), in PLA2 reaction buffer (20 mM Tris-HCl [pH 8.0], 2 mM CaCl2, and 1 mM dithiothreitol) containing 20 µM NBD-C6-HPC for 0.5 h using 96 replicate wells of 384-well plates for each condition. PLA2 activity was measured by fluorescence intensity with excitation and emission filters of 485 nm and 530 nm, respectively, and reporting results as relative fluorescence units (RFU). (
Screening and characterization of hits from the LOPAC™ and Spectrum™ libraries
Next, we undertook a pilot screen of a chemical library (LOPAC™) enriched in bioactive molecules to test the performance of the HTS assay (
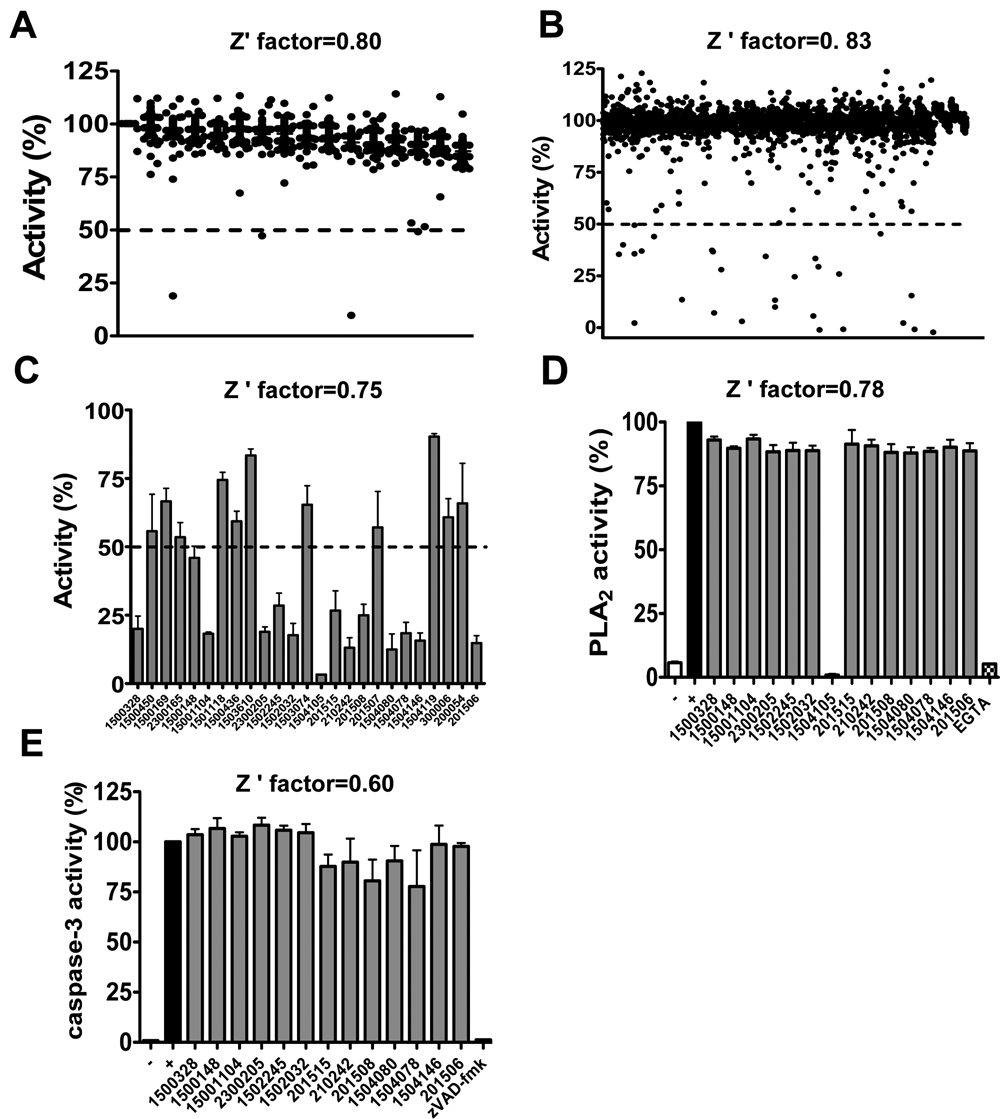
High-throughput screening (HTS) for Atg4B inhibitors—pilot library screens. (
We therefore also screened the Spectrum™ library, which consists of 2000 bioactive compounds. Again, the HTS assay was performed at room temperature for 1 h. After screening and confirmation, 14 of initial 25 hits were confirmed, thus representing a confirmed hit rate of 0.7% (
We also tested the hits with LCB-PLA2 reporter assay in kinetic mode using covalent inhibitor iodoacetamide (IAA) as a positive control, which attacks the active site cysteine of Atg4B (see
The hits were then cherry picked for dose-response testing (
IC50 Determination for Screening Hits Using the LC3B-PLA2-Based Assay
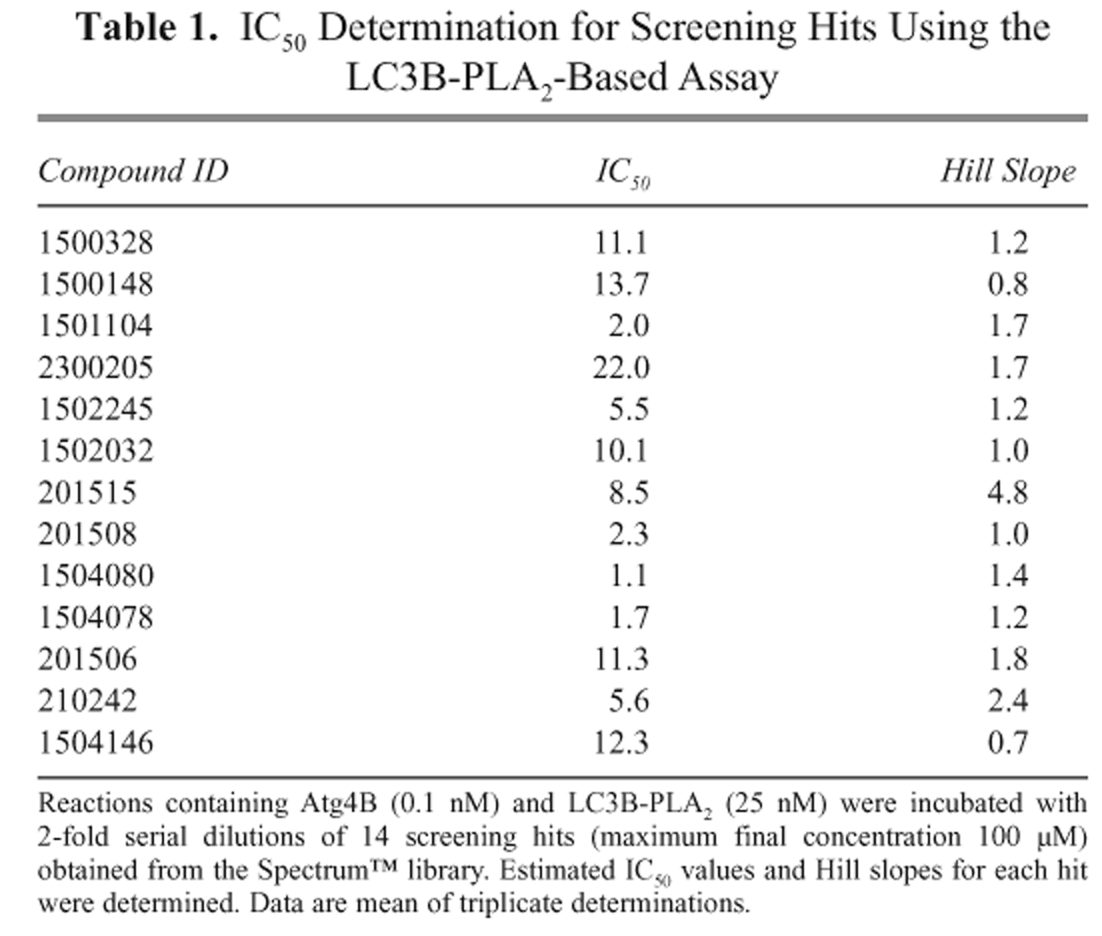
Reactions containing Atg4B (0.1 nM) and LC3B-PLA2 (25 nM) were incubated with 2-fold serial dilutions of 14 screening hits (maximum final concentration 100 µM) obtained from the Spectrum™ library. Estimated IC50 values and Hill slopes for each hit were determined. Data are mean of triplicate determinations.
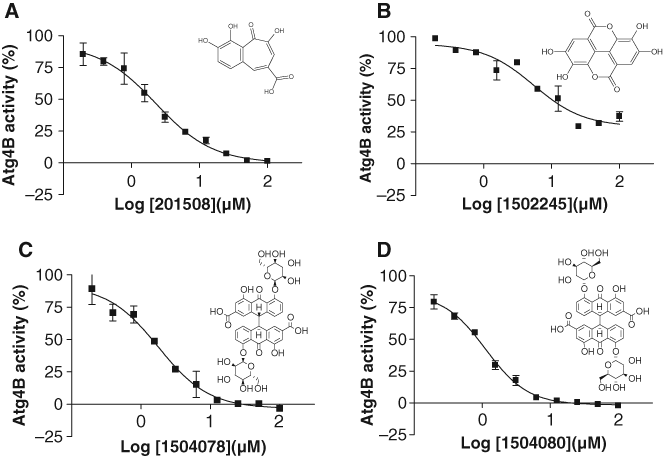
Dose-dependent inhibition of Atg4B by screening hits. Reactions containing Atg4B (0.1 nM) and LC3B-PLA2 (25 nM) were incubated with 2-fold serial dilutions of screening hits (maximum final concentration 100 µM), including (
Discussion
Autophagins play a crucial role in conjugation and deconjugation of Atg8 paralogs to autophagic vesicles and are indispensable for the process of autophagy. 31 Chemical inhibitors of autophagy are largely lacking, thus representing a void in the armamentarium of experimental approaches available to study this process. In this study, we have devised an HTS fluorogenic assay for measuring activity of autophagin-1 (Atg4B). The assay performs robustly in the 384-well environment with Z′ factors consistently >0.7. The signal-to-noise ratio and reproducibility of the assay are sustained for at least 5 h, which is appropriate for HTS applications. Moreover, acceptable hit rates were obtained in pilot screens of 3280 bioactive compounds at ~10 µM, with confirmed hit rates of 0.23% to 0.70%. Finally, the expression plasmids and methods for bacterial expression that we have described for recombinant Atg4B (protease) and LC3B-PLA2 (substrate) proteins provide excellent yields (~6 mg/L culture), thus making large-scale chemical library screening entirely feasible.
PLA2 has been used previously in other protease applications because of the inhibitory effect that N-terminal appendages have on this enzyme. 26 When expressed as a fusion protein with a cleavable N-terminus, PLA2 serves as a convenient reporter for monitoring protease activity. For example, ubiquitin-PLA2 fusion protein is cleaved by deubiquitin enzymes (DUBs) and efficiently produces active PLA2. The LC3B-PLA2 fusion protein substrate that we have recently described and characterized represents the first application of the PLA2 fusion protein approach to autophagy proteases. 19 Compared to AFC (7-amino-4-trifluoromethyl coumarin)–conjugated tetrapeptide substrates that we investigated in a previous study, 19 the catalytic efficiency of the LC3B-PLA2 substrate is 5.7 × 105 higher (kcat/Km 5.26 × 105 M−1/s−1). In this regard, the crystal structure of the LC3/Atg4B complex has shown that full-length LC3B induces conformational changes in Atg4B that promote formation of the active catalytic site, 32 thus presumably explaining why our LC3B-PLA2 protein substrate is far superior to peptide substrates. Thus, the LC3B-PLA2 substrate is not only more physiological but is also much more efficient from the perspective of enzyme kinetics.
We have also begun to create additional PLA2 fusion proteins with other autophagin substrates appended, including GATE-16 and GABARAP-L1, which can serve as alternatives to LC3B-PLA2 for some autophagins or as secondary assay reagents for independent confirmation of hits. In this regard, Atg4 members have differential substrate specificities. For example, Atg4A mainly cleaves GATE-16, whereas Atg4D cleaves both GATE-16 and GABARAP-L1. 20,22 Thus, GATE-16-PLA2 fusion protein may provide a suitable substrate for Atg4A and Atg4D assay development. Indeed, we have generated and successfully tested a GATE-16-PLA2 fusion protein for measuring activity of recombinant Atg4A, 19 using essentially the same assay conditions reported here (data not shown).
After counterscreening and elimination of false positives using a PLA2 counterscreen, 4 compounds were identified from among 3280 bioactive molecules, showing concentration-dependent inhibitory activity (IC50 <10 µM) against Atg4B but not an alternative class of cysteine protease, caspase-3. Further testing using our autophagin-1 HTS assay in the presence or absence of DTT eliminated 2 compounds (1504078 and 1504080) that showed additive inhibition in the presence of DTT, indicating that hydrogen superoxide generation in vitro may be partially involved in inhibition of Atg4B. 29,30 Three of the compounds (1502245, 1504078, and 1504080) are also reported hits in other HTS assays reported in the PubChem database, whereas another compound (201508) inhibits Shp2 tyrosine phosphatase, 33 showing promiscuous activity. In addition, these compounds have structures that are not promising for lead optimization, with 2 compounds representing polyphenols containing carboxylates (with or without internal cyclization to form lactone rings; 201508, 1502245) and 2 compounds representing stereoisomers of a natural product senoside glycosylated with pyranose sugars (1504078 and 1504080). Although none of these compounds is an attractive candidate for undertaking structure-activity relation (SAR) studies toward potent and selective autophagin inhibitors, these pilot screening results nevertheless provide proof-of-concept evidence that the HTS assay described here is capable of identifying compounds with the potential for selectivity against autophagins relative to other classes of cysteine proteases.
Autophagy plays cytoprotective roles in some cancer-relevant contexts. 29,30,32 In this regard, inhibition of autophagy has been reported to sensitize some tumor cells to chemotherapy, suggesting that chemical inhibitors of autophagins may provide novel candidates for cancer drug discovery. Silencing Atg5 or Atg7 enhances apoptosis in chronic myeloid leukemia (CML) cells treated with imatinib mesylate (IM). 34 Autophagy inhibitor, 3-MA, or siRNA against Atg7 also augments 5-fluorouracil (5-FU)–induced apoptosis in colon cancer cells in vitro and in vivo. 35 Inhibiting autophagy with the nonselective PI3K inhibitor wortmannin (which crossreacts on the class III PI3K of autophagy, Vps34), as well as siRNAs targeting Vps34, Beclin1, or Atg5, appears to be synergistic with gossypol, a natural product that inhibits Bcl-2 family proteins, in human breast cancer cell MCF-7. 36 In these cancer cell culture models, inhibition of autophagy synergizes with cytotoxic anticancer drugs, and thus it has been suggested that autophagy allows cancer cells to survive the stress of chemotherapy. Thus, we anticipate that Atg4B-specific inhibitors may have tumor growth inhibitory activity, as well as an ability to improve cytotoxic responses to chemotherapy. It is hoped that applications of the HTS assay reported here for screening large compound libraries will provide attractive chemical scaffolds for the pursuit of a novel class of anticancer agents based on inhibition of autophagy.
Footnotes
Acknowledgements
This work was supported by grants from National Institutes of Health (R01-AI-082629, R03-MH-090871, U54HG005033) and a fellowship grant to Dr. Shu from the SASS foundation for Medical Research (Rosyln, NY). We thank Tessa Siegfried and Melanie Hanaii for manuscript preparation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.